

Abstract
The urea cycle and glutamine synthetase (GS) are the two main pathways for waste nitrogen removal and their deficiency results in hyperammonemia. Here, we investigated the efficacy of liver-specific GS overexpression for therapy of hyperammonemia. To achieve hepatic GS overexpression, we generated a helper-dependent adenoviral (HDAd) vector expressing the murine GS under the control of a liver-specific expression cassette (HDAd-GS). Compared to mice injected with a control vector expressing an unrelated reporter gene (HDAd-AFP), wild-type mice with increased hepatic GS showed reduced blood ammonia levels and a concomitant increase of blood glutamine after intraperitoneal injections of ammonium chloride, whereas blood urea was unaffected. Moreover, injection of HDAd-GS reduced blood ammonia levels at baseline and protected against acute hyperammonemia following ammonia challenge in a mouse model with conditional hepatic deficiency of carbamoyl phosphate synthetase 1 (Cps1), the initial and rate-limiting step of ureagenesis. In summary, we found that upregulation of hepatic GS reduced hyperammonemia in wild-type and Cps1-deficient mice, thus confirming a key role of GS in ammonia detoxification. These results suggest that hepatic GS augmentation therapy has potential for treatment of both primary and secondary forms of hyperammonemia.
Keywords: urea cycle disorders, glutamine synthetase, helper-dependent adenoviral vectors, hyperammonemia, carbamoyl phosphate synthetase 1 deficiency
INTRODUCTION
The glutamate-ammonia ligase enzyme (GLUL), typically known as glutamine synthetase (GS) (E.C. 6.3.1.2), is a cytosolic enzyme that catalyzes ATP-dependent production of glutamine from glutamate and ammonia. GS is highly conserved within living organisms and its expression and activity have been detected in several tissues including liver and brain (mostly in astrocytes) (Adeva et al 2012). As a serial pathway, together with the urea cycle enzymes expressed in the peri-portal region, peri-venous GS is a major ammonia detoxification system (Haussinger 1990; Meijer et al 1990). Both systems are required for complete ammonia detoxification (Haussinger 1990). In mice, liver-specific deletion of GS results in hyperammonemia leading to neuronal and behavioral abnormalities (Qvartskhava et al 2015). Similar to inherited urea cycle disorders, patients with the ultra-rare genetic GS deficiency have chronic hyperammonemia in addition to decreased levels of glutamine in body fluids (Haberle et al 2005). Moreover, GS expression is reduced in patients with liver cirrhosis and chronic hyperammonemia (Gebhardt and Reichen 1994). Taken together, evidence both in mice and humans illustrate a key role of GS in ammonia homeostasis leading to the concept that the urea cycle and GS contribute equally to ammonia detoxification (Hakvoort et al 2017). Based on these considerations, we reasoned that GS augmentation therapy can be an effective treatment against hyperammonemia, a life-threatening condition requiring the development of more effective therapeutic approaches (Matoori and Leroux 2015).
MATERIALS AND METHODS
Mouse procedures.
Male wild-type (WT) 6-week-old C57BL/6 (Charles River Laboratories) and adult female transgenic Cps1tm1c/tm1c mice (Khoja et al 2018) were randomly assigned to treatment groups. Investigators were not blinded to allocation during experiments and outcome assessment. All intravenous (i.v.) injections were performed by retro-orbital injections under isoflurane anesthesia. Vectors were prepared in sterile pharmaceutical-grade saline. WT and Cps1tm1c/tm1c mice were injected with a dose of 1×1013 viral particles (vp)/kg of HDAd-GS or HDAd-AFP (α-fetoprotein) as a control. The ammonium intraperitoneal (i.p.) challenge in WT mice was performed at 4 weeks post-vector injection as described previously (Soria et al 2018). Briefly, mice were overnight fasted before i.p. injections of 10 mmol/kg of ammonium chloride (Merck) dissolved in water. Three days after the injections of HDAd expressing either AFP or GS, Cps1tm1c/tm1c mice were injected with 1.5×1010 genome copies (gc)/mouse of AAV8 expressing Cre recombinase under the control of the thyroxine binding globulin promoter (TBG, liver-specific) (University of Pennsylvania Vector Core, Philadelphia, PA). Cps1tm1c/tm1c mice were weighed daily and monitored for any signs of poor health. In Cps1tm1c/tm1c mice the i.p. ammonia challenge was also performed 4 weeks after the injections of the HDAd vectors but with a lower dose of i.p. 5 mmol/kg ammonium chloride (Fisher-Scientific) and without prior fasting. Blood samples were collected by retro-orbital bleedings at the indicated time-points. Mice were sacrificed by cervical dislocation or isoflurane overdose. Serum or plasma and liver samples were harvested and stored at −80°C until analyses.
Helper-dependent adenoviral (HDAd) vectors.
