

Abstract
High hyperdiploidy (HD) is the most common cytogenetic subtype of childhood acute lymphoblastic leukemia (ALL), and a higher incidence of HD has been reported in ALL patients with congenital cancer syndromes. We assessed the frequency of predisposing germline mutations in 57 HD-ALL patients from the California Childhood Leukemia Study (CCLS) via targeted sequencing of cancer-relevant genes. Three out of 57 patients (5.3%) harbored confirmed germline mutations that were likely causal, in NBN, ETV6, and FLT3, with an additional 6 patients (10.5%) harboring putative predisposing mutations that were rare in unselected individuals (<0.01% allele frequency in the Exome Aggregation Consortium, ExAC) and predicted functional (scaled CADD score ≥20) in known or potential ALL predisposition genes (SH2B3, CREBBP, PMS2, MLL, ABL1, and MYH9). Three additional patients carried rare and predicted damaging germline mutations in GAB2, a known activator of the ERK/MAPK and PI3K/AKT pathways and binding partner of PTPN11-encoded SHP2. The frequency of rare and predicted functional germline GAB2 mutations was significantly higher in our patients (2.6%) than in ExAC (0.28%, P=4.4×10−3), an observation that was replicated in ALL patients from the TARGET project (P=0.034). We cloned patient GAB2 mutations and expressed mutant proteins in HEK293 cells and found that frameshift mutation P621fs led to reduced SHP2 binding and ERK1/2 phosphorylation but significantly increased AKT phosphorylation, suggesting possible RAS-independent leukemogenic effects. Our results support a significant contribution of rare, high penetrance germline mutations to HD-ALL etiology, and pinpoint GAB2 as a putative novel ALL predisposition gene.
INTRODUCTION
The etiology of acute lymphoblastic leukemia (ALL), the most common childhood cancer, is multifactorial, involving a combination of genetic predisposition, early-life infectious exposures, and environmental toxins (reviewed in ref 1). Genome-wide association studies (GWAS) have identified common genetic variants associated with ALL risk. 2–8 Recent studies of familial ALL have revealed rare predisposing germline mutations in the hematopoietic regulator genes PAX5, SH2B3, ETV6, and IKZF1, 9–15 and next-generation sequencing in large numbers of de novo childhood ALL patients has identified at least 4% with likely functional germline mutations. 16,17 Elucidating the genetic component of ALL will be essential for risk stratification, prevention, and early detection of disease.
High hyperdiploidy (HD), characterized by a nonrandom gain of chromosomes (51–68 chromosomes at diagnosis), is the most common cytogenetic subtype of childhood ALL. Common genetic variants in CEBPE, ARID5B, PIP4K2A, and BMI1 are more strongly associated with HD than other ALL subtypes. 3,5,18–20 Moreover, the high frequency of the HD subtype in ALL patients harboring rare germline ETV6 mutations 9 and in patients with Noonan syndrome, a RASopathy caused by germline mutations in RAS pathway genes including PTPN11, 21 supports a prominent role for heritable predisposition in HD-ALL risk.
Here, we characterized predisposing germline mutations in a set of 57 HD-ALL patients, through targeted sequencing of >500 cancer-relevant genes, and identified GAB2 as a putative novel predisposition gene.
MATERIALS AND METHODS
Ethics Statement
This study was reviewed and approved by institutional review committees at the University of California Berkeley, the California Department of Public Health (CDPH), and all collaborating institutions. Written informed consent was obtained from all parents of participants.
Study subjects
Subjects were enrolled in the California Childhood Leukemia Study (CCLS), as previously described. 22 Targeted sequencing data were available for 57 HD-ALL patients diagnosed from 1995–2015, which were selected for a recent investigation of somatic alterations based on the absence of KRAS and NRAS hotspot mutations detectable by Sanger sequencing and absence of common gene copy-number deletions, and the availability of diagnostic bone marrow samples (Supporting Information Figure S1). 22 Of the 57 patients, 48% were Hispanic and 36% were non-Hispanic white (the remainder included a mixture of African-American, Asian, and other ethnicities). Median age-at-diagnosis was 4.0 years (range: 1.1–13.1). DNA was extracted from diagnostic leukemia bone marrow samples using the QIAamp DNA Blood Mini Kit (QIAGEN, Germany).
Determination of immuno-phenotype and high hyperdiploid status
Flow cytometry profiles were used to determine the ALL immune-phenotype, with those expressing CD10 or CD19 (≥20%) classed as B-lineage ALL, as described previously. 23 FISH or G-banding were used to determine ploidy, with HD patients classified as having >50 chromosomes.
