

Abstract
Endolymphatic sac tumors (ELSTs) are rare, slowly growing temporal bone neoplasms which show a high association with von Hippel-Lindau (VHL) syndrome. The immunohistochemistry evaluation of these papillary-cystic neoplasms frequently raises the differential diagnosis with renal cell carcinoma, among other metastatic neoplasms, whether in VHL patients or not. A cohort of 26 patients with ELSTs were evaluated for histologic features, immunohistochemistry findings, and association with VHL. Standard immunohistochemistry evaluation was performed. Sixteen females and 10 males ranging in age from 10 to 69 years (mean 44; VHL mean: 32) at initial presentation, comprised the cohort of patients. Most (86%) experienced hearing changes or inner ear symptoms (vertigo, dizziness), with an average duration of symptoms for 39 months (range 2–240 months). The tumors were an average of 2.9 cm (range 0.4–8 cm), with 14 left, 11 right sided and one bilateral tumor. Nine patients had documented VHL, with 3 patients having a concurrent or subsequent clear cell renal cell carcinoma. Patients were followed an average of 6.2 years (available in 24 patients): 19 alive without disease, 7.5 years; 2 dead without disease, 1.2 years; and 3 alive with disease, 3.1 years. The neoplastic cells show the following immunohistochemistry findings: AE1/AE3, EMA, CK7, CAIX, GLUT1, VEGF: 100% of cases tested were positive; pax-8: 85% of cases positive; CD10 and RCC: 0% of cases reactive. Based on this cohort of 26 patients with ELST, 9 of whom had VHL, the strong pax-8 and CAIX should be used in conjunction with negative CD10 and RCC to help exclude a metastatic renal cell carcinoma. As CAIX is an enzyme overexpressed in hypoxia and hypoxia inducible factor is what VHL protein regulates, this is an expected, although previously unreported finding. Whether part of VHL or not, VHL mutations may be a somatic rather than germline finding in the tumors, a possible further explanation for the CAIX reaction.
Keywords: Endolymphatic sac tumor, Immunohistochemistry, CAIX, pax-8, Von Hippel-Lindau syndrome, Renal cell carcinoma, Differential diagnosis
Introduction
Endolymphatic sac tumors (ELSTs) are a very rare, low grade tumor of endolymphatic sac origin [1], showing a well-developed association with von Hippel-Lindau (VHL) syndrome [1–5]. The endolymphatic duct arises in the posteroinferior part of the inner ear vestibule, passes through the bony canal of the vestibular aqueduct and widens in the superior–inferior plane into the endolymphatic sac, which traverses the temporal bone to end blindly on the posterior wall of the temporal bone [6]. While endolymphatic sac tumor is the preferred terminology, it is important to recognize the wide diversity of names applied to this tumor over the years: papillary adenoma of endolymphatic sac [7], papillary endolymphatic sac tumor [8], aggressive papillary adenoma [9], aggressive papillary middle-ear tumor [10], invasive papillary cystadenoma [11], low grade papillary adenomatous tumors of the temporal bone [12], endolymphatic sac papillary tumor [13, 14], papillary neoplasm of the endolymphatic sac [4], low grade adenocarcinoma [15], endolymphatic sac carcinoma [16], adenocarcinoma of endolymphatic sac [17], low-grade malignant endolymphatic sac tumor [18], and low grade adenocarcinoma of probable endolymphatic sac origin (AKA, Heffner tumor) [1, 19]. Just from the wide diversity of names employed, it can be inferred that the tumor causes a significant diagnostic difficulty and is frequently confused with other papillary tumors on histopathologic grounds. Autopsy studies have shown a 1 millimeter papillary glandular neoplasm, completely confined to the endolymphatic duct, thus supporting the tumor’s origin in the endolymphatic sac [6, 20]. Incorrect interpretation of the diagnosis results in significant morbidity and mortality to the patients, especially when the relationship to VHL syndrome is not further explored. Immunohistochemistry studies have been performed to aid in the differential diagnosis, but strong reactivity with pax-8 and CAIX, two markers that are strongly positive in many clear cell renal cell carcinomas (ccRCC), has further contributed to the diagnostic confusion. This clinical and immunohistochemistry evaluation was undertaken in an effort to highlight some of these differential diagnostic considerations and attempt to help resolve them in an evaluation of 26 ELSTs.
Materials and Methods
All patient records with temporal bone surgical cases performed between 2000 and 2017 at the 12 Southern California Permanente Medical Group hospitals were analyzed. Six cases were identified. In addition, the consultation files and surgical pathology records of the collaborating authors were reviewed for cases of endolymphatic sac tumors, with an additional 20 cases retrieved. Materials within the files were supplemented by a review of the patient demographics (sex, age, and race); symptoms and physical findings and duration at presentation; imaging studies, surgical pathology, and operative reports were reviewed when available, with follow-up information obtained by direct written or oral communication with the referring pathologist, patient’s physician, oncology data services and tumor registries, or the patient (patient’s family member[s]). Follow-up data included information regarding presence of recurrent disease, treatment modalities used, and the current patient status. Cases in general were submitted as consultations to several of the authors, who conducted this study as a retrospective review, without actually treating the patients. Submitted diagnoses by the primary pathologists included middle ear adenoma, paraganglioma, aggressive papillary adenocarcinoma, metastatic carcinoma, ependymoma and adenoma. This clinical investigation was conducted in accordance and compliance with an Internal Review Board authorization (#5968) performed under the direction of Southern California Permanente Medical Group.
