

Abstract
Reversible covalency, achieved with, for instance, highly electron-deficient olefins, offers a compelling strategy to design chemical probes and drugs that benefit from the sustained target engagement afforded by irreversible compounds, while avoiding permanent protein modification that persists following unfolding and/or proteolytic processing. So far, reversible covalency has mainly been evaluated for cysteine residues in individual kinases and the broader potential for this strategy to engage cysteines across the proteome remains unexplored. Here we describe a mass-spectrometry-based platform that integrates gel filtration (GF) with activity-based protein profiling (ABPP) to assess cysteine residues across the human proteome for both irreversible and reversible interactions with small-molecule electrophiles. Using this method, we identify numerous cysteine residues from diverse protein classes that are reversibly engaged by cyanoacrylamide fragment electrophiles, revealing the broad potential for reversible covalency as a strategy for chemical probe discovery.
Keywords: proteomics, reversible covalency, ABPP, reactive cysteines, α-cyanoacrylamides
Graphical Abstract:
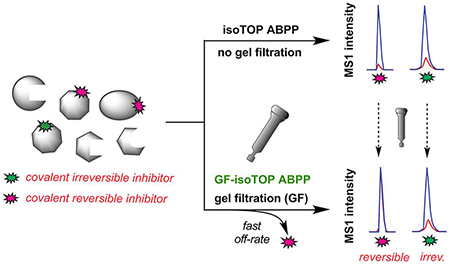
A chemical proteomic platform that integrates gel filtration (GF) with activity-based protein profiling (ABPP) provides a method to assess small-molecule electrophiles for reversible versus irreversible interactions with cysteine residues in native biological systems, revealing the broad potential for reversible covalency as a strategy for chemical probe discovery.
Chemical probes and drugs that operate by a covalent irreversible mechanism have several potentially advantageous properties, including increased duration of action, reduced pharmacokinetic sensitivity, and the potential for improved potency at otherwise shallow small-molecule binding pockets.[1–4] A number of FDA-approved drugs act by a covalent irreversible mechanism, including multiple recently approved kinase inhibitors used to treat diverse cancers.[5–7] These compounds react with non-catalytic cysteine residues in the active sites of target kinases like EGFR and BTK. Despite the remarkable success of drugs that act by a covalent irreversible mechanism, concerns remain about the potential safety and immunogenicity risks associated with the chemical modification of proteins in vivo, especially for drugs that require higher doses for efficacy, which may increase the adduction of off-target proteins.[8–9]
Advanced chemical proteomic methods have emerged to facilitate the characterization and optimization of target selectivity for covalent, irreversible drugs in vitro[10–12] and in vivo.[13] These methods, combined with additional strategies – including the design of reactive groups with i) tempered intrinsic electrophilicity,[14–16] ii) metabolic vulnerabilities that attenuate reactivity,[17] and iii) covalent reversible mechanisms of action[18–23] have expanded the optionality for design of advanced chemical probes and drugs that covalently bind to proteins.[24–25] The third strategy, which has a rich history of success for targeting catalytic serines/threonines in the active sites of hydrolases/proteases (e.g., α-ketoamides (serine[26–28]), boronic acids (targeting serine and threonine)[29], cyanamides[30–32]) has more recently been extended to cysteine (e.g., α-cyanoacrylamide[18–22], reversible formation of Meisenheimer complexes[33]), and lysine (e.g., 2-acetyl arylboronic acids[34]) residues. Optimized covalent reversible electrophiles have potential advantages of preserving the pharmacological benefits of extended on-target residence time associated with irreversibly acting compounds, while possibly also i) achieving greater selectivity through avoidance of weaker-binding (and, consequently, rapidly disassociating) off-targets, and ii) minimizing risk for idiosyncratic toxicity that may be caused by permanent modification of proteins.
