

SUMMARY
As a Ca2+-activated lipid scramblase and ion channel that mediates Ca2+ influx, TMEM16F relies on both functions to facilitate extracellular vesicle generation, blood coagulation, and bone formation. How a bona fide ion channel scrambles lipids remains elusive. Our structural analyses revealed the coexistence of an intact channel pore and PIP2-dependent protein conformation changes leading to membrane distortion. Correlated to the extent of membrane distortion, many tightly bound lipids are slanted. Structure-based mutagenesis studies further reveal that neutralization of some lipid-binding residues or those near membrane distortion specifically alters the onset of lipid scrambling, but not Ca2+ influx, thus identifying features outside of channel pore that are important for lipid scrambling. Together, our studies demonstrate that membrane distortion does not require open hydrophilic grooves facing the membrane interior and provide further evidence to suggest separate pathways for lipid scrambling and ion permeation.
In Brief
TMEM16F is a calcium-activated ion channel and lipid scramblase linked to the bleeding disorder Scott syndrome. Feng et al. examine cryo-EM structures of TMEM16F with or without Ca2+ ions and PIP2 nanodisc supplementation and identify structural features for lipid binding and membrane distortion critical for lipid scrambling activity.
Graphical Abstract
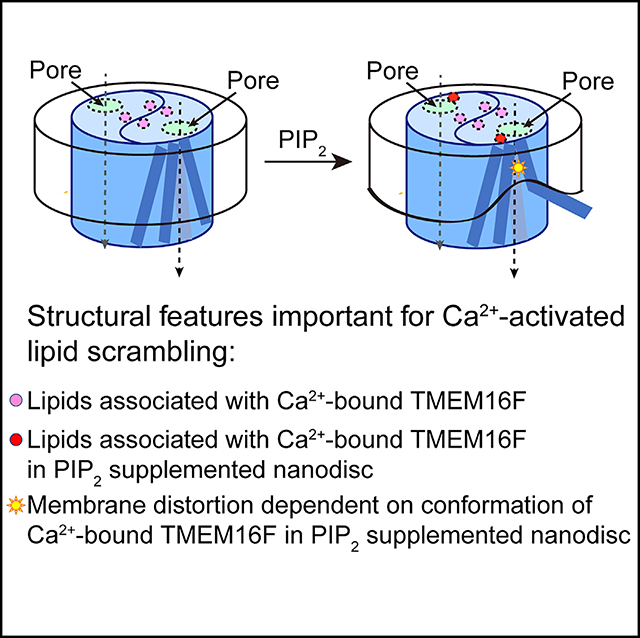
INTRODUCTION
Linked to the human bleeding disorder Scott syndrome (Boisseau et al., 2018; Suzuki et al., 2010), TMEM16F is a Ca2+-activated scramblase as well as ion channel that mediates Ca2+ influx (Alvadia et al., 2019; Han et al., 2019; Watanabe et al., 2018; Yang et al., 2012). The dual function of this protein is critical for exposing phosphatidylserine (PS) (Zwaal et al., 2004) and generating and releasing of extracellular vesicles (EVs) by the platelets (György et al., 2011; Raposo and Stoorvogel, 2013; Sims et al., 1989; Whitlock and Hartzell, 2017). These processes play important roles in bone formation (Ehlen et al., 2013; Ousingsawat et al., 2015b) and anti-inflammatory response of neutrophils (Headland et al., 2015) as well as blood coagulation (Fujii et al., 2015; Wolf, 1967; Yang et al., 2012; Zwaal et al., 2004). For a channel that is activated by Ca2+ and mediates Ca2+ influx—a positive feedback that needs to be held in check to prevent excessive increase of intracellular Ca2+ level, TMEM16F has a small single-channel conductance (Yang et al., 2012). Moreover, TMEM16F channel activity is reduced by Ca2+-induced degradation of phosphatidylinositol-(4, 5)-bisphosphate (PIP2) (Ye et al., 2018), and its ion selectivity shifts dynamically with rising Ca2+ levels (Ye et al., 2019); this dynamic shift may contribute to the time-dependent change and variations of ion selectivity revealed in whole-cell recordings (Bricogne et al., 2019; Grubb et al., 2013; Nguyen et al., 2019; Ousingsawat et al., 2015a, 2015b; Shimizu et al., 2013; Yu et al., 2015). The structural basis of TMEM16F modulation by Ca2+ and PIP2 remains an open and interesting question.