Specific information about the exact location, laterality, and tumor size (greatest dimension in cm) were documented (imaging reports and/or surgical pathology material). Hematoxylin and eosin-stained slides from all cases were reviewed to document specific histologic features. Immunophenotypic analysis was performed in cases with sufficient suitable material by a standardized Envision™ or Ventana method (as previously described [21]) employing 4 µm-thick, formalin fixed, paraffin embedded sections. Table 1 documents the pertinent, commercially available immunohistochemical antibody panel used. The analysis was performed on a single representative block for each primary tumor. However, the biopsies were often small, yielding a limited amount of tissue for additional examination, thus prioritizing immunohistochemistry studies for diagnostic clarity was paramount. Epitope retrieval was performed, as required by the manufacturer guidelines. Standard positive controls were used throughout, with serum used as the negative control. The antibody reactions were graded as positive or negative; positives were separated into strong, moderate and weak; diffuse (> 50%), patchy (10–50%) or focal (< 10%); and by pattern (cytoplasmic, membrane, nuclear, nuclear and cytoplasmic). Staining equivalent reactions would be absent to weak (0 to 1 +), moderate (2 + to 3 +) and strong (4 +) staining.
Table 1.
Immunohistochemical panel
| Antigen | Source | Result | %/# of cases |
|---|---|---|---|
| Cytokeratin (AE1/AE3) | Dako | P, S, D, C | 100% (16/16) |
| Epithelial membrane antigen (EMA) (E29) | Ventana | P, S, D, C | 100% (12/12) |
| CK7 (OV-TL-12/30) | Dako | P, S, D, C | 100% (20/20) |
| CAIX (CA9) | Cell Marque | P, S, D, M | 100% (25/25) |
| pax-8 | LifeSpan BioSciences | P, S, D, N | 85% (22/26) |
| CD10 (SP67) | Ventana | Non-reactive | 0% (0/16) |
| RCC | Vector Laboratories | Non-reactive | 0% (0/15) |
P positive, S strong, D diffuse, C cytoplasmic, M membrane, N nuclear
Statistical evaluation was performed using a standard statistics software package with categorical variables analyzed using Chi square tests and Fisher’s Exact tests to compare observed and expected frequency distributions. Comparison of means between groups was made with independent t-tests (including 1-tailed and 2-tailed tests with degrees of freedom) or one-way analysis of variance, depending on whether there were two groups or more than two groups, respectively. Confidence intervals of 95% were generated for all positive findings. The alpha level was set at p < 0.05.
Results
Clinical Findings
The patients included 16 women and 10 men (1.6:1; Table 2), ranging in age from 10 to 69 years (mean 44.1 years). Women were slightly older than men (46 vs 41, years), but this was not a statistically significant difference. During evaluation, nine patients were identified to be part of VHL syndrome, while 16 patients were not. Importantly, in many of these cases genetic testing was not performed or not offered to the patients, interpreted by the treating physicians to not be a significant association. When the patients who had documented VHL were separated from patients without documented VHL, the mean age at presentation was 32 vs 51 years, respectively, a finding that was statistically significant (p = 0.008). There were 13 whites, 8 African Americans, and two Asians, while the race/ethnicity was unknown in three patients. This finding represents a disproportionate representation of African Americans when compared to the general population.
Table 2.
Clinical characteristics
| Clinical characteristics | Number (n = 26) |
|---|---|
| Sex | |
| Females | 16 |
| Males | 10 |
| Race | |
| White | 13 |
| Black | 8 |
| Asian | 2 |
| Unknown | 3 |
| VHL | |
| Documented | 9 |
| Undocumented | 17 |
| Age (in years) | |
| Range | 10–69 |
| Mean | 44 |
| Median | 44 |
| Females (range 10–69 years) (mean) | 46 |
| Males (range 14–65 years) (mean) | 41 |
| VHL patients (range 10–53) (mean) (p = 0.008) | 32 |
| Undocumented VHL patients (range 32–69) (mean) | 51 |
| Symptoms | |
| Hearing changes (sensorineural loss) | 19 |
| Vertigo, dizziness or balance changes | 12 |
| Tinnitus | 10 |
| CNS symptoms | 8 |
| Pain | 6 |
| Asymptomatic | 0 |
| Duration of symptoms (in months) | |
| Range | 2–240 |
| Mean | 39 |
| Females (p = 0.087) | 22.9 |
| Males | 64.4 |
| VHL patients | 34.8 |
| Undocumented VHL patients (p = 0.626) | 26.7 |
| Laterality | |
| Left | 14 |
| Right | 11 |
| Bilateral | 1 |
| Size (cm) (pT) | |
| Range | 0.4–8.0 |
| Mean | 2.9 |
| Median | 2.9 |
| Females (p = 0.273) | 3.6 |
| Males | 2.6 |
| VHL patients (p = 0.126) | 2 |
| Undocumented VHL patients | 3.6 |
Patients presented with symptoms of a relatively long duration (mean 39 months), without any significant difference between patients with and without documented VHL (p = 0.626) nor between males and females (p = 0.087). The vast majority of patients presented with hearing changes, and specifically sensorineural hearing loss, although symptoms were not known in two of the patients (Table 2). Inner ear symptoms, including vertigo, dizziness, ataxia or balance changes/loss were reported in 12 of the patients, while tinnitus was seen in 10. Central nervous system symptoms, including headaches, tingling, weakness or muscle spasms of the face were reported in eight patients. Ear drainage, tongue weakness and atrophy, numbness and even slurred speech were also document in one patient each. Importantly, while symptoms were unknown in two patients, none of the remaining patients were asymptomatic. In the patients who had VHL, patients had clear cell renal cell carcinoma (n = 3), hemangioblastoma (n = 3), pheochromocytoma, epididymis cyst, pancreatic cysts, small cerebellar lesions, and Meniere disease, among other stigmata of the disease.
Only one patient (a 14 year old male VHL patient) reported bilateral tumors, with 14 tumors identified only on the left and 11 tumors identified only on the right. Imaging studies identified permeative and destructive masses that involved the temporal bone, centered along the posterior face of the petrous part of the bone to include the retrolabyrinthine region, although centering was size dependent. Calcifications within a spiculated mass were noted. By MR, the tumors presented as an inhomogeneous hyperintense mass often with heterogenous enhancement (Fig. 1). Imaging studies provided the best size determination, with the tumors ranging from 0.4 up to 8 cm (mean 2.9 cm). There was no statistically significant difference in tumor size at presentation between the VHL patients and those without documented VHL (mean 2.3 vs 3.6 cm, respectively; p = 0.126) nor between females and males (mean 3.6 vs 2.6, respectively; p = 0.273).