Most of the methods described to date for characterizing covalent reversible electrophiles are target-specific, often employing recombinantly expressed proteins, and to our knowledge, strategies to evaluate reversible covalency on a proteome-wide scale have not yet been described. Establishing a robust method to profile the landscape of protein targets of covalent reversible electrophiles in native biological systems would enable the optimization of compound selectivity, as well as the discovery of additional proteins amenable to this form of pharmacological perturbation. Here, we describe a quantitative method that combines gel filtration (GF) with activity-based protein profiling (ABPP) to evaluate the proteome-wide target landscape of α-cyanoacrylamide fragments as a prototype cysteine-directed covalent reversible electrophile.
We adapted a competitive isoTOP-ABPP (isotopic tandem orthogonal proteolysis-ABPP) method, which has been used to quantify the interactions of cysteine[11] and lysine[10] residues with covalent irreversible electrophilic fragments, to evaluate the covalent reversible interactions of α-cyanoacrylamide fragments with cysteine residues in the human proteome (Fig. 1A). We hypothesized that introducing a GF step after fragment treatment could distinguish fragments that reversibly versus irreversibly bind to cysteines, as the former, but not latter, events should show substantially reduced competitive isoTOP-ABPP ratios, or R values (DMSO-treated/fragment-treated), following GF (Fig. 1B).
Figure 1.

isoTOP-ABPP (A) and GF-isoTOP-ABPP (B) for proteome-wide evaluation of reactivity and reversibility of cysteine-directed electrophilic compounds.
The human Ramos B cell line proteome was prepared and treated with DMSO, α-chloroacetamide fragment 1, or one of two α-cyanoacrylamides (2 or 3) (Fig. 2A). α-Chloroacetamide 1 was chosen because this electrophilic fragment has been found to show broad reactivity with cysteines in the human proteome, enabling its deployment as a “scout” fragment to discover druggable cysteines at protein-protein interfaces[11, 35] and that support E3 ligase-mediated protein degradation.[36] The electron-withdrawing nitrile group on the α-cyanoacrylamide of the corresponding 6-methoxy-tetrahydroquinoline fragments 2 and 3 elevates the reactivity of the Michael acceptor towards nucleophilic addition at the β-carbon compared to the corresponding acrylamide group and also increases the acidity of the Cα–H bond due to stabilization of the α-carbanion, rendering the reaction reversible.[18–20] α-Cyanoacrylamides have been used to create potent and selective kinase inhibitors that act by a covalent reversible mechanism.[18–22] In most of these cases, however, α-cyanoacrylamides were appended to high-affinity binding elements targeting the kinase ATP pocket. The extent to which the hyper-electrophilic α-cyanoacrylamide group can reversibly bind to cysteine residues in other proteins across the human proteome remains unknown.
Figure 2.

Proteome-wide assessment of reversiblity of cysteine-electrophilic compound interactions by GF-isoTOP-ABPP. (A) Structures of covalent irreversible (1) and covalent reversible (2 and 3) electrophiles used in the study. (B) Bar graph showing cysteines that are liganded irreversibly (purple) or reversibly (green) by compounds 1–3. (C) Scatter plot comparisons of isoTOP-ABPP R values for cysteines before and after GF. The color coding matches that used in part B to designate cysteines that are reversibly or non-reversibly liganded by compounds 1–3. Red line denotes limit of reversibility (R ≥ 4 pre-GF and ΔR ≥ 50% post-GF). Identity line (Rpre-GF = Rpost-GF) is dotted grey. Cysteines that were not liganded (R < 4 pre-GF) are depicted in grey.
Following treatment with compounds (500 μM each, 1h) or DMSO, Ramos cell proteome samples were split in half, with one portion undergoing GF on a Zeba Spin Desalting Column column (7K MWCO, 2 mL) to remove compounds. Both gel-filtered and unfiltered samples were then treated separately with an iodoacetamide (IA)-alkyne probe (100 μM, 1h), which broadly reacts with cysteine residues, and analyzed by isoTOP-ABPP to identify compound-sensitive cysteines. In total, more than 5000 cysteines were quantified on 2499 proteins (Supplementary Table 1) and individual sites were considered: 1) liganded, if they displayed R values ≥ 4 (≥ 75% reduction in IA-alkyne labeling) before GF, and 2) reversibly liganded, if the reduction in R value (ΔR) following GF was ≥ 2 fold (≥ 50%).