Germline mutation characterization
Targeted sequencing of 538 cancer-relevant genes using the UCSF500 Cancer Gene Panel was performed in diagnostic bone marrow (i.e. tumor) DNA from 57 HD B-ALL patients, for which we previously reported the somatic mutation results. 22 Here, we evaluated putative functional germline mutations from tumor sequencing data as follows. After exclusion of single nucleotide polymorphisms (SNPs) by filtering out variants present in dbSNP database or with a minor allele frequency above 0.01% (i.e. >0.0001) in the Exome Aggregation Consortium (ExAC) Database, 24 we filtered for predicted-functional mutations using the Combined Annotation Dependent Depletion (CADD) tool version 1.3 with a CADD Phred score threshold of ≥20 (i.e. top 1% deleterious variants in the genome), as recommended for discovery of potentially causal variants. 25
Targeted sequencing data were not available from matched germline DNA samples. We capitalized on extremely high sequencing coverage (mean read depth = 596X) to predict “likely germline” mutations based on mutant allele fraction (MAF) ≥0.44. The MAF of each mutation was adjusted based on chromosome copy number, which was determined using CNVkit, 26 with whole chromosome gains identified using mean log2 copy ratio and visual confirmation of copy number scatter plots.
Manual curation of variants was performed to identify putative predisposing mutations. In addition to determining the frequency of mutations in unselected individuals (i.e. ExAC) and their predicted functional effects using CADD, we cross-checked our list of variants and genes against three publicly available databases: 1) ClinVar 27 was used to identify the clinical significance of previously reported mutations; 2) The Online Mendelian Inheritance in Man (OMIM) database (https://omim.org) was used to determine human phenotypes associated with germline mutations in each gene, such as cancer predisposition; and 3) The Catalogue of Somatic Mutations in Cancer (COSMIC) database 28 was searched to identify mutations identified previously as somatic mutations in ALL or other tumor types (Supporting Information Table S1). Mutations that were rare/absent in ExAC and predicted functional by CADD were classed as putatively causal if identified in known or suspected ALL predisposition genes and/or in genes known to be somatically altered in ALL tumors (Supporting Information Table S2), and/or genes mutated in at least 3 out of 57 (~5%) patients, and PCR and Sanger sequencing were performed for validation in patients with available germline DNA (Supporting Information Figure S1). We also assessed whether mutations were located on chromosomes that were gained, and whether the mutant or reference allele was present on the duplicated homologue.
To compare the frequency of rare and predicted functional mutations in candidate ALL predisposition genes in the 57 HD-ALL patients with the frequency of such mutations in unselected individuals, we downloaded variant call format (VCF) files containing individual-level variant data for 2,504 individuals from the 1000 Genomes Project (Phase 3). 29 We limited to coding variants in the 79 genes included in our list of ALL candidate predisposition genes (Supporting Information Table S2), and to rare and predicted functional variants as described above. Chi-squared tests were used to test for a significant difference between the frequency of rare and predicted functional germline mutations in the 57 HD-ALL patients and the 2,504 individuals from the 1000 Genomes Project.
Confirmation of germline status by Sanger sequencing
PCR and Sanger sequencing of a subset of mutations of interest (n = 37) was performed in patients with available remission or neonatal bloodspot DNA, to confirm germline status of putative causal mutations and to assess the accuracy of our germline prediction methods (primer pairs available upon request). We prioritized validation of mutations in known or candidate ALL predisposition genes, or in genes recurrently mutated (n>2) amongst the 57 patients (Supporting Information Table S1).
We also carried out Sanger sequencing of FLT3 hotspot mutation loci (exons 14, 16, and 20) in a set of 100 independent children with clinically suspected RASopathies who tested negative for Noonan syndrome-causative genes (leukemia status unknown), 30,31 to investigate whether germline FLT3 mutations account for a proportion of such patients (primer pairs available upon request).
GAB2 mutation frequency analysis
Prior to testing whether the frequency of GAB2 mutations in our HD-ALL patients was significantly higher than the frequency in ExAC, we re-filtered variants based on an allele frequency >0.01% in a pooled set of our HD-ALL patients, 309 ALL patients from TARGET (see below), plus the ExAC database. To assess the frequency of rare and predicted-functional germline GAB2 mutations in an independent set of ALL patients, we downloaded germline whole exome sequencing data (of remission DNA) from 309 ALL patients from the TARGET (Therapeutically Applicable Research to Generate Effective Treatments) Project 32 (dbGaP, gap accession: phs000464). The Genome Analysis Toolkit (GATK) tool UnifiedGenotyper was used to call single nucleotide variants and indels from the 309 BAM files, limited to the GAB2 gene region (chr11:77,926,336–78,128,868, hg19). We removed variants with read depth <10, genotype score <15, and variants present in dbSNP or with allele frequency >0.01% in the pooled set of data from ExAC and ALL patients (our study plus TARGET). To reduce false positive calls, only variants with mapping quality (MQ) scores >40, Fisher Strand (FS) <60, and Quality by Depth (QD) scores >2.0 were retained. Predicted-functional mutations were those with CADD Phred scores ≥20.