Fig. 1.
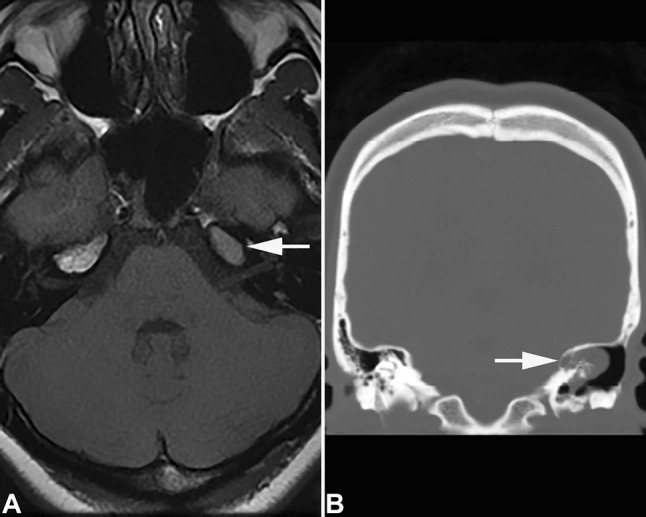
Imaging studies of an endolymphatic sac tumor. A This axial T1 weighted MR demonstrates an intermediate signal, 16 mm soft tissue density mass (arrow) in the region of the petrous apex (23 year old female VHL patient). B This coronal computed tomography image shows a partly calcified moderately heterogenous 30 mm mass (arrow) involving the posterior region of the left petrous bone (53 year old male VHL patient)
Pathologic Features
Microscopic
All of the tumors showed an unencapsulated, destructive mass associated with bone invasion and/or bone remodeling (Fig. 2). The tumor shows a biphasic appearance of papillary and glandular profiles. The papillary structures were simple, coarse and broad, with fibrovascular cores coursing through the center of the papillae. The cystic spaces focally contained fluid and/or extravasated erythrocytes. In some areas, acinar or follicular-type spaces were filled with inspissated material that simulated colloid of thyroid follicular epithelial cells (Fig. 3). The neoplastic cells were usually arranged as a single layer of low cuboidal to columnar epithelial cells. There was clear to lightly eosinophilic, granular cytoplasm with indistinct cell borders or membranes (Fig. 3), surrounding centrally placed small, round, hyperchromatic nuclei. Sometimes the glandular appearance was more difficult to see (Fig. 4), where there was a more solid appearance, along with histiocytes and degeneration. Tumor necrosis was not seen, while mitotic activity was inconspicuous, without atypical forms. A background of cholesterol clefts from blood breakdown was focally present in the background.
Fig. 2.
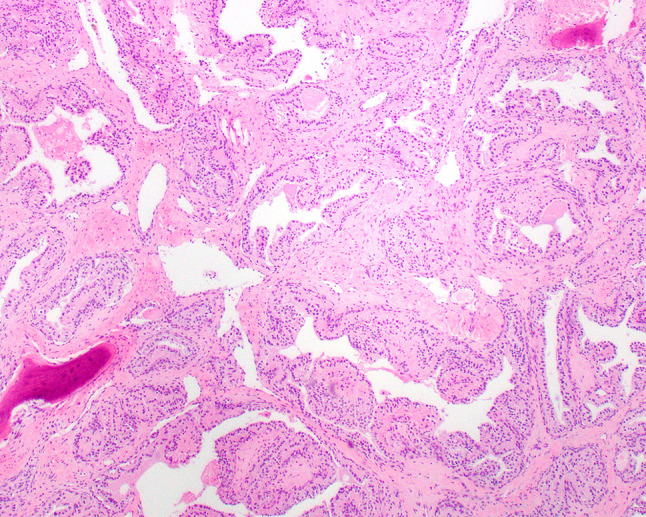
Hematoxylin and eosin stained tumor shows remodeled spicules of bone with broad, coarse papillae
Fig. 3.
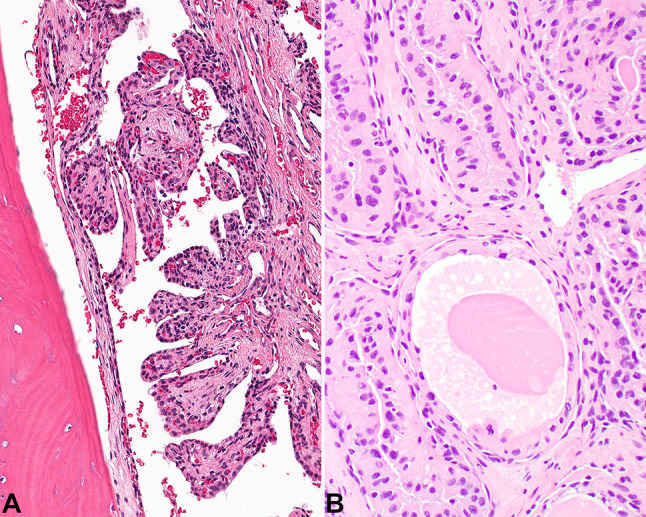
A The coarse papillae are broad with well developed fibrovascular cores. B Inspissated material is noted within an area that resembles a thyroid follicle
Fig. 4.