Both chloroacetamide 1 and α-cyanoacrylamide 2 showed broad reactivity profiles, with each electrophilic fragment liganding more than 100 cysteines in the Ramos cell proteome (Fig. 2B, Supplementary Fig. 1A and 1B, and Supplementary Table 2). α-Cyanoacrylamide 3, on the other hand, was much less reactive with the cysteine proteome, likely reflecting the sterically obstructive impact of the larger tBu capping group (Fig. 2B, Supplementary Fig. 1C, and Supplementary Table 2). The vast majority of cysteines liganded by 2 and 3 were found to be reversible, while a much smaller fraction of apparently reversible interactions were observed for 1 (Fig. 2B and C).
A comparison of the target landscape of 1 and 2 revealed a striking number of cysteines that were preferentially liganded by one of the two fragments (Fig. 3A, B–D, and Supplementary Table 3). However, this difference in target interactions is unlikely to contribute to the distinct reversibility profiles displayed by 1 and 2, as cysteines liganded by both fragments generally showed reversible interactions exclusively with fragment 2 (e.g., see REEP5_C18 in Fig. 3E and other examples in Supplementary Fig. 2). We also note that most of the cysteines preferentially liganded by 2 did not interact with the analogous acrylamide fragment SI-1 (Supplementary Fig. 1), indicating that the greater intrinsic electrophilicity of 2 contributed to its broader reactivity profile with the cysteine proteome (Supplementary Fig. 1B). We confirmed the respective reactivity profiles of REEP5_C18 with 1 and 2, and the selective reversibility of the latter interaction by gel-based ABPP, using recombinantly expressed wild type and C18A mutant forms of this protein (Fig. 3F and Supplementary Fig. 3).
Figure 3.

Comparison of protein targets of chloroacetamide 1 and α-cyanoacrylamide 2. (A) Scatter plot showing pre-GF targets of 1 (blue) and 2 (red), with overlapping targets shown in purple. Areas of high selectivity for individual compounds (> 3-fold) are shaded. (B–E) Representative MS1 spectra showing examples of cysteines that were preferentially liganded by compounds 1 or 2 – (B) C113 of PIN1, (C) C757 of IPO7, (D) C95 of UCHL3 – or generally liganded by both – (E) C18 of REEP5 (E). Examples of reversible (C, E) and irreversible (D) liganding with 2 are shown. (F) Fluorescent gel and Western blot confirmation of non-reversible and reversible interactions of C18 of REEP5 with 1 and 2, respectively. Top, gel-based ABPP of HEK293T cells expressing recombinant REEP5, REEP5_C18A or empty vector (mock, M) treated with DMSO, 1, or 2 with and without GF and then subsequently labeled with an alkyne analogue of 1 (1-alkyne) and conjugated to an azide-rhodamine tag by copper-catalyzed azide-alkyne cycloaddition chemistry for visualization (see SI for details). Bottom, recombinant protein expression was confirmed by anti-FLAG Western blotting.
The cysteines liganded by 2 were broadly distributed across different protein classes, including proteins such as transcriptional regulators and adaptors that have historically represented challenging targets for chemical probe development (Fig. 4 and Supplementary Table 4). Interestingly, while most cysteines interacted with 2 in a reversible manner, there were compelling examples of cysteines that maintained engagement with 2 post-GF, including some cysteines, which were not targeted by 1 (despite its greater overall cysteine reactivity profile across the proteome). A prominent example was the catalytic cysteine (C95) in the ubiquitin hydrolase UCHL3 (Fig. 3D). We speculate that these cases reflect a binding interaction that is sufficiently strong to preserve 2-cysteine interactions following removal of excess free compound. Consistent with this hypothesis, we confirmed that PRN629, an optimized α-cyanoacrylamide inhibitor of BTK, also maintained target engagement post-GF (Supplementary Fig. 4).