Mouse GS cDNA was synthesized by Life Technologies (Carlsbad, CA). HDAd-GS and HDAd-AFP vectors both bear a liver-specific expression cassette (Brunetti-Pierri et al 2006) driving the expression of either mouse GS or baboon AFP, respectively. HDAd vectors were produced as previously reported (Palmer and Ng 2003) in 116 cells regularly tested and found negative for Mycoplasma by quantitative real time PCR.
Analyses of serum and plasma samples.
Serum or plasma ammonia levels were measured by an ammonia colorimetric assay kit (BioVision Incorporated; Cat# K370–100) or (Abcam; Cat# ab83360), respectively. Serum or plasma glutamine concentrations were measured by a colorimetric assay kit (BioVision Incorporated; Cat# K556–100). Serum urea content was determined by an assay kit based on Jung’s method (BioVision Incorporated; Cat# K376–100). For blood urea nitrogen (BUN), 10 μl of plasma per sample were pipetted into cups and placed into a Vet Excel Clinical Chemistry Analyzer (Alfa Wassermann Diagnostic Technologies, West Caldwell, NJ). Calculation of urea nitrogen concentration was performed automatically by the clinical chemistry system. Hepatic ATP determination was performed by a fluorometric assay kit according to the manufacturer’s instructions (BioVision Incorporated; Cat# K354–100). Liver lysates were made by homogenization in the corresponding hydrolysis buffer using a TissueLyser LT (Qiagen). Hepatic ATP levels were normalized for protein concentrations determined by Bradford Reagent (Bio-Rad).
Western blotting.
Liver specimens were homogenized in RIPA buffer in the presence of complete protease inhibitor cocktail (Sigma), incubated for 20 min at 4°C and centrifuged at 13,200 rpm for 10 min. Pellets were discarded and cell lysates were used for western blots. Total protein concentration in cellular extracts was measured using the Bradford Reagent (Bio-Rad). Protein extracts were separated by SDS–PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes. Blots were blocked with TBS-Tween-20 containing 5% non-fat milk for 1 h at room temperature followed by incubation with primary antibody overnight at 4°C. Primary antibodies were: rabbit anti-GS (Abcam; Cat# ab16802), rabbit anti-OAT (Abcam; Cat# ab137679), and rabbit anti-p115 (Marra et al 2001). Proteins of interest were detected with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG antibody (GE Healthcare). Peroxidase substrate was provided by ECL Western Blotting Substrate kit (Pierce). Densitometric analyses of the westernblotting bands were performed using ImageJ Software.
Immunofluorescence.
Immunofluorescence was performed as previously described (Khoja et al 2018). Briefly, liver sections were fixed in 10% neutral buffered formalin, stored in 70% ethanol and embedded in paraffin blocks followed by cutting the sections at 4μm thickness. Sections were then deparaffinized in xylene and rehydrated in serial ethanol washes. Antigen retrieval was performed in 10 mM sodium citrate buffer pH 6.0, after which the sections were permeabilized in 0.1% Triton X-100 and blocked with 10% normal goat serum. Slides were incubated with anti-CPS1 antibody (Abcam; Cat# ab45956) and anti-GLUL antibody (Abcam; Cat# ab64613), both at 1 μg/ml in the blocking buffer overnight at 4°C. Secondary antibody staining was performed at room temperature for 1 hour using Alexa Fluor 594 goat anti-rabbit (Invitrogen A-11012) and Alexa Fluor 488 goat anti-mouse (Invitrogen A-11001) antibodies. Cell nuclei were counterstained with DAPI using VECTASHIELD Antifade Mounting Medium. Visualization of the stained sections was performed using Olympus IX71 Fluorescence Microscope.
RNA extraction and real time PCR.
Individual liver pieces were homogenized and RNA was extracted by column purification (Roche; Cat# 11828665001). Purified RNA was used to generate cDNA (Roche; Cat# 04379012001) for qPCR with BioRad SYBR Green (Bio-Rad; Cat# 1725270) and IQ2 iCycler. β-actin gene was used as endogenous control. Fold changes were calculated using the DDCt method. Cps1 primers were: forward CACCAATTTCCAGGTGACCA; reverse TACTGCTTTAGGCGGCCTTT; Glul primers were: forward CCACTTGAACAAAGGCATCA; reverse GTCCTTCTCCGGTACCATCA; Actb primers were: CTAAGGCCAACCGTGAAAAG; reverse ACCAGAGGCATACAGGGACA.
Statistical Analyses.
Data are expressed as means ± SEM. A two-tailed unpaired Student’s t test was performed when comparing two groups of mice. One-way ANOVA and Tukey’s post-hoctests were performed when comparing more than two groups relative to a single factor. Two-way ANOVA and Tukey’s post-hoc tests or Sidak’s multiple tests were performed when comparing two groups relative to two factors. No statistical methods were used to predetermine the sample size. A p value < 0.05 was considered statistically significant.
RESULTS
Upregulation of hepatic GS protects wild-type mice against acute hyperammonemia.