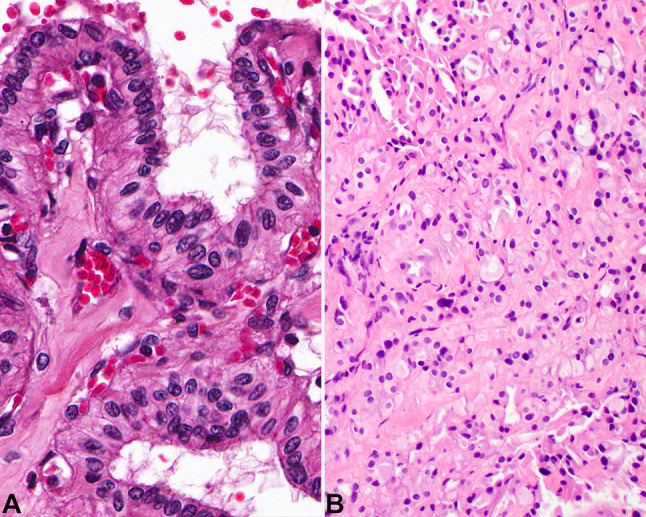
A The cytoplasm is lightly eosinophilic to cleared with bland nuclei. B A more solid growth pattern is seen in this tumor, with small gland-like lumen noted
Immunohistochemistry
The neoplastic cells were strongly reactive with pancytokeratin (AE1/AE3), EMA, and CK7 (Table 1). All of the tumors tested stained strongly and diffusely, along the membrane for CAIX (Fig. 5), while 85% of cases stained positive in the nuclei with pax-8 (Fig. 5). Importantly, all tested cases were non-reactive with CD10 and RCC (Fig. 6). While not performed in all cases, GLUT1, CAM5.2, VEGF and CK5/6 were also positive in the tumors studied. Vimentin was also strongly positive in the neoplastic cells, while CD117, P504S, TTF1, p63, synaptophysin, napsin, GATA3, thyroglobulin, WT1 and CD34 were negative (these stains had been performed by contributing pathologists prior to consultation, and thus were not formally evaluated with known antibodies, dilutions and techniques). S100 protein showed a patchy, nuclear and cytoplasmic reactivity in 5 of 7 cases tested.
Fig. 5.
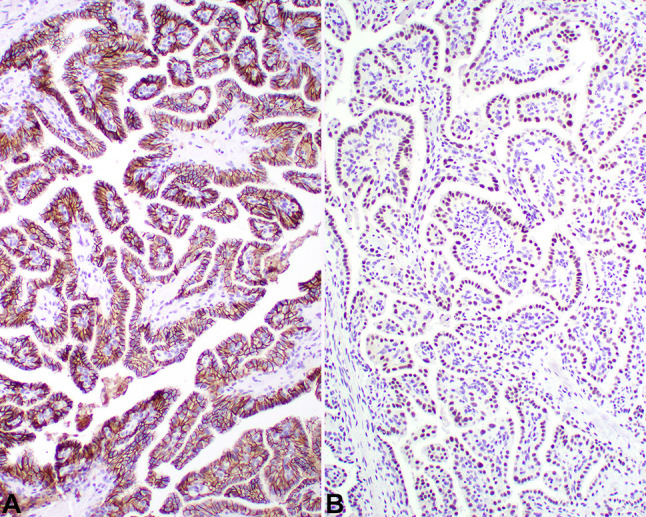
A There is a strong, diffuse, membranous reaction with CAIX. B There is a strong, diffuse, nuclear reaction with pax-8
Fig. 6.
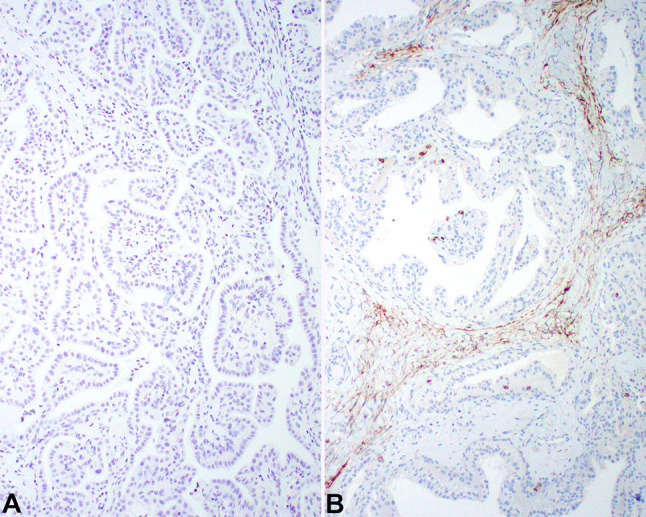
The neoplastic cells show a lack of reactivity with A RCC and B CD10
Clinical Treatment and Patient Outcome
All of the patients were treated with surgery, although one patient received pre-surgical embolization and one patient had biopsy only before definitive surgery. In all of the patients in whom follow-up was available (n = 24), 19 were alive with no evidence of residual disease at last follow-up (mean 6.2 years; range 0.1–23.8 years). Three patients were alive, but with disease, either persistent or recurrent at last follow-up (mean 3.1 years; range 0.5 to 6.1 years). Two patient were dead, without disease, although one patient died from a complication of vocal cord paralysis as a result of the surgical intubation (2.2 years after initial presentation, the persistence was operated on again). None of the patients in this series reported loss of cranial nerve function or neurological function as a post-surgical complication.
Discussion
The connection between an ELST and VHL syndrome was first postulated by a surgeon who observed the autopsy findings of a 24 year old patient who died as a result of biopsy complications of a temporal bone cystic and papillary neoplasm, and was documented to have intra-abdominal lesions of VHL syndrome [22], although not formally proposed until the 1980s [1, 7, 10] and ultimately confirmed as part of the disorder in the late 1990s [3].
VHL syndrome is an autosomal-dominant inherited disease, affecting about 1 in 36,000 people, caused by germline mutations in the tumor suppressor gene VHL, mapped to 3p26–p25. With more than 1500 known mutations, mutations generally result in inactivation of the wild-type copy of the VHL gene [23]. Tumors generally develop after biallelic VHL inactivation (two hit hypothesis), which leads to up-regulation of hypoxic response and tumor formation in a number of different organs, including those in the endolymphatic sac [20, 23]. pVHL is critical in regulating the proteolytic degradation of the α subunits of HIF-1 and HIF-2 transcriptions factors with a ubiquitin ligase protein complex that binds the α subunits of HIF-1 and HIF-2 transcription factors that allows for ubiquitination and proteosomal degradation (Fig. 7) [24, 25]. When normoxic conditions apply, the HIF-α subunits are rapidly degraded, but oxygen is essential as a co-factor that modify the HIF-α subunits for pVHL binding [26, 27]. In tumors, the pVHL is either absent or inactive (conceptually similar to hypoxia), and thus HIF-1 and HIF-2 are stabilized and activate the hypoxic gene response which oversees a large number of target genes, including vascular endothelial growth factor (VEGF), Cyclin-D1, platelet derived growth factor (PDGFß), CAIX and GLUT1 [28].