To assess the frequency of predicted-functional, rare germline GAB2 mutations in unselected individuals, we downloaded all nonsynonymous and loss-of-function GAB2 variants from the ExAC database minus TCGA subjects, 24 for a total of 53,105 individuals (106,210 chromosomes). For direct comparison with data from our HD-ALL patients and TARGET ALL patients, we removed variants with allele frequency >0.01% in the pooled set of ExAC plus ALL patients, and carried out CADD score analysis, retaining only variants with Phred scores ≥20. Fisher’s exact tests were carried out in R to test for significant differences in GAB2 mutation allele frequency in ALL patients, separately for our study and for TARGET ALL patients, compared with the allele frequency in ExAC.
Site-directed mutagenesis of GAB2 mutations
We transfected HEK293 cells with GAB2 clones expressing either the S592F, P621fs, or R154Q mutations to assess their effects on RAS/MAPK and AKT signaling pathways. Site-directed mutagenesis (SDM) was used to generate the two missense mutations S592F and R154Q using the Agilent QuikChange Lightning SDM Kit, with primer pairs designed to introduce each mutation (primer sequences available on request). In brief, the GAB2 OmicsLink expression clone plasmid with CMV promoter (EX-N0090-M02, GeneCopoeia, Inc., Rockville, MD) was grown overnight at 37°C in LB broth containing ampicillin, and plasmid DNA extracted using the Qiagen Plasmid Midi prep kit. Mutagenesis of plasmid DNA was carried out for each mutation, followed by transformation of XL10-Gold ultracompetent cells, which were subsequently grown on LB-ampicillin agar plates and incubated overnight at 37°C. Single clones were picked from each plate and grown overnight at 37°C in LB broth containing ampicillin. Plasmid DNA was then extracted as above, and Sanger sequencing performed to confirm successful insertion of mutations.
A custom-designed plasmid containing the GAB2 coding sequencing with the frameshift P621fs mutation was synthesized using the GeneArt™ Plasmid Construction Service (Thermo Fisher Scientific).
Cell culture and transfection
HEK293 cells were obtained from American Type Culture Collection (ATCC) and cultured at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) with 4.5 g/L glucose supplemented with 5% (v/v) fetal bovine serum (FBS), 100 IU/mL penicillin, and 100 µg/mL streptomycin. Cells were regularly tested by PCR to exclude mycoplasma contamination. HEK293 cells were transfected by calcium phosphate precipitation as previously described. 33 Cells were grown for 24h after transfection and serum-starved overnight using serum-free DMEM where indicated. Starved cells were stimulated with EGF (25 ng/ml; Invitrogen) before being harvested.
Immunoprecipitations and immunoblotting
All cell lysates were prepared as previously described. 33 Briefly, cells were washed three times with ice-cold phosphate-buffered saline (PBS) and lysed in BLB (10 mM K3PO4, 1 mM EDTA, 5 mM EGTA, 10 mM MgCl2, 50 mM β-glycerophosphate, 0.5% Nonidet P-40, 0.1% Brij 35, 0.1% deoxycholic acid, 1 mM sodium orthovanadate [Na3VO4], 1 mM phenylmethylsulfonyl fluoride, and a cOmplete protease inhibitor cocktail tablet [Roche]). For immunoprecipitations, cell lysates were incubated with the indicated antibodies for 2h, followed by a 1h incubation with protein A-Sepharose CL-4B beads (GE Healthcare). Immunoprecipitates were washed three times in lysis buffer and beads were eluted and boiled in 2×reducing sample buffer (5×buffer is 60 mM Tris-HCl, pH 6.8, 25% glycerol, 2% SDS, 14.4 mM 2-mercaptoethanol, 0.1% bromophenol blue). Eluates and total cell lysates were subjected to 8–12% SDS-PAGE, and resolved proteins were transferred onto polyvinylidene difluoride (PVDF) membranes for immunoblotting.
RESULTS
Analysis of tumor sequencing data in 57 HD-ALL patients highlighted 43 likely germline mutations in genes of interest; 33/37 (89%) mutations in patients with available constitutive DNA were confirmed germline in origin (Supporting Information Table S1). One of the four non-germline mutations, confirmed instead as somatic, was a TP53 mutation with MAF=0.84, likely due to copy-neutral loss of heterozygosity (LOH) at chromosome 17p. The other three somatic mutations likely had a high mutant allele fraction in patients with very high blast count. Twenty-eight (49%) out of 57 HD-ALL patients harbored at least one rare and predicted functional germline mutation in genes of interest. A similar frequency was observed in 2,504 unselected individuals from the 1000 Genomes Project (51.6%, P=0.81), suggesting that the majority of mutations identified in the HD-ALL patients may not contribute to leukemia predisposition.