HDAd vectors can efficiently deliver genes to hepatocytes to mediate long-term, high-level transgene expression without inducing chronic toxicity (Brunetti-Pierri and Ng 2011) and have been successfully used in mouse models of a variety of inherited liver diseases (Pastore et al 2013; Castello et al 2016). To investigate the potential of hepatic GS augmentation as a therapeutic approach for hyperammonemia, we constructed an HDAd vector expressing the murine GS under the control of a liver-specific expression cassette (HDAd-GS) (Fig. 1A). Compared to C57BL/6 WT control mice injected with HDAd vector encoding the unrelated nontoxic, nonimmunogenic AFP reporter gene under the control of the same liver-specific expression cassette, administration of the vector expressing GS resulted in increased hepatic GS protein expression levels by western blot (band densitometric densities of HDAd-AFP: 1.00 ± 0.05 AU vs. HDAd-GS: 1.88 ± 0.20 AU; p = 0.022, two-tailed unpaired Student’s t test; n = 3/group) (Fig. 1B). Despite no changes in serum ammonia at baseline, mice with hepatic GS upregulation showed a 39% reduction in serum ammonia at 0.5 h after i.p. ammonium chloride challenge (p<0.05; Fig. 1C). The reduction of blood ammonia was accompanied by a concomitant increase in serum glutamine levels in mice with hepatic GS overexpression compared to HDAd-AFP-injected controls (Fig. 1D) whereas serum urea was unaffected (Fig. 1E). Hepatic content of ATP was similar between HDAd-GS injected mice and AFP-treated controls (Supplementary Fig. 1), suggesting that no changes in energy levels were induced by GS-overexpression. Taken together, these results suggest that liver-specific GS overexpression improved ammonia detoxification through an increase in hepatic glutamine synthesis in WT mice with acute hyperammonemia.
Fig 1. Hepatic GS overexpression protects wild-type mice against acute hyperammonemia.
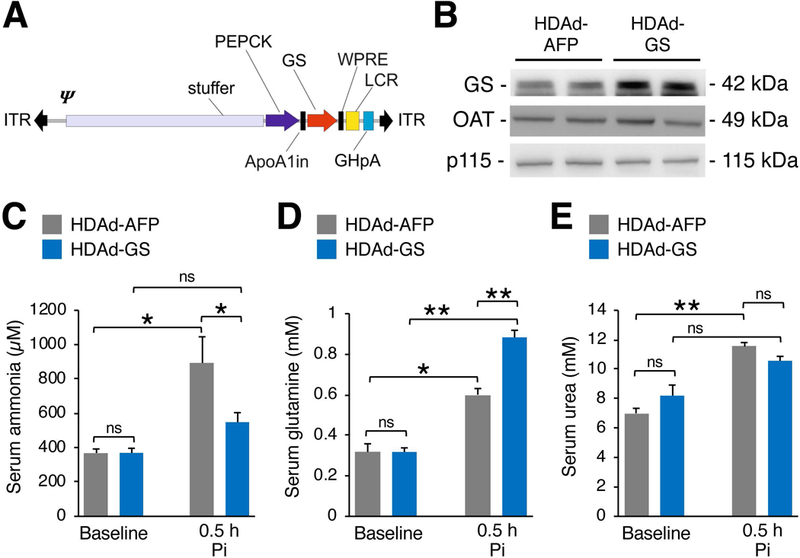
(A) A helper dependent adenovirus was produced to express murine GS under control of the PEPCK promoter (HDAd-GS). The expression cassette also contains inverted terminal repeats (ITR), a packaging signal (Ψ), the ApoA1 intron (ApoA1in), the hepatic locus control region (LCR), and a growth hormone polyadenylation signal (GHpA). (B) Western blot of GS in livers of HDAd-GS injected C57BL/6 wild-type (WT) mice compared to controls injected with a control vector expressing an unrelated reporter gene (HDAd-AFP) at twelve weeks post-vector injection. (C-E) Serum ammonia, glutamine, and urea at baseline and 0.5 h after i.p. injections of ammonium chloride (10 mmol/kg) in WT mice injected with HDAd-GS or HDAd-AFP vector. Ornithine aminotransferase (OAT) and p115 were used as loading controls. All values are shown as averages ± S.E.M. (n ≥ 9/group). *p<0.05; **p< 0.01 (Two-way ANOVA). Abbreviations: Pi= post-injection; ns= not significant; GS= glutamine synthetase; AFP= α-fetoprotein.
Liver-specific GS overexpression improved ammonia detoxification in Cps1-deficient mice.