Fig. 7.

A diagramatic representation of the function of the VHL gene. HIF hypoxia inducible factor; OH hydroxlation group
The proceedings from the International Endolymphatic Sac Tumor Registry [5], show ELSTs have a prevalence of about 3.6% in VHL syndrome. Further VHL germline mutations were identified in 39% of apparently sporadic ELSTs, highlighting that 32% of patients with VHL present with ELSTs as the initial presentation of VHL. Thus, genetic testing of all patients who present with ELSTs is highly recommended [5, 29–31]. Somatic and germline mutation analysis of ELSTs and controls has been performed, with documentation of specific VHL mutations and allelic deletions, although quite diverse given the large genotype variations seen in VHL syndrome [20, 31–36]. Examples including missense mutations in exon 1 of the VHL gene (p.Asp92Gly), point mutations in exon 1, deletion in exon 1 (p.Val62GlyfsX66), and missense mutations in exon 3 (p.Leu158Pro) [2, 31, 34–37]. The exact type of VHL mutation might predict the aggressiveness of the disorder and hence tumor management [31].
The clinical findings of an average age in the fifth decade, with a female to male ratio of 1.6:1 in patients who present with audiovestibular symptoms (sensorineural hearing loss, tinnitus and vertigo), is similar to the findings presented in the literature [5, 18, 29, 30, 33, 38]. Although unknown, there does seem to be a proclivity for tumors to develop on the left rather than the right [18]. Imaging studies confirm a destructive, lytic bony lesion involving the endolymphatic sac region as a vascularized tumor involving the petrous bone, frequently containing calcifications, but as the size of the tumor expands, a more extensive bony involvement is noted [8, 31, 33]. Flow voids are noted in larger tumors, while increased signal intensity at unenhanced T1-weighted MR imaging is common [8].
The histology shows a characteristic papillary and glandular architecture, comprised of relatively simple, coarse papillary structures, showing only limited branching, with well-developed fibrovascular cores. The epithelial cells show a glandular arrangement in other areas, sometimes with a very well developed inspissated secretory material or blood, which can mimic clear cell renal cell carcinoma or papillary thyroid carcinoma. The tumor cells are low cuboidal to columnar with delicate to cleared cytoplasm surrounding round to oval nuclei with coarse to heavy chromatin distribution. Bone expansion is nearly always seen, although the bone is remodeled rather than truly destroyed [1–3, 39].
As described above, the protein VHL is important to the regulation of HIF-1α. Normally, during conditions of low oxygen tension, degradation of HIF-1 is inhibited by oxygen-dependent hydroxylation of the protein that prevents interaction of HIF-1 with pVHL ubiquitin ligase complex [40]. But when VHL is altered, mutated or absent, HIFα and HIFß combine to result in gene activation, of which CAIX is one of several factors produced. CAIX is a member of the carbonic anhydrase family (carbonic anhydrase IX), and as a transmembrane protein, catalyzes the conversion of carbon dioxide to bicarbonate and protons, critical to ion transport and pH maintenance during hypoxia. Hypoxia activates hypoxia inducible factor-1 (HIF-1), which in turn upregulates CAIX [41, 42]. In renal carcinoma cells that are defective for VHL, up-regulation of CAIX is associated with loss of regulation by hypoxia, consistent with the critical function of pVHL in the regulation of HIF-1 [42]. Since ELST is one of the tumors within the VHL syndrome spectrum, strong, membranous/cytoplasmic immunoreactivity with CAIX is to be expected, as shown in this cohort of patients. However, this strong reactivity raises the differential diagnostic consideration with ccRCC even more so. Likewise, it is well known that pax-8 expression is found in many normal and neoplastic tissues, including epididymis, rete testis, fallopian tumor and ovary, pancreatic islets and lymph nodes, while commonly found in bladder adenocarcinoma, seminoma, endometrial adenocarcinoma, ovarian serous carcinoma, renal cell carcinoma, oncocytoma, neuroendocrine tumors, parathyroid carcinoma and papillary, follicular and medullary thyroid carcinoma [43–49]. During embryogenesis, pax-2 and pax-8 transcription factors play a redundant role by together regulating inner ear development, associated with otic placode induction, where they are required to maintain otic differentiation [50, 51]. Both pax-2 and pax-8 are noted in hair cell development of the vestibular end organ (otic progenitor cells) during gestational weeks 8–12 [51, 52]. Thus, in the neoplastic setting of a tumor arising from this zone in later life, it is perhaps an explanation for pax-8 expression, and why it is so strongly expressed in ELSTs, as seen in 85% of the tumors studied. By immunohistochemistry, CK-pan, CAM5.2, CK7, CK8, CK19, HIF-1α, VEGF, and CAIX have been shown to be increased in ELSTs [40, 53–55]. while transthyretin (a marker for choroid plexus papilloma) is reported to be negative [54].
The differential diagnosis histologically includes metastatic ccRCC, papillary thyroid carcinoma or follicular thyroid carcinoma, while choroid plexus papilloma, papillary ependymoma, papillary meningioma, paraganglioma and even middle ear adenoma are less frequently considered [56]. Centering of the tumors within the cerebellopontine angle, tumors with psammoma bodies and whorling, along with unique immunohistochemistry findings (such as CK5/6, synaptophysin, progesterone receptors) help to exclude the less common considerations of choroid plexus papilloma, papillary ependymoma and papillary meningioma. The lack of any neuroendocrine markers in ELSTs along with the overall zellballen arrangement seen in paraganglioma, remove paraganglioma from the histologic differential diagnosis, although often it is a consideration by imaging studies. However, as patients with VHL syndrome frequently develop ccRCC, a neoplasm with clear or lightly eosinophilic cytoplasm in the temporal bone may represent metastasis. The ccRCC would be immunoreactive with CAIX and pax-8, and so the strong reactivity with these markers in ELST does not help to make the separation. The addition of negative reactions with CD10 and RCC, however, would help to make the separation between these tumor types.