Germline mutations in a putatively novel predisposition gene GAB2
Three genes, ARID1B, KDR, and GAB2, were mutated in >2 patients, with confirmed germline mutations in 3 patients each (Supporting Information Table S1 and Figure S2). We focused on GAB2 as two of the mutations (P621fs and S592F) were absent in ExAC and predicted highly deleterious (CADD scores >30), and GAB2 is an important binding partner of the PTPN11-encoded protein SHP2. 34 The P621fs frameshift mutation results in removal of serine 623, a phosphorylation target of ERK, and a SHP2 binding site at Y643, with an additional 56 codons added to the 3’ end of the GAB2 protein (Supporting Information Figures S3 and S4). The S592F mutation results in conversion from an uncharged polar serine to a hydrophobic, nonpolar phenylalanine.
The allele frequency of rare, predicted functional GAB2 mutations in our HD-ALL patients (3/114 chromosomes, 2.6%) was significantly higher than in ExAC (301/106193, 0.28%, P = 4.4 × 10−3). An additional 5 rare, predicted functional germline GAB2 mutations were identified in germline sequencing data from 309 ALL patients from the TARGET project, including frameshift mutation K357fs that results in a premature stop codon at c.377 (Supporting Information Table S3). Allele frequency of GAB2 mutations in TARGET (0.81%) was again significantly higher than in ExAC (P = 0.034). Only one of the five TARGET ALL patients had the HD subtype (Supporting Information Table S3), suggesting germline GAB2 mutations may not specifically predispose HD-ALL. The six missense mutations identified across patients in our study and TARGET were distributed across GAB2, with no indication of hotspots, suggesting potential loss-of-function effects. GAB2 is loss-of-function intolerant, with a probability of loss-of-function intolerance (pLI) score of 1.0, supporting a role for germline GAB2 mutations in disease predisposition. 24
GAB2 is a scaffolding protein that forms complexes with both SHP2 (encoded by PTPN11) and the p85 subunit of phosphoinositide 3-kinase (PI3K). Therefore, we investigated the downstream effects of each of the 3 HD-ALL patient GAB2 mutations on the MAPK/ERK and PI3K/AKT signaling pathways, in HEK293 cells. Following EGF stimulation, the S592F and R154Q mutations showed no significant effects (Supporting Information Figure S5). However, P621fs resulted in reduced SHP2 binding and significantly decreased ERK1/2 phosphorylation, but in significantly increased AKT phosphorylation, potentially through increased binding with p85 (Figure 1).
Figure 1. Effects of GAB2 frameshift mutation P621fs on ERK/MAPK and PI3K/AKT signaling pathways.
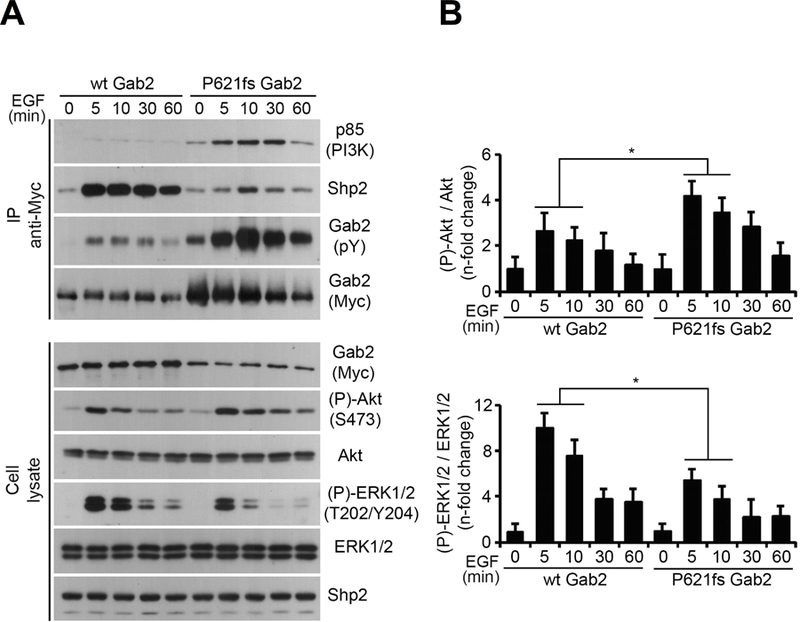
(A) HEK293 cells were transfected with MYC-tagged wild-type GAB2 or the GAB2 P621fs mutant. Cells were serum starved overnight and stimulated with EGF (25 ng/ml) over a time course (0 to 60 min). Immunoprecipitated (IP) GAB2 was then assayed for phosphorylation with an anti-phosphotyrosine antibody, as well as p85 and SHP2 association. Total and phosphorylated forms of AKT and ERK1/2 were assayed by immunoblotting on total cell lysates, as well as GAB2 and SHP2 levels. (B) Densitometric analysis of AKT phosphorylation at Ser473 was normalized to total AKT levels and expressed as fold changes over a time course of EGF stimulation. Densitometric analysis of ERK1/2 phosphorylation at T202/Y204 was normalized to total ERK1/2 levels and expressed as fold changes over a time course of EGF stimulation. Unpaired Student’s t tests were used to compare results of three replicate experiments at each time point between mutant and wildtype GAB2, with statistically significant changes indicated by asterisks (*, P < 0.05).