To investigate whether increased hepatic GS expression is effective for therapy of hyperammonemia due to urea cycle disorders, we injected HDAd-GS in mice with deficiency of carbamoyl phosphate synthetase 1 (CPS1), the initial and rate limiting step in ureagenesis (Khoja et al 2018). Deletion of the Cps1 locus was achieved in adult transgenic Cps1tm1c/tm1c mice by i.v. injection of a low dose of serotype 8 adeno-associated viral (AAV8) vector expressing the Cre recombinase inducing non-lethal increase in plasma ammonia (Khoja et al 2018). Cre-mediated reduction in hepatic Cps1 mRNA levels was similar in mice injected with HDAd expressing either AFP or GS (Fig. 2A). Immunofluorescence on livers with CPS1 antibody also showed similar reduction of CPS1 levels in mice expressing either AFP or GS when compared to WT animals (Fig. 2B). The decrease in blood urea nitrogen (BUN) compared to WT controls was also similar among the two groups (Fig. 2C). As expected, livers of HDAd-GS injected mice showed increased GS expression compared to HDAd-AFP controls (p<0.01; Fig. 3A) and immunofluorescence confirmed that GS signal was expanded outside the peri-venous region in mice receiving HDAd-GS whereas GS expression was confined to the central vein in HDAd-AFP-injected mice (Fig. 3B). Moreover, mice treated with HDAd-GS showed an increased plasma glutamine/ammonia ratio (p<0.05; Fig. 3C), suggesting that increased glutamine synthesis is responsible for the reduced blood ammonia induced by HDAd-GS. Cps1-deficient mice injected with HDAd-GS showed reduced serum ammonia at baseline and after i.p. challenge with ammonium chloride (Fig. 4). Baseline plasma ammonia levels were reduced to wild-type levels in Cps1-deficient mice injected with HDAd-GS (Fig. 4). Reduced ammonia levels were also detected at 20 min and 60 min post-ammonia challenge although these levels were higher than wild-type mice (Fig. 4). Taken together, these in vivo data support the therapeutic potential of hepatic GS augmentation for treatment of acute hyperammonemia in an inherited ammonia detoxification disorder.
Fig. 2. Expression of CPS1 in livers of HDAd-GS and HDAd-AFP injected mice.
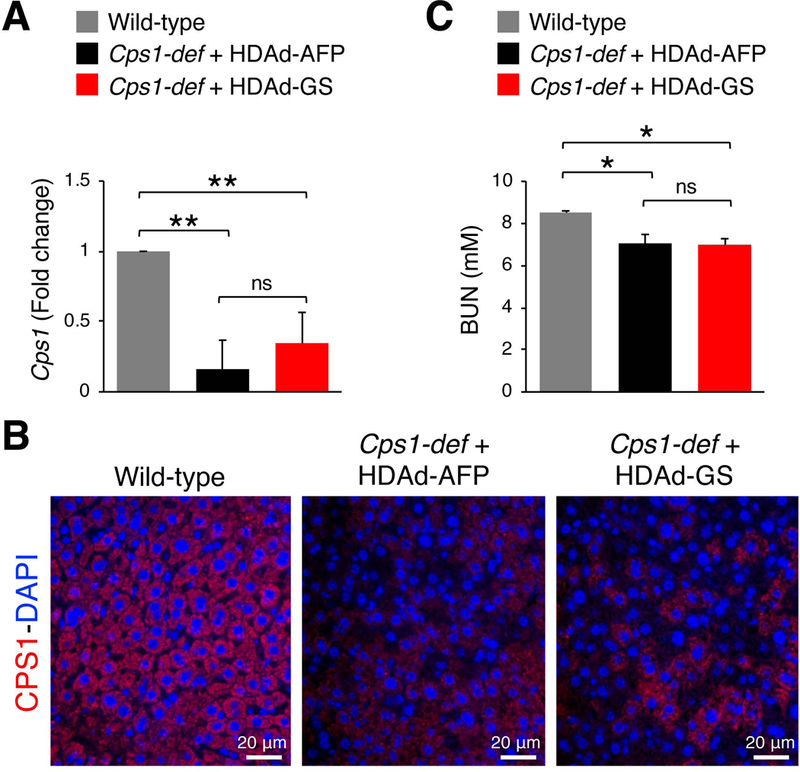
(A) Cps1 mRNA expression in livers harvested at day twenty-eight (D28) post-vector administration in Cre-mediated Cps1-deficient (Cps1-def) mice injected with HDAd-GS or HDAd-AFP compared to control wild-type animals (n = 5/group). **p< 0.01 (One-way ANOVA). (B) Representative images of immunofluorescence microscopy for CPS1 (red) in livers harvested at D28 post-vector administration in Cps1-def mice injected with HDAd-GS or HDAd-AFP compared to control wild-type animals. (C) BUN in plasma harvested at day twenty-eight (D28) post-vector administration in Cre-mediated Cps1-deficient (Cps1-def) mice injected with HDAd-GS or HDAd-AFP compared to control wild-type animals (n = 5/group). *p< 0.05 (One-way ANOVA). Data represent averages ± S.E.M. Abbreviations: ns= not significant; BUN= blood urea nitrogen; D= day.
Fig. 3. Expression of GS in livers of HDAd-GS and HDAd-AFP injected mice.