In general, the smaller the tumor at the time of removal, the lower the postoperative morbidity, where extensive surgery for much larger tumors is associated with disruption of the fallopian canal, jugular foramen, petrous apex, cerebrospinal fluid leak or involvement of the posterior cranial fossa, with concurrent loss of cranial nerve function (specifically VII, IX, and X) [29, 33, 38, 57, 58]. Thus, while many clinicians seem to eschew genetic counseling or testing in these patients, identification of the potential genetic relationship and removing the tumors at the earliest, and therefore smallest size possible, is advocated [5, 31, 33, 57, 58].
Conclusion
ELSTs are frequently the initial presenting finding in a patient who later is documented to have VHL. As such, genetic counseling and additional evaluation is warranted in all patients who have an ELST, especially when patients are young at initial presentation or have bilateral tumors. In limited biopsies or tumors that show significant clear cell change and hemorrhage in the background, confirming the diagnosis of an ELST with positive reactions to CAIX and/or pax-8 with concurrent negative reactions with CD10 and/or RCC would help to confirm the diagnosis and help to exclude other tumors in the histological differential diagnosis.
Acknowledgements
Presented at the 106th Annual Meeting of the United States and Canadian Academy of Pathology, Vancouver, British Columbia, Canada, March, 2018.
Compliance with Ethical Standards
Conflict of interest
All authors declare that they have no conflict of interest as it relates to this research project. The opinions or assertions contained herein are the private views of the author and are not to be construed as official or as reflecting the views of Southern California Permanente Medical Group.
Ethical Approval
All procedures performed in this retrospective data analysis involving human participants were in accordance with the ethical standards of the institutional review board (IRB #5968), which did not require informed consent.
References
- 1.Heffner DK. Low-grade adenocarcinoma of probable endolymphatic sac origin A clinicopathologic study of 20 cases. Cancer. 1989;64:2292–2302. doi: 10.1002/1097-0142(19891201)64:11<2292::AID-CNCR2820641119>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 2.Skalova A, Sima R, Bohus P, Curik R, Lukas J, Michal M. Endolymphatic sac tumor (aggressive papillary tumor of middle ear and temporal bone): report of two cases with analysis of the VHL gene. Pathol Res Pract. 2008;204:599–606. doi: 10.1016/j.prp.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 3.Manski TJ, Heffner DK, Glenn GM, et al. Endolymphatic sac tumors. A source of morbid hearing loss in von Hippel-Lindau disease. JAMA. 1997;277:1461–1466. doi: 10.1001/jama.1997.03540420057030. [DOI] [PubMed] [Google Scholar]
- 4.Delisle MB, Uro E, Rouquette I, Yardeni E, Rumeau JL. Papillary neoplasm of the endolymphatic sac in a patient with von Hippel-Lindau disease. J Clin Pathol. 1994;47:959–961. doi: 10.1136/jcp.47.10.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bausch B, Wellner U, Peyre M, et al. Characterization of endolymphatic sac tumors and von Hippel-Lindau disease in the International Endolymphatic Sac Tumor Registry. Head Neck. 2016;38(Suppl 1):E673–E679. doi: 10.1002/hed.24067. [DOI] [PubMed] [Google Scholar]
- 6.Michaels L. Origin of endolymphatic sac tumor. Head Neck Pathol. 2007;1:104–111. doi: 10.1007/s12105-007-0016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hassard AD, Boudreau SF, Cron CC. Adenoma of the endolymphatic sac. J Otolaryngol. 1984;13:213–216. [PubMed] [Google Scholar]
- 8.Mukherji SK, Albernaz VS, Lo WW, et al. Papillary endolymphatic sac tumors: CT, MR imaging, and angiographic findings in 20 patients. Radiology. 1997;202:801–808. doi: 10.1148/radiology.202.3.9051037. [DOI] [PubMed] [Google Scholar]
- 9.Folker RJ, Meyerhoff WL, Rushing EJ. Aggressive papillary adenoma of the cerebellopontine angle: case report of an endolymphatic sac tumor. Am J Otolaryngol. 1997;18:135–139. doi: 10.1016/S0196-0709(97)90103-4. [DOI] [PubMed] [Google Scholar]
- 10.Gaffey MJ, Mills SE, Fechner RE, Intemann SR, Wick MR. Aggressive papillary middle-ear tumor. A clinicopathologic entity distinct from middle-ear adenoma. Am J Surg Pathol. 1988;12:790–797. doi: 10.1097/00000478-198810000-00009. [DOI] [PubMed] [Google Scholar]
- 11.Meyer JR, Gebarski SS, Blaivas M. Cerebellopontine angle invasive papillary cystadenoma of endolymphatic sac origin with temporal bone involvement. AJNR Am J Neuroradiol. 1993;14:1319–1321. [PMC free article] [PubMed] [Google Scholar]
- 12.Lavoie M, Morency RM. Low-grade papillary adenomatous tumors of the temporal bone: report of two cases and review of the literature. Mod Pathol. 1995;8:603–608. [PubMed] [Google Scholar]
- 13.Bisceglia M, D’Angelo VA, Wenig BM. Endolymphatic sac papillary tumor (Heffner tumor) Adv Anat Pathol. 2006;13:131–138. doi: 10.1097/00125480-200605000-00005. [DOI] [PubMed] [Google Scholar]
- 14.Yu SJ, Chen YD, Gao F, Qiu XG, Chang H. Endolymphatic sac papillary tumor: a case report. Chin Med J (Engl) 2011;124:3828–3829. [PubMed] [Google Scholar]