Germline mutations in known leukemia genes
The majority of remaining mutations were of unknown significance; however, three patients harbored known likely causal germline mutations that we confirmed via Sanger sequencing (Supporting Information Figure S2): i) frameshift mutation Ala32fs in the Nijmegen breakage syndrome gene NBN that was reported “pathogenic” in ClinVar in a patient with immunodeficiency, microcephaly, and a hereditary cancer-predisposing syndrome. Heterozygous mutations in NBN have previously been associated with childhood ALL 35 and other malignancies; 36 ii) ETV6 mutation R49C recently reported as ALL predisposition-related in a patient with HD-ALL; 9 and iii) a FLT3 mutation we recently reported at Y842C, a known somatic mutation hotspot locus in the kinase domain activation loop, which was the first implication of a germline FLT3 mutation in ALL. 22 Germline mutations in RAS pathway genes, including PTPN11, cause congenital syndromes that predispose to childhood leukemia, thus we explored whether germline FLT3 mutations might associate with such RASopathies, as this had not been previously investigated. However, sequencing of FLT3 hotspot mutation loci did not reveal any germline mutations in an independent set of 100 patients with genetically undiagnosed RASopathies. 31
One patient harbored an apparent homozygous mutation in the DNA mismatch repair gene PMS2, predicted likely pathogenic in ClinVar, although constitutive material was not available to confirm this mutation as germline. Confirmed germline and putative predisposing mutations in SH2B3, CREBBP, KMT2A (MLL), ABL1, and MYH9 were identified in 5 additional patients (Supporting Information Table S1), although further work is required to confirm their role in ALL predisposition.
Finally, we identified 8 germline mutations that resided on gained chromosomes, including an ARID5B missense mutation E864K in which the mutant allele was duplicated (Supporting Information Table S1). Indeed, for 6 out of 8 germline mutations the mutant allele was duplicated; however, this was not significantly greater than expected in a binomial test (P=0.14, one-sided).
DISCUSSION
Here, we report findings from the first investigation of predisposing germline mutations in HD-ALL. We discovered that at least 15% of patients harbored rare and putatively functional germline mutations in known or suspected ALL predisposition genes, suggesting that genetic predisposition may comprise a large component of HD-ALL etiology.
Previous studies demonstrated enrichment of HD in ALL patients with germline predisposing mutations, for example in ETV6. 9 In children with Noonan syndrome harboring germline mutations in PTPN11 or other RAS pathway genes, 76% of those that developed ALL were of the HD subtype. 21 We found a relatively high proportion of HD-ALL patients carrying rare and putatively functional germline mutations, across a range of RAS pathway, epigenetic regulatory, DNA repair, and other leukemia-related genes, suggesting that the manifestation of HD-ALL may depend on a child’s genetic predisposition. This is supported by the stronger association of SNPs in ARID5B, CEBPE, PIP4K2A, and BMI1 with HD-ALL than with other ALL subtypes. 3,5,18–20 Of interest, the low hypodiploid subtype, characterized by 32–39 chromosomes, is strongly associated with germline mutations in TP53. 37,38 Thus, there may be a stronger genetic predisposition for ALL characterized by chromosomal aneuploidies compared to other cytogenetic subtypes. The cellular stress induced by gene copy number gains or losses may require a genetic predisposition for increased cell survival and/or proliferation to enable aneuploid preleukemic clones to persist until acquisition of secondary mutations and progression to overt leukemia. Alternatively, specific germline predisposition factors may influence the likelihood of mitotic catastrophe events that result in aneuploidy.
Likely causal germline mutations were identified in NBN, FLT3, and ETV6, of which only germline ETV6 mutations have previously been associated with predisposition to high hyperdiploid ALL, 9 although somatic FLT3 mutations are more common in HD than in other ALL subtypes. 39,40 Germline mutations in RAS pathway genes cause congenital syndromes that predispose to childhood leukemia, thus we explored whether germline mutations in FLT3, an upstream activator of RAS signaling, might also associate with such RASopathies. However, we did not detect any germline FLT3 hotspot mutations in a set of patients with genetically undiagnosed RASopathies, suggesting that the phenotypic consequences may be limited to hematopoiesis.
Common variants in ARID5B contribute to risk of HD-ALL, and ARID5B SNP risk alleles are preferentially gained in HD-ALL tumors. 41 Although rare variation in this gene has not previously been implicated in ALL predisposition, it was interesting to discover a rare missense mutation in ARID5B in which the mutant allele was duplicated in the leukemia, supporting a potential leukemogenic role for this germline mutation.