Figure 4.

Functional classes of proteins with cysteines that are liganded by compound 2.
In summary, we have developed a chemical proteomic platform to globally evaluate reversible covalency of cysteine-reactive electrophilic compounds. Building upon our experience in mapping reactive cysteines on a proteome-wide scale,[11, 35, 37] we have shown that introducing a GF step after electrophilic compound treatment and prior to IA-alkyne exposure and chemical proteomic workup can illuminate cysteines that interact with compounds in a reversible manner. We used the described platform to evaluate the proteomic reactivity of the hyper-electrophilic α-cyanoacrylamide group, revealing a strikingly broad potential to engage cysteines across diverse protein classes, in many cases with selectivity over a structurally related α-chloroacetamide. These data indicate that even the presumably modest degree of binding affinity afforded by the 6-methoxy-tetrahydroquinoline fragment recognition group is sufficient to stabilize a large number of cysteine-α-cyanoacrylamide interactions in native proteomes. That most of these interactions are reversed following GF, unlike the PRN629-BTK interaction, indicates future studies could use the persistent blockade of IA-reactivity following GF as a convenient assay to evaluate analogue compounds for improved potency of binding to specific targets of interest. As one qualification to the approach, we should note that some proteins, such as those that are part of dynamic complexes or that require small molecule/metal cofactors for stability, may unfold following GF and produce profiles that are accordingly challenging to interpret for ligand interactions. We found, for instance, that several cysteines showing apparently reversible engagement by α-chloroacetamide 1 are in ribosomal proteins (Supplementary Table 2), and it is possible that these proteins undergo complex disassembly (or unfolding) following GF to expose a greater fraction of cysteines for labeling by the IA-alkyne probe. This caveat notwithstanding, we envision that the chemical proteomic platform described herein could be applied to additional cell systems and electrophilic chemotypes to create a comprehensive map of cysteines amenable to reversible covalency for chemical probe and drug development, as well as to other nucleophilic amino acid residues and corresponding reversible covalent chemistries.[23]
Supplementary Material
Acknowledgements
We gratefully acknowledge NIH (CA231991). This work was supported by the National Cancer Institute (CA212467 to V.M.C) and by the American Cancer Society (PF-15-142-01-CDD to B.W.Z.). We thank Yan Lou and Leonard Sung for help with fragment synthesis and characterization. E.V.V. was supported by the Life Sciences Research Foundation (Pfizer Fellow). We thank Chris Joslyn for help with Gateway cloning.
References
- [1].Singh J, Petter RC, Baillie TA, Whitty A, Nat. Rev. Drug Discov 2011, 10, 307–317. [DOI] [PubMed] [Google Scholar]
- [2].Liu QS, Sabnis Y, Zhao Z, Zhang TH, Buhrlage SJ, Jones LH, Gray NS, Chem. Biol 2013, 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bauer RA, Drug Discov. Today 2015, 20, 1061–1073. [DOI] [PubMed] [Google Scholar]