- 15.Malhotra S, Rao RV, Valiathan M, Mathew M, Nayak DR, Raja A. Low-grade adenocarcinoma of endolymphatic sac origin. Am J Otolaryngol. 2006;27:362–365. doi: 10.1016/j.amjoto.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Monedero Martinez-Pardo E, Navia Alvarez P. Endolymphatic sac carcinoma: a case report. Radiologia. 2011;53:483–484. doi: 10.1016/j.rx.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 17.Mukherji SK, Castillo M. Adenocarcinoma of the endolymphatic sac: imaging features and preoperative embolization. Neuroradiology. 1996;38:179–180. doi: 10.1007/BF00604815. [DOI] [PubMed] [Google Scholar]
- 18.Wang HQ, Jie L, Shi HY. Clinicopathological features of low-grade malignant endolymphatic sac tumors. Pathol Res Pract. 2018;214:431–435. doi: 10.1016/j.prp.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 19.Batsakis JG, El-Naggar AK. Papillary neoplasms (Heffner’s tumors) of the endolymphatic sac. Ann Otol Rhinol Laryngol. 1993;102:648–651. doi: 10.1177/000348949310200815. [DOI] [PubMed] [Google Scholar]
- 20.Glasker S, Lonser RR, Tran MG, et al. Effects of VHL deficiency on endolymphatic duct and sac. Cancer Res. 2005;65:10847–10853. doi: 10.1158/0008-5472.CAN-05-1104. [DOI] [PubMed] [Google Scholar]
- 21.Andreasen S, Therkildsen MH, Grauslund M, Friis-Hansen L, Wessel I, Homoe P. Activation of the interleukin-6/Janus kinase/STAT3 pathway in pleomorphic adenoma of the parotid gland. Apmis. 2015;123:706–715. doi: 10.1111/apm.12407. [DOI] [PubMed] [Google Scholar]
- 22.Bellairs JA, Gluth MB. A histopathological connection between a fatal endolymphatic sac tumour and von Hippel-Lindau disease from 1960. J Laryngol Otol. 2018;132:75–78. doi: 10.1017/S0022215117001888. [DOI] [PubMed] [Google Scholar]
- 23.Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet. 2011;19:617–623. doi: 10.1038/ejhg.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamura T, Koepp DM, Conrad MN, et al. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science. 1999;284:657–661. doi: 10.1126/science.284.5414.657. [DOI] [PubMed] [Google Scholar]
- 25.Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 26.Ivan M, Kondo K, Yang H, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 27.Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 28.Kaelin WG., Jr Treatment of kidney cancer: insights provided by the VHL tumor-suppressor protein. Cancer. 2009;115:2262–2272. doi: 10.1002/cncr.24232. [DOI] [PubMed] [Google Scholar]
- 29.Wick CC, Manzoor NF, Semaan MT, Megerian CA. Endolymphatic sac tumors. Otolaryngol Clin North Am. 2015;48:317–330. doi: 10.1016/j.otc.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 30.Timmer FC, Neeskens LJ, van den Hoogen FJ, et al. Endolymphatic sac tumors: clinical outcome and management in a series of 9 cases. Otol Neurotol. 2011;32:680–685. doi: 10.1097/MAO.0b013e318215992b. [DOI] [PubMed] [Google Scholar]
- 31.Codreanu CM, Duet M, Hautefort C, et al. Endolymphatic sac tumors in von Hippel-Lindau disease: report of three cases. Otol Neurotol. 2010;31:660–664. doi: 10.1097/MAO.0b013e3181d8d863. [DOI] [PubMed] [Google Scholar]
- 32.Rao Q, Zhou J, Wang JD, et al. Endolymphatic sac tumor with von Hippel-Lindau disease: report of a case with analysis of von Hippel-Lindau gene and review. Ann Diagn Pathol. 2010;14:361–364. doi: 10.1016/j.anndiagpath.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Kim HJ, Butman JA, Brewer C, et al. Tumors of the endolymphatic sac in patients with von Hippel-Lindau disease: implications for their natural history, diagnosis, and treatment. J Neurosurg. 2005;102:503–512. doi: 10.3171/jns.2005.102.3.0503. [DOI] [PubMed] [Google Scholar]
- 34.Vortmeyer AO, Huang SC, Koch CA, et al. Somatic von Hippel-Lindau gene mutations detected in sporadic endolymphatic sac tumors. Cancer Res. 2000;60:5963–5965. [PubMed] [Google Scholar]
- 35.Vortmeyer AO, Choo D, Pack S, Oldfield E, Zhuang Z. VHL gene inactivation in an endolymphatic sac tumor associated with von Hippel-Lindau disease. Neurology. 2000;55:460. doi: 10.1212/WNL.55.3.460. [DOI] [PubMed] [Google Scholar]
- 36.Vortmeyer AO, Choo D, Pack SD, Oldfield E, Zhuang Z. von Hippel-Lindau disease gene alterations associated with endolymphatic sac tumor. J Natl Cancer Inst. 1997;89:970–972. doi: 10.1093/jnci/89.13.970-a. [DOI] [PubMed] [Google Scholar]
- 37.Hamazaki S, Yoshida M, Yao M, et al. Mutation of von Hippel-Lindau tumor suppressor gene in a sporadic endolymphatic sac tumor. Hum Pathol. 2001;32:1272–1276. doi: 10.1053/hupa.2001.28961. [DOI] [PubMed] [Google Scholar]
- 38.Kim HJ, Hagan M, Butman JA, et al. Surgical resection of endolymphatic sac tumors in von Hippel-Lindau disease: findings, results, and indications. Laryngoscope. 2013;123:477–483. doi: 10.1002/lary.23646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bell D, Gidley P, Levine N, Fuller GN. Endolymphatic sac tumor (aggressive papillary tumor of middle ear and temporal bone): sine qua non radiology-pathology and the University of Texas MD Anderson Cancer Center experience. Ann Diagn Pathol. 2011;15:117–123. doi: 10.1016/j.anndiagpath.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 40.Jensen RL, Gillespie D, House P, Layfield L, Shelton C. Endolymphatic sac tumors in patients with and without von Hippel-Lindau disease: the role of genetic mutation, von Hippel-Lindau protein, and hypoxia inducible factor-1alpha expression. J Neurosurg. 2004;100:488–497. doi: 10.3171/jns.2004.100.3.0488. [DOI] [PubMed] [Google Scholar]