We identified GAB2, another upstream activator of RAS signaling as well as PI3K/AKT signaling, as a putative novel ALL predisposition gene. GAB2 is a member of the GRB2-associated binder adaptor/scaffolding protein family, and is an important binding partner of the PTPN11-encoded protein SHP2. 34 Binding of GAB2 to SHP2 and to the p85 regulatory subunit of PI3K results in activation of the RAS/MAPK and PI3K/AKT signaling pathways respectively (reviewed in ref 42). Gain-of-function somatic and germline PTPN11 mutations play a role in HD-ALL tumorigenesis and predisposition, respectively; therefore, we predicted that germline GAB2 mutations may function via increased SHP2 binding and concomitant RAS pathway activation. However, we found that the P621fs frameshift mutation, which was absent in healthy populations and which removes a SHP2 binding site at Y643, resulted in reduced SHP2 binding and RAS/MAPK signaling but in increased PI3K/AKT activation in HEK293 cells, potentially through increased binding with p85. Activated PI3K/AKT signaling plays a crucial role in prevention of apoptosis. 43 HD leukemia cells are particularly prone to apoptosis and among the most treatable leukemias during induction therapy; 44 thus a subset of HD tumors may persist via reduced apoptosis through activation of the PI3K/AKT pathway.
Additional evidence is required, in particular from studies of familial leukemia, to confirm GAB2 as a novel ALL predisposition gene. Though only the P621fs mutation demonstrated functional effects, our analyses of patient GAB2 mutations were limited to HEK293 cells and to assessment of RAS/MAPK and PI3K/AKT pathways, and future studies should assess effects in cell types more relevant to HD-ALL and explore potential effects on JAK-STAT signaling. 45 Given the well-recognized role of gain-of-function PTPN11 mutations in ALL, discovery of loss-of-function GAB2 mutations seemed counterintuitive; however, leukemia has been reported in children with LEOPARD syndrome, caused by dominant negative PTPN11 mutations, 46,47 suggesting that RAS downregulation may contribute to leukemia risk in some patients.
We discovered a relatively high frequency of rare and predicted functional germline mutations in HD-ALL. A caveat of this study is that patients were selected for sequencing based on an absence of somatic KRAS/NRAS hotspot mutations as tested by screening via conventional PCR-Sanger sequencing, originally to enrich for discovery of novel somatic mutations. 22 Two-thirds of the 57 patients, however, still harbored somatic RAS pathway mutations, including subclonal KRAS/NRAS mutations not detectable by Sanger sequencing in addition to several mutations in FLT3 and PTPN11. Thus, our germline mutation results were unlikely to have been biased by the patient selection criteria. Another limitation was the use of tumor-only sequencing data to identify germline mutations. Although the extremely high sequencing depth enabled us to predict putative germline mutations with relatively high specificity (false positive rate ~10%), we may have missed germline mutations that resided in regions of somatic copy number loss; however, given the low frequency of somatic deletions in HD-ALL, 48 and that the 57 HD-ALL patients were selected based on their lacking any of the 8 most common gene deletions, this is unlikely to have significantly affected our findings. Our analyses were limited to known ALL-related genes and recurrently mutated genes within the targeted sequencing results; thus, studies using whole exome or genome sequencing of large numbers of patients are required to obtain a more accurate frequency of predisposing mutations in HD-ALL, especially given the range of different genes that likely contribute. Furthermore, although patient outcomes were not available in this study, future studies should assess whether predisposing germline mutations may affect risk of relapse in HD-ALL patients, which comprise a large proportion of overall relapsed ALL. 49
Supplementary Material
ACKNOWLEDGMENTS
A subset of biospecimens and/or data used in this study were obtained from the California Biobank Program, (SIS request # 26), Section 6555(b), 17 CCR. The California Department of Public Health is not responsible for the results or conclusions drawn by the authors of this publication. For recruitment of subjects enrolled in the California Childhood Leukemia Study (CCLS), the authors gratefully acknowledge the clinical investigators at the following collaborating hospitals: University of California Davis Medical Center (Dr. Jonathan Ducore), University of California San Francisco (Drs. Mignon Loh and Katherine Matthay), Children’s Hospital of Central California (Dr. Vonda Crouse), Lucile Packard Children’s Hospital (Dr. Gary Dahl), Children’s Hospital Oakland (Drs. James Feusner and Carla Golden), Kaiser Permanente Roseville (formerly Sacramento) (Drs. Kent Jolly and Vincent Kiley), Kaiser Permanente Santa Clara (Drs. Carolyn Russo, Alan Wong, and Denah Taggart), Kaiser Permanente San Francisco (Dr. Kenneth Leung), Kaiser Permanente Oakland (Drs. Daniel Kronish and Stacy Month), California Pacific Medical Center (Dr. Louise Lo), Cedars-Sinai Medical Center (Dr. Fataneh Majlessipour), Children’s Hospital Los Angeles (Dr. Cecilia Fu), Children’s Hospital Orange County (Dr. Leonard Sender), Kaiser Permanente Los Angeles (Dr. Robert Cooper), Miller Children’s Hospital Long Beach (Dr. Amanda Termuhlen), University of California, San Diego Rady Children’s Hospital (Dr. William Roberts), and University of California, Los Angeles Mattel Children’s Hospital (Dr. Theodore Moore). The authors additionally thank the families for their participation in the CCLS (formerly known as the Northern California Childhood Leukemia Study).