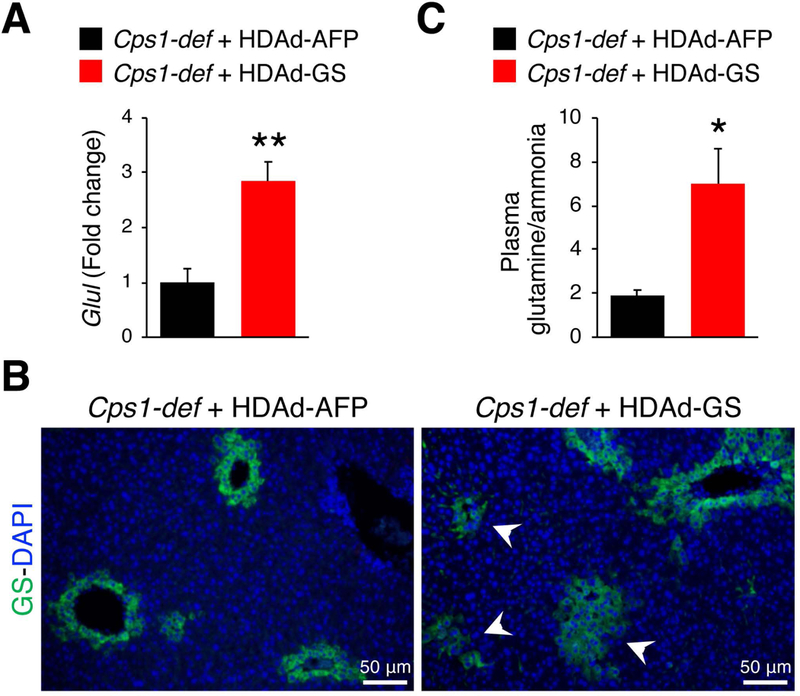
(A) Glul real time PCR on mRNA extracted from livers of Cps1-deficient (Cps1-def) mice injected with HDAd-GS or HDAd-AFP at day twenty-eight (D28). **p< 0.01 (unpaired Student’s t test) (n = 5/group). (B) Hepatic immunostaining for GS (green) in livers of Cps1-def mice injected with HDAd-AFP or HDAd-GS harvested at D28. White arrow heads point to hepatocytes with GS expression outside the peri-venous area. (C) Plasma glutamine/ammonia ratio at D28 in Cps1-def mice injected with HDAd-GS or HDAd-AFP. *p< 0.05 (unpaired Student’s t test) (n = 5/group). Data represent averages ± S.E.M.
Fig 4. Liver-specific GS overexpression improved ammonia detoxification in Cps1-deficient mice.
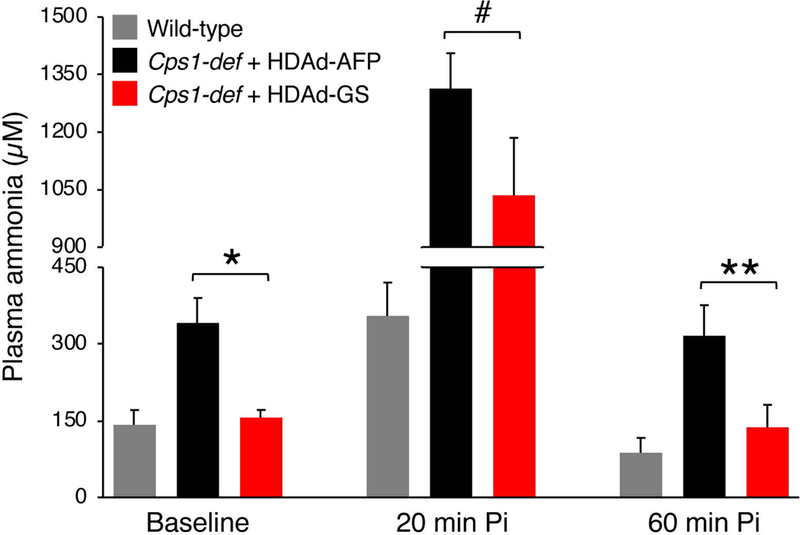
Plasma ammonia in Cps1-deficient (Cps1-def) mice injected with HDAd-GS (n = 9) or HDAd-AFP vector (n = 10) at baseline, 20 min and 60 min after i.p. injections of ammonium chloride (5 mmol/kg). #p≤ 0.05; *p< 0.05; **p< 0.01 (Two-way ANOVA; p= 0.0016). Control ammonia-treated wild-type mice are also included (n = 5). Data represent averages ± S.E.M. Abbreviations: Pi= post-injection.
DISCUSSION
This study supports the concept that liver GS plays a key role in ammonia homeostasis and provides a proof-of-concept that hepatic GS upregulation improves ammonia detoxification. Either chemical (Haussinger 1983; Haussinger and Gerok 1984) or genetic (Qvartskhava et al 2015; Hakvoort et al 2017) inhibition of hepatic GS expression/activity resulted in increased blood ammonia, clearly showing an important contribution of GS in ammonia detoxification.