- [4].Johnson DS, Weerapana E, Cravatt BF, Future Med Chem 2010, 2, 949–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, Chaves J, Wierda WG, Awan FT, Brown JR, Hillmen P, Stephens DM, Ghia P, Barrientos JC, Pagel JM, Woyach J, Johnson D, Huang J, Wang X, Kaptein A, Lannutti BJ, Covey T, Fardis M, McGreivy J, Hamdy A, Rothbaum W, Izumi R, Diacovo TG, Johnson AJ, Furman RR, 2016, 374, 323–332. [DOI] [PMC free article] [PubMed]
- [6].Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, Buggy JJ, Proc Natl Acad Sci U S A 2010, 107, 13075–13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Maione P, Rossi A, Bareschino M, Sacco PC, Schettino C, Casaluce F, Sgambato A, Gridelli C, Current pharmaceutical design 2014, 20, 3894–3900. [DOI] [PubMed] [Google Scholar]
- [8].Mah R, Thomas JR, Shafer CM, Bioorg. Med. Chem. Lett 2014, 24, 33–39. [DOI] [PubMed] [Google Scholar]
- [9].Nakayama S, Atsumi R, Takakusa H, Kobayashi Y, Kurihara A, Nagai Y, Nakai D, Okazaki O, Drug metabolism and disposition: the biological fate of chemicals 2009, 37, 1970–1977. [DOI] [PubMed] [Google Scholar]
- [10].Hacker SM, Backus KM, Lazear MR, Forli S, Correia BE, Cravatt BF, Nature chemistry 2017, 9, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Backus KM, Correia BE, Lum KM, Forli S, Horning BD, Gonzalez-Paez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, Wolan DW, Cravatt BF, Nature 2016, 534, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chen YC, Zhang C, Genes & cancer 2016, 7, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Niessen S, Dix MM, Barbas S, Potter ZE, Lu S, Brodsky O, Planken S, Behenna D, Almaden C, Gajiwala KS, Ryan K, Ferre R, Lazear MR, Hayward MM, Kath JC, Cravatt BF, Cell chemical biology 2017, 24, 1388–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chang JW, Cognetta AB 3rd, Niphakis MJ, Cravatt BF, ACS chemical biology 2013, 8, 1590–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Barf T, Covey T, Izumi R, van de Kar B, Gulrajani M, van Lith B, van Hoek M, de Zwart E, Mittag D, Demont D, Verkaik S, Krantz F, Pearson PG, Ulrich R, Kaptein A, J. Pharmacol. Exp. Ther 2017, 363, 240–252. [DOI] [PubMed] [Google Scholar]
- [16].Shindo N, Fuchida H, Sato M, Watari K, Shibata T, Kuwata K, Miura C, Okamoto K, Hatsuyama Y, Tokunaga K, Sakamoto S, Morimoto S, Abe Y, Shiroishi M, Caaveiro JMM, Ueda T, Tamura T, Matsunaga N, Nakao T, Koyanagi S, Ohdo S, Yamaguchi Y, Hamachi I, Ono M, Ojida A, Nat Chem Biol 2019, 15, 250–258. [DOI] [PubMed] [Google Scholar]
- [17].Zaro BW, Whitby LR, Lum KM, Cravatt BF, J. Am. Chem. Soc 2016, 138, 15841–15844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Serafimova IM, Pufall MA, Krishnan S, Duda K, Cohen MS, Maglathlin RL, McFarland JM, Miller RM, Frodin M, Taunton J, Nat. Chem. Biol 2012, 8, 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Miller RM, Paavilainen VO, Krishnan S, Serafimova IM, Taunton J, J. Am. Chem. Soc 2013, 135, 5298–5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Krishnan S, Miller RM, Tian BX, Mullins RD, Jacobson MP, Taunton J, J. Am. Chem. Soc 2014, 136, 12624–12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].London N, Miller RM, Krishnan S, Uchida K, Irwin JJ, Eidam O, Gibold L, Cimermancic P, Bonnet R, Shoichet BK, Taunton J, Nat. Chem. Biol 2014, 10, 1066–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bradshaw JM, McFarland