Funding information: This work was supported by the 500 Cancer Gene Panel Pilot Award from the UCSF Helen Diller Family Comprehensive Cancer Center (A.J.D.), and by Alex’s Lemonade Stand Foundation ‘A’ Awards (A.J.D., K.M.W.). This work was supported by research grants from the National Institutes of Health (R01 CA155461 to J.L.W., R01 CA175737 to J.L.W., R01 ES009137 to C.M., R24 ES028524 to C.M., and P01 ES018172 to C.M.) and the Environmental Protection Agency (RD83451101 to C.M.), United States. This work was also supported by operating grants from the Canadian Institutes of Health Research (PJT-152995 and MOP-142374 to P.P.R.). P.P.R. is supported by a FRQS Senior investigator career award. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health and the EPA.
Footnotes
Competing Interests: The authors have nothing to disclose.
Data Availability Statement: The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1.Wiemels J Perspectives on the causes of childhood leukemia. Chem Biol Interact 2012;196(3):59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Migliorini G, Fiege B, Hosking FJ, et al. Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood 2013;122(19):3298–3307. [DOI] [PubMed] [Google Scholar]
- 3.Papaemmanuil E, Hosking FJ, Vijayakrishnan J, et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet 2009;41(9):1006–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sherborne AL, Hosking FJ, Prasad RB, et al. Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet 2010;42(6):492–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trevino LR, Yang W, French D, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet 2009;41(9):1001–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vijayakrishnan J, Kumar R, Henrion MY, et al. A genome-wide association study identifies risk loci for childhood acute lymphoblastic leukemia at 10q26.13 and 12q23.1. Leukemia 2017;31(3):573–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiemels JL, Walsh KM, de Smith AJ, et al. GWAS in childhood acute lymphoblastic leukemia reveals novel genetic associations at chromosomes 17q12 and 8q24.21. Nat Commun 2018;9(1):286-017-02596-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu H, Yang W, Perez-Andreu V, et al. Novel susceptibility variants at 10p12.31–12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst 2013;105(10):733–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moriyama T, Metzger ML, Wu G, et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: A systematic genetic study. Lancet Oncol 2015;16(16):1659–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noetzli L, Lo RW, Lee-Sherick AB, et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet 2015;47(5):535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perez-Garcia A, Ambesi-Impiombato A, Hadler M, et al. Genetic loss of SH2B3 in acute lymphoblastic leukemia. Blood 2013;122(14):2425–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shah S, Schrader KA, Waanders E, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet 2013;45(10):1226–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Topka S, Vijai J, Walsh MF, et al. Germline ETV6 mutations confer susceptibility to acute lymphoblastic leukemia and thrombocytopenia. PLoS Genet 2015;11(6):e1005262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshida N, Sakaguchi H, Muramatsu H, et al. Germline IKAROS mutation associated with primary immunodeficiency that progressed to T-cell acute lymphoblastic leukemia. Leukemia 2017;31(5):1221–1223. [DOI] [PubMed] [Google Scholar]
- 15.Zhang MY, Churpek JE, Keel SB, et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet 2015;47(2):180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Walsh MF, Wu G, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med 2015;373(24):2336–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grobner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;555(7696):321–327. [DOI] [PubMed] [Google Scholar]
- 18.Wiemels JL, de Smith AJ, Xiao J, et al. A functional polymorphism in the CEBPE gene promoter influences acute lymphoblastic leukemia risk through interaction with the hematopoietic transcription factor ikaros. Leukemia 2016;30(5):1194–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walsh KM, de Smith AJ, Chokkalingam AP, et al. Novel childhood ALL susceptibility locus BMI1-PIP4K2A is specifically associated with the hyperdiploid subtype. Blood 2013;121(23):4808–4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Smith AJ, Walsh KM, Francis SS, et al. BMI1 enhancer polymorphism underlies chromosome 10p12.31 association with childhood acute lymphoblastic leukemia. Int J Cancer 2018. [DOI] [PMC free article] [PubMed]
- 21.Cave H, Caye A, Strullu M, et al. Acute lymphoblastic leukemia in the context of RASopathies. Eur J Med Genet 2016;59(3):173–178. [DOI] [PubMed] [Google Scholar]