In the present study, we overexpressed GS in liver by the hepatotropic HDAd vectors and we observed improved ammonia detoxification in WT mice with acute hyperammonemia. The increased ammonia detoxification was associated with enhanced glutamine synthesis without changes in serum urea. Consistent with this data, increased activity of muscle GS induced by ornithine-phenylacetate treatment or muscle-directed gene transfer reduced blood ammonia levels in rat models of chronic liver failure (Jover-Cobos et al 2014) and acute hyperammonemia (Torres-Vega et al 2015), respectively. Nevertheless, we showed for the first time that increased hepatic GS is effective at reducing hyperammonemia. Despite finding GS protein localization is expanded in the peri-portal zone, hepatic energy stores were unaffected, suggesting that exogenous GS augmentation does not affect other functions of the liver. Whether a residual activity of the urea cycle is needed for GS augmentation therapy to be effective cannot be established because these studies were performed in mice with residual CPS1 activity. Nevertheless, this study supports that GS upregulation might be a therapeutic option for primary hyperammonemia in the deficiency of CPS1, the most severe urea cycle defect due to a defect in the initial incorporation of ammonia into intermediate compounds of the urea cycle (Diez-Fernandez et al 2014). Available treatments for this disease, such as protein-restricted diet, supplementation with urea cycle intermediates, or ammonia scavenger drugs are often insufficient, and liver transplantation is the only available effective therapeutic option (Leonard and McKiernan 2004).
Secondary hyperammonemia is defined as an increase in blood ammonia driven by inhibition of ureagenesis as consequence of accumulation of toxic metabolites or by substrate deficiencies (Haberle 2013). The most common examples of this type of hyperammonemia are organic acidemias in which urea synthesis is impaired by reduced production of N-acetylglutamate (NAG), the allosteric activator of ureagenesis and CPS1 inhibition (Stewart and Walser 1980; Aires et al 2011; Adeva et al 2012). However, secondary hyperammonemia also occurs in fatty acid oxidation defects, carnitine-associated deficiency, lysinuric protein intolerance, and other disorders (Haberle 2013). It is likely that all these conditions can be ameliorated by enhancement of hepatic glutamine synthesis. In addition, hepatic GS upregulation might be effective in either acute or chronic acquired liver disease resulting in reduced urea cycle capacity (Nielsen et al 2007; Olde Damink et al 2009; Thomsen et al 2014). Moreover, treatments aiming at increasing GS activity can be combined with available treatments such as low protein diet, ammonia scavengers, or the recently described activation of hepatic autophagy (Soria et al 2018) to obtain more effective control of blood ammonia levels. Interestingly, improved ammonia control can be obtained by supplementation with glutamate (or its precursors) or phenylacetate to enhance ammonia removal by increased synthesis and clearance of glutamine, respectively (Hakvoort et al 2017).
Hepatic GS upregulation might be also achieved by systemic mRNA delivery, a therapeutic strategy that has been successfully used for correction on an inborn error of liver metabolism (An et al 2017). Moreover, GS augmentation therapy might be achieved by uploading of GS onto lipid- or polymeric-based nanoparticles (Yu et al 2016) for systemic delivery or with membrane derived from natural cells such as erythrocytes or white blood cells (Narain et al 2017). In addition, ammonia detoxification may be further improved by vesicles uploaded with recombinant GS enzyme through for liposome-supported peritoneal dialysis (Forster et al 2014).
Small molecule drugs upregulating hepatic GS expression are another attractive approach. Interestingly, glucocorticoids are known to positively regulate expression of GS at the transcriptional level in multiple tissues (Lie-Venema et al 1998). Dexamethasone was indeed found to be effective in mice at increasing ammonia detoxification (Hakvoort et al 2017). However, overexpression of GS in astrocytes is likely to be detrimental because it could lower the threshold of brain toxicity since cerebral edema depends on glutamine synthesis in astrocytes (Takahashi et al 1991). Hence, treatments with low glucocorticoid doses or other drugs upregulating GS in livers but not in astrocytes are desirable.
Current guidelines for management of patients with suspected or confirmed diagnosis of urea cycle disorders recommend avoidance of steroids because they induce protein catabolism and can trigger hyperammonemia (Haberle et al 2012). This recommendation is based on anecdotal experience in patients with acute decompensation following steroid treatments (Summar et al 2005). The results of our study suggest that GS upregulation is protective against hyperammonemia although the increased GS expression in astrocytes is likely to be detrimental.
In summary, our data show that liver-specific GS augmentation improved ammonia detoxification in WT and Cps1-deficient mice thus confirming a key role of GS in ammonia homeostasis. Hence, hepatic GS augmentation therapy has potential for treatment of both primary and secondary forms of hyperammonemia.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. A. Iuliano from the Telethon Institute of Genetics and Medicine (TIGEM) Bioinformatics Core for assistance with statistical analyses and Dr. M.A. De Matteis (TIGEM) for kindly providing the anti-p115 antibody. This work was supported by the European Research Council (IEMTx) (N.B.-P.); Fondazione Telethon Italy Grants TCBP37TELC and TCBMT3TELD (to N.B.-P.); and the US National Institutes of Health NIH (5R21NS091654–02 to G.S.L.). L.R.S. was partially supported by the Dulbecco Telethon Institute International Mobility for Postdoctoral Research Training (DTI-IMPORT) Marie Skłodowska-Curie COFUND program.