- 41.Sedlakova O, Svastova E, Takacova M, Kopacek J, Pastorek J, Pastorekova S. Carbonic anhydrase IX, a hypoxia-induced catalytic component of the pH regulating machinery in tumors. Front Physiol. 2014;4:400. doi: 10.3389/fphys.2013.00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wykoff CC, Beasley NJ, Watson PH, et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000;60:7075–7083. [PubMed] [Google Scholar]
- 43.Laury AR, Perets R, Piao H, et al. A comprehensive analysis of PAX8 expression in human epithelial tumors. Am J Surg Pathol. 2011;35:816–826. doi: 10.1097/PAS.0b013e318216c112. [DOI] [PubMed] [Google Scholar]
- 44.Tacha D, Zhou D, Cheng L. Expression of PAX8 in normal and neoplastic tissues: a comprehensive immunohistochemical study. Appl Immunohistochem Mol Morphol. 2011;19:293–299. doi: 10.1097/PAI.0b013e3182025f66. [DOI] [PubMed] [Google Scholar]
- 45.Ozcan A, Shen SS, Hamilton C, et al. PAX 8 expression in non-neoplastic tissues, primary tumors, and metastatic tumors: a comprehensive immunohistochemical study. Mod Pathol. 2011;24:751–764. doi: 10.1038/modpathol.2011.3. [DOI] [PubMed] [Google Scholar]
- 46.Hu Y, Hartmann A, Stoehr C, et al. PAX8 is expressed in the majority of renal epithelial neoplasms: an immunohistochemical study of 223 cases using a mouse monoclonal antibody. J Clin Pathol. 2012;65:254–256. doi: 10.1136/jclinpath-2011-200508. [DOI] [PubMed] [Google Scholar]
- 47.Ordonez NG. Value of PAX8, PAX2, napsin A, carbonic anhydrase IX, and claudin-4 immunostaining in distinguishing pleural epithelioid mesothelioma from metastatic renal cell carcinoma. Mod Pathol. 2013;26:1132–1143. doi: 10.1038/modpathol.2013.34. [DOI] [PubMed] [Google Scholar]
- 48.Mentrikoski MJ, Wendroth SM, Wick MR. Immunohistochemical distinction of renal cell carcinoma from other carcinomas with clear-cell histomorphology: utility of CD10 and CA-125 in addition to PAX-2, PAX-8, RCCma, and adipophilin. Appl Immunohistochem Mol Morphol. 2014;22:635–641. doi: 10.1097/PAI.0000000000000004. [DOI] [PubMed] [Google Scholar]
- 49.Magers MJ, Udager AM, Chinnaiyan AM, et al. Comprehensive Immunophenotypic Characterization of Adult and Fetal Testes, the Excretory Duct System, and Testicular and Epididymal Appendages. Appl Immunohistochem Mol Morphol. 2016;24:e50–e68. doi: 10.1097/PAI.0000000000000326. [DOI] [PubMed] [Google Scholar]
- 50.Mackereth MD, Kwak SJ, Fritz A, Riley BB. Zebrafish pax8 is required for otic placode induction and plays a redundant role with Pax2 genes in the maintenance of the otic placode. Development. 2005;132:371–382. doi: 10.1242/dev.01587. [DOI] [PubMed] [Google Scholar]
- 51.Johnson Chacko L, Pechriggl EJ, Fritsch H, et al. Neurosensory differentiation and innervation patterning in the human fetal vestibular end organs between the gestational weeks 8–12. Front Neuroanat. 2016;10:111. doi: 10.3389/fnana.2016.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.DeJonge RE, Liu XP, Deig CR, Heller S, Koehler KR, Hashino E. Modulation of Wnt signaling enhances inner ear organoid development in 3D culture. PLoS ONE. 2016;11:e0162508. doi: 10.1371/journal.pone.0162508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horiguchi H, Sano T, Toi H, Kageji T, Hirokawa M, Nagahiro S. Endolymphatic sac tumor associated with a von Hippel-Lindau disease patient: an immunohistochemical study. Mod Pathol. 2001;14:727–732. doi: 10.1038/modpathol.3880380. [DOI] [PubMed] [Google Scholar]
- 54.Megerian CA, Pilch BZ, Bhan AK, McKenna MJ. Differential expression of transthyretin in papillary tumors of the endolymphatic sac and choroid plexus. Laryngoscope. 1997;107:216–221. doi: 10.1097/00005537-199702000-00014. [DOI] [PubMed] [Google Scholar]
- 55.Megerian CA, McKenna MJ, Nuss RC, et al. Endolymphatic sac tumors: histopathologic confirmation, clinical characterization, and implication in von Hippel-Lindau disease. Laryngoscope. 1995;105:801–808. doi: 10.1288/00005537-199508000-00006. [DOI] [PubMed] [Google Scholar]
- 56.Du J, Wang J, Cui Y, et al. Clinicopathologic study of endolymphatic sac tumor (ELST) and differential diagnosis of papillary tumors located at the cerebellopontine angle. Neuropathology. 2015;35:410–420. doi: 10.1111/neup.12200. [DOI] [PubMed] [Google Scholar]
- 57.Zanoletti E, Girasoli L, Borsetto D, Opocher G, Mazzoni A, Martini A. Endolymphatic sac tumour in von Hippel-Lindau disease: management strategies. Acta Otorhinolaryngol Ital. 2017;37:423–429. doi: 10.14639/0392-100X-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bastier PL, de Mones E, Marro M, et al. Endolymphatic sac tumors: experience of three cases. Eur Arch Otorhinolaryngol. 2013;270:1551–1557. doi: 10.1007/s00405-012-2298-7. [DOI] [PubMed] [Google Scholar]