- 22.de Smith AJ, Ojha J, Francis SS, et al. Clonal and microclonal mutational heterogeneity in high hyperdiploid acute lymphoblastic leukemia. Oncotarget 2016;7(45):72733–72745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aldrich MC, Zhang L, Wiemels JL, et al. Cytogenetics of hispanic and white children with acute lymphoblastic leukemia in california. Cancer Epidemiol Biomarkers Prev 2006;15(3):578–581. [DOI] [PubMed] [Google Scholar]
- 24.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536(7616):285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46(3):310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol 2016;12(4):e1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landrum MJ, Lee JM, Riley GR, et al. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014;42(Database issue):D980–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015;43(Database issue):D805–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.1000 Genomes Project Consortium, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature 2015;526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ceremsak JJ, Yu A, Esquivel E, et al. Germline RRAS2 mutations are not associated with noonan syndrome. J Med Genet 2016. [DOI] [PubMed]
- 31.Kouz K, Lissewski C, Spranger S, et al. Genotype and phenotype in patients with noonan syndrome and a RIT1 mutation. Genet Med 2016;18(12):1226–1234. [DOI] [PubMed] [Google Scholar]
- 32.Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018;555(7696):371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Houles T, Gravel SP, Lavoie G, et al. RSK regulates PFK-2 activity to promote metabolic rewiring in melanoma. Cancer Res 2018. [DOI] [PubMed]
- 34.Gu H, Pratt JC, Burakoff SJ, Neel BG. Cloning of p97/Gab2, the major SHP2-binding protein in hematopoietic cells, reveals a novel pathway for cytokine-induced gene activation. Mol Cell 1998;2(6):729–740. [DOI] [PubMed] [Google Scholar]
- 35.Mosor M, Ziolkowska I, Pernak-Schwarz M, Januszkiewicz-Lewandowska D, Nowak J. Association of the heterozygous germline I171V mutation of the NBS1 gene with childhood acute lymphoblastic leukemia. Leukemia 2006;20(8):1454–1456. [DOI] [PubMed] [Google Scholar]
- 36.Seemanova E, Jarolim P, Seeman P, et al. Cancer risk of heterozygotes with the NBN founder mutation. J Natl Cancer Inst 2007;99(24):1875–1880. [DOI] [PubMed] [Google Scholar]
- 37.Holmfeldt L, Wei L, Diaz-Flores E, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 2013;45(3):242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qian M, Cao X, Devidas M, et al. TP53 germline variations influence the predisposition and prognosis of B-cell acute lymphoblastic leukemia in children. J Clin Oncol 2018;36(6):591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang P, Kang M, Xiao A, et al. FLT3 mutation incidence and timing of origin in a population case series of pediatric leukemia. BMC Cancer 2010;10:513-2407-10-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andersson A, Paulsson K, Lilljebjorn H, et al. FLT3 mutations in a 10 year consecutive series of 177 childhood acute leukemias and their impact on global gene expression patterns. Genes Chromosomes Cancer 2008;47(1):64–70. [DOI] [PubMed] [Google Scholar]
- 41.Studd JB, Vijayakrishnan J, Yang M, Migliorini G, Paulsson K, Houlston RS. Genetic and regulatory mechanism of susceptibility to high-hyperdiploid acute lymphoblastic leukaemia at 10p21.2. Nat Commun 2017;8:14616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wohrle FU, Daly RJ, Brummer T. Function, regulation and pathological roles of the gab/DOS docking proteins. Cell Commun Signal 2009;7:22–811X-7–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/akt and apoptosis: Size matters. Oncogene 2003;22(56):8983–8998. [DOI] [PubMed] [Google Scholar]
- 44.Ito C, Kumagai M, Manabe A, et al. Hyperdiploid acute lymphoblastic leukemia with 51 to 65 chromosomes: A distinct biological entity with a marked propensity to undergo apoptosis. Blood 1999;93(1):315–320. [PubMed] [Google Scholar]
- 45.Arnaud M, Crouin C, Deon C, Loyaux D, Bertoglio J. Phosphorylation of Grb2-associated binder 2 on serine 623 by ERK MAPK regulates its association with the phosphatase SHP-2 and decreases STAT5 activation. J Immunol 2004;173(6):3962–3971. [DOI] [PubMed] [Google Scholar]
- 46.Laux D, Kratz C, Sauerbrey A. Common acute lymphoblastic leukemia in a girl with genetically confirmed LEOPARD syndrome. J Pediatr Hematol Oncol 2008;30(8):602–604. [DOI] [PubMed] [Google Scholar]
- 47.Ucar C, Calyskan U, Martini S, Heinritz W. Acute myelomonocytic leukemia in a boy with LEOPARD syndrome (PTPN11 gene mutation positive). J Pediatr Hematol Oncol 2006;28(3):123–125. [DOI] [PubMed] [Google Scholar]
- 48.de Smith AJ, Kaur M, Gonseth S, et al. Correlates of prenatal and early-life tobacco smoke exposure and frequency of common gene deletions in childhood acute lymphoblastic leukemia. Cancer Res 2017;77(7):1674–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Irving JA, Enshaei A, Parker CA, et al. Integration of genetic and clinical risk factors improves prognostication in relapsed childhood B-cell precursor acute lymphoblastic leukemia. Blood 2016;128(7):911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.