ABBREVIATIONS
- AAV
Adeno-associated virus
- AFP
Alpha-fetoprotein
- AU
Arbitrary Units
- BUN
Blood urea nitrogen
- CPS1
Carbamoyl Phosphate Synthetase 1
- gc
Genome copies
- Glul
Glutamate-ammonia ligase (gene name)
- GS
Glutamine Synthetase
- HDAd
Helper-Dependent Adenoviral vector
- i.p.
Intraperitoneal
- i.v.
Intravenous
- NAG
N-acetylglutamate
- OAT
Ornithine aminotransferase
- Pi
Post-injection
- vp
Viral particles
- WT
Wild-type
Footnotes
COMPLIANCE WITH ETHICS GUIDELINES
Conflict of interest: L.R. Soria, M. Nitzahn, A. De Angelis, S. Khoja, S. Attanasio, P. Annunziata, D.J. Palmer, P. Ng, and N. Brunetti-Pierri have no conflicts of interest to declare.
G.S. Lipshutz has served as a consultant to Audentes Therapeutics unrelated to the studies conducted herein.
Informed consent: This article does not contain any studies with human subjects performed by any of the authors.
Animal rights: Mouse procedures were performed in accordance with regulations and were authorized by both the Italian Ministry of Health and the IACUC of David Geffen School of Medicine at UCLA, Los Angeles, United States.
REFERENCES
- Adeva MM, Souto G, Blanco N, Donapetry C (2012) Ammonium metabolism in humans. Metabolism 61: 1495–1511. [DOI] [PubMed] [Google Scholar]
- Aires CC, van Cruchten A, Ijlst L, et al. (2011) New insights on the mechanisms of valproate-induced hyperammonemia: inhibition of hepatic N-acetylglutamate synthase activity by valproyl-CoA. J Hepatol 55: 426–434. [DOI] [PubMed] [Google Scholar]
- An D, Schneller JL, Frassetto A, et al. (2017) Systemic Messenger RNA Therapy as a Treatment for Methylmalonic Acidemia. Cell Rep 21: 3548–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunetti-Pierri N, Ng P (2011) Helper-dependent adenoviral vectors for liver-directed gene therapy. Hum Mol Genet 20: R7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunetti-Pierri N, Ng T, Iannitti DA, et al. (2006) Improved hepatic transduction, reduced systemic vector dissemination, and long-term transgene expression by delivering helper-dependent adenoviral vectors into the surgically isolated liver of nonhuman primates. Hum Gene Ther 17: 391–404. [DOI] [PubMed] [Google Scholar]
- Castello R, Borzone R, D’Aria S, Annunziata P, Piccolo P, Brunetti-Pierri N (2016) Helper-dependent adenoviral vectors for liver-directed gene therapy of primary hyperoxaluria type 1. Gene Ther 23: 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez-Fernandez C, Hu L, Cervera J, Haberle J, Rubio V (2014) Understanding carbamoyl phosphate synthetase (CPS1) deficiency by using the recombinantly purified human enzyme: effects of CPS1 mutations that concentrate in a central domain of unknown function. Mol Genet Metab 112: 123–132. [DOI] [PubMed] [Google Scholar]
- Forster V, Signorell RD, Roveri M, Leroux JC (2014) Liposome-supported peritoneal dialysis for detoxification of drugs and endogenous metabolites. Sci Transl Med 6: 258.ra. [DOI] [PubMed] [Google Scholar]
- Gebhardt R, Reichen J (1994) Changes in distribution and activity of glutamine synthetase in carbon tetrachloride-induced cirrhosis in the rat: potential role in hyperammonemia. Hepatology 20: 684–691. [PubMed] [Google Scholar]
- Haberle J (2013) Clinical and biochemical aspects of primary and secondary hyperammonemic disorders. Arch Biochem Biophys 536: 101–108. [DOI] [PubMed] [Google Scholar]
- Haberle J, Boddaert N, Burlina A, et al. (2012) Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis 7: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberle J, Gorg B, Rutsch F, et al. (2005) Congenital glutamine deficiency with glutamine synthetase mutations. N Engl J Med 353: 1926–1933. [DOI] [PubMed] [Google Scholar]
- Hakvoort TB, He Y, Kulik W, et al. (2017) Pivotal role of glutamine synthetase in ammonia detoxification. Hepatology 65: 281–293. [DOI] [PubMed] [Google Scholar]
- Haussinger D (1983) Hepatocyte heterogeneity in glutamine and ammonia metabolism and the role of an intercellular glutamine cycle during ureogenesis in perfused rat liver. Eur J Biochem 133: 269–275. [DOI] [PubMed] [Google Scholar]