JM, Paavilainen VO, Bisconte A, Tam D, Phan VT, Romanov S, Finkle D, Shu J, Patel V, Ton T, Li XY, Loughhead DG, Nunn PA, Karr DE, Gerritsen ME, Funk JO, Owens TD, Verner E, Brameld KA, Hill RJ, Goldstein DM, Taunton J, Nat. Chem. Biol 2015, 11, 525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bandyopadhyay A, Gao JM, Curr. Opin. Chem. Biol 2016, 34, 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Potashman MH, Duggan ME, J Med Chem 2009, 52, 1231–1246. [DOI] [PubMed] [Google Scholar]
- [25].Baillie TA, Angew. Chem.-Int. Edit 2016, 55, 13408–13421. [DOI] [PubMed] [Google Scholar]
- [26].Perni RB, Almquist SJ, Byrn RA, Chandorkar G, Chaturvedi PR, Courtney LF, Decker CJ, Dinehart K, Gates CA, Harbeson SL, Heiser A, Kalkeri G, Kolaczkowski E, Lin K, Luong YP, Rao BG, Taylor WP, Thomson JA, Tung RD, Wei YY, Kwong AD, Lin C, Antimicrob. Agents Chemother 2006, 50, 899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Venkatraman S, Bogen SL, Arasappan A, Bennett F, Chen K, Jao E, Liu YT, Lovey R, Hendrata S, Huang YH, Pan WD, Parekh T, Pinto P, Popov V, Pike R, Ruan S, Santhanam B, Vibulbhan B, Wu WL, Yang WY, Kong JS, Liang X, Wong J, Liu R, Butkiewicz N, Chase R, Hart A, Agrawal S, Ingravallo P, Pichardo J, Kong R, Baroudy B, Malcolm B, Guo ZY, Prongay A, Madison V, Broske L, Cui XM, Cheng KC, Hsieh YS, Brisson JM, Prelusky D, Korfmacher W, White R, Bogdanowich-Knipp S, Pavlovsky A, Bradley P, Saksena AK, Ganguly A, Piwinski J, Girijavallabhan V, Njoroge FG, J. Med. Chem 2006, 49, 6074–6086. [DOI] [PubMed] [Google Scholar]
- [28].Boger DL, Sato H, Lerner AE, Hedrick MP, Fecik RA, Miyauchi H, Wilkie GD, Austin BJ, Patricelli MP, Cravatt BF, Proc Natl Acad Sci U S A 2000, 97, 5044–5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Adams J, Kauffman M, Cancer Invest 2004, 22, 304–311. [DOI] [PubMed] [Google Scholar]
- [30].Berteotti A, Vacondio F, Lodola A, Bassi M, Silva C, Mor M, Cavalli A, ACS medicinal chemistry letters 2014, 5, 501–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mathieu C, Degrande E, Vascular health and risk management 2008, 4, 1349–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Augeri DJ, Robl JA, Betebenner DA, Magnin DR, Khanna A, Robertson JG, Wang A, Simpkins LM, Taunk P, Huang Q, Han S-P, Abboa-Offei B, Cap M, Xin L, Tao L, Tozzo E, Welzel GE, Egan DM, Marcinkeviciene J, Chang SY, Biller SA, Kirby MS, Parker RA, Hamann LG, J. Med. Chem 2005, 48, 5025–5037. [DOI] [PubMed] [Google Scholar]
- [33].Pearson RJ, Blake DG, Mezna M, Fischer PM, Westwood NJ, McInnes C, Cell chemical biology 2018, 25, 1107–1116. [DOI] [PubMed] [Google Scholar]
- [34].Akcay G, Belmonte MA, Aquila B, Chuaqui C, Hird AW, Lamb ML, Rawlins PB, Su N, Tentarelli S, Grimster NP, Su Q, Nat Chem Biol 2016, 12, 931–936. [DOI] [PubMed] [Google Scholar]
- [35].Bar-Peled L, Kemper EK, Suciu RM, Vinogradova EV, Backus KM, Horning BD, Paul TA, Ichu TA, Svensson RU, Olucha J, Chang MW, Kok BP, Zhu Z, Ihle NT, Dix MM, Jiang P, Hayward MM, Saez E, Shaw RJ, Cravatt BF, Cell 2017, 171, 696–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhang X, Crowley VM, Wucherpfennig TG, Dix MM, Cravatt BF, 2018, 443804.
- [37].Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, Cravatt BF, Nature 2010, 468, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.