- Haussinger D (1990) Nitrogen metabolism in liver: structural and functional organization and physiological relevance. Biochem J 267: 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussinger D, Gerok W (1984) Hepatocyte heterogeneity in ammonia metabolism: impairment of glutamine synthesis in CCl4 induced liver cell necrosis with no effect on urea synthesis. Chem Biol Interact 48: 191–194. [DOI] [PubMed] [Google Scholar]
- Jover-Cobos M, Noiret L, Lee K, et al. (2014) Ornithine phenylacetate targets alterations in the expression and activity of glutamine synthase and glutaminase to reduce ammonia levels in bile duct ligated rats. J Hepatol 60: 545–553. [DOI] [PubMed] [Google Scholar]
- Khoja S, Nitzahn M, Hermann K, et al. (2018) Conditional disruption of hepatic carbamoyl phosphate synthetase 1 in mice results in hyperammonemia without orotic aciduria and can be corrected by liver-directed gene therapy. Mol Genet Metab 124: 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard JV, McKiernan PJ (2004) The role of liver transplantation in urea cycle disorders. Mol Genet Metab 81 Suppl1: S74–78. [DOI] [PubMed] [Google Scholar]
- Lie-Venema H, Hakvoort TB, van Hemert FJ, Moorman AF, Lamers WH (1998) Regulation of the spatiotemporal pattern of expression of the glutamine synthetase gene. Prog Nucleic Acid Res Mol Biol 61: 243–308. [DOI] [PubMed] [Google Scholar]
- Marra P, Maffucci T, Daniele T, et al. (2001) The GM130 and GRASP65 Golgi proteins cycle through and define a subdomain of the intermediate compartment. Nat Cell Biol 3: 1101–1113. [DOI] [PubMed] [Google Scholar]
- Matoori S, Leroux JC (2015) Recent advances in the treatment of hyperammonemia. Adv Drug Deliv Rev 90: 55–68. [DOI] [PubMed] [Google Scholar]
- Meijer AJ, Lamers WH, Chamuleau RA (1990) Nitrogen metabolism and ornithine cycle function. Physiol Rev 70: 701–748. [DOI] [PubMed] [Google Scholar]
- Narain A, Asawa S, Chhabria V, Patil-Sen Y (2017) Cell membrane coated nanoparticles: next-generation therapeutics. Nanomedicine (Lond) 12: 2677–2692. [DOI] [PubMed] [Google Scholar]
- Nielsen SS, Grofte T, Tygstrup N, Vilstrup H (2007) Cirrhosis and endotoxin decrease urea synthesis in rats. Hepatol Res 37: 540–547. [DOI] [PubMed] [Google Scholar]
- Olde Damink SW, Jalan R, Dejong CH (2009) Interorgan ammonia trafficking in liver disease. Metab Brain Dis 24: 169–181. [DOI] [PubMed] [Google Scholar]
- Palmer D, Ng P (2003) Improved system for helper-dependent adenoviral vector production. Mol Ther 8: 846–852. [DOI] [PubMed] [Google Scholar]
- Pastore N, Blomenkamp K, Annunziata F, et al. (2013) Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha-1-anti-trypsin deficiency. EMBO Mol Med 5: 397–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qvartskhava N, Lang PA, Görg B, et al. (2015) Hyperammonemia in gene-targeted mice lacking functional hepatic glutamine synthetase. Proc Natl Acad Sci U S A 112: 5521–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria LR, Allegri G, Melck D, et al. (2018) Enhancement of hepatic autophagy increases ureagenesis and protects against hyperammonemia. Proc Natl Acad Sci U S A 115: 391–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart PM, Walser M (1980) Failure of the normal ureagenic response to amino acids in organic acid-loaded rats. Proposed mechanism for the hyperammonemia of propionic and methylmalonic acidemia. J Clin Invest 66: 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summar ML, Barr F, Dawling S, et al. (2005) Unmasked adult-onset urea cycle disorders in the critical care setting. Crit Care Clin 21: S1–8. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Koehler RC, Brusilow SW, Traystman RJ (1991) Inhibition of brain glutamine accumulation prevents cerebral edema in hyperammonemic rats. Am J Physiol 261: H825–829. [DOI] [PubMed] [Google Scholar]
- Thomsen KL, Gronbaek H, Glavind E, et al. (2014) Experimental nonalcoholic steatohepatitis compromises ureagenesis, an essential hepatic metabolic function. Am J Physiol Gastrointest Liver Physiol 307: G295–301. [DOI] [PubMed] [Google Scholar]
- Torres-Vega MA, Vargas-Jeronimo RY, Montiel-Martinez AG, et al. (2015) Delivery of glutamine synthetase gene by baculovirus vectors: a proof of concept for the treatment of acute hyperammonemia. Gene Ther 22: 58–64. [DOI] [PubMed] [Google Scholar]
- Yu M, Wu J, Shi J, Farokhzad OC (2016) Nanotechnology for protein delivery: Overview and perspectives. J Control Release 240: 24–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.