TMEM16F is functionally distinct from its closely related family member TMEM16A (Dang et al., 2017; Paulino et al., 2017), which is a Ca2+-activated Cl¯channel with no scramblase activity (Yu et al., 2015) but somewhat similar to the fungal scramblases nhTMEM16 and afTMEM16 (Brunner et al., 2014; Falzone et al., 2019; Kalienkova et al., 2019) that may permeate both cations and anions (Falzone et al., 2018). Structures of these fungal scramblases have a half-open subunit cavity with a hydrophilic groove open to the interior of the lipid bilayer. Such configuration is compatible with the lipid-scrambling model, in which the charged headgroup of lipid goes through this open groove while being scrambled, the so-called “credit card” model (Pomorski and Menon, 2006) for lipid scrambling (Bethel and Grabe, 2016; Jiang et al., 2017; Lee et al., 2018; Whitlock and Hartzell, 2016). Furthermore, membrane distortion was observed at the site of this open groove (Falzone et al., 2019; Kalienkova et al., 2019). Such distortion is likely caused by the open groove and could reduce the energy barrier of scrambling a lipid from one leaflet to the other, further strengthening the credit card model. However, recent findings that the fungal afTMEM16 scramblase can flip lipids with headgroups larger than the width of the open groove raises the possibility of “out-of-the-groove” lipid scrambling (Malvezzi et al., 2018), which is likely also facilitated by the membrane distortion (Falzone et al., 2019). The dual functionality of channel and scramblase of TMEM16F and its fungal homologs (Falzone et al., 2018) raises intriguing questions: how might an open groove permeate ions? If TMEM16F has a channel pore similar to the enclosed pore of TMEM16A, how might the membrane be distorted to facilitate lipid scrambling without an open hydrophilic groove facing the membrane interior?
We combined structural and mutagenesis studies to explore the possibility that lipid scrambling by TMEM16F does not require an open hydrophilic groove. Using single-particle electron cryo-microscopy (cryo-EM), we determined four structures of TMEM16F under different physiological conditions. Our structural analyses reveal an enclosed channel pore in all these structures for different functional states and PIP2-dependent membrane distortion and thinning. Such membrane distortions vary with different protein conformations, and they are accompanied with tightly bound lipids that are in slanted orientations. Mutations of some residues that either bind lipids or are associated with membrane distortion impact the onset of lipid scrambling, but not Ca2+ influx, thus establishing a connection between membrane distortion and lipid scrambling. The specific effects of these mutations further suggest that lipid scrambling and ion permeation are not always correlated. Together, our studies reveal that membrane distortion and lipid scrambling may take place without an open groove and further suggest that lipid scrambling and ion permeation do not share the same pathway.
RESULTS
Ca2+ Induces Conformational Changes on the Structure of TMEM16F
Recombinant mouse TMEM16F was expressed in HEK293 cells and purified in digitonin with added lipids. We determined two cryo-EM structures of TMEM16F in digitonin, without and with Ca2+ (Figures 1 and S1; Table S1). The overall resolutions of both structures are better than 4 Å. The dimeric architecture of TMEM16F resembles that of TMEM16A with ten transmembrane segments (TM1–10). Unexpectedly, a single Ca2+ ion is resolved in the density map of Ca2+-bound TMEM16F, which is coordinated by N621 (and likely E624 as well) on TM6, E670 on TM7, and D703 on TM8 (Figure 1C). Comparison of these structures revealed that Ca2+ binding induced a conformational change in the TM6, from a helix that is slightly bent at G615 to a straight helix (Figure 1G). Another noticeable change involves the extracellular loops (Figure 1H); Ca2+ binding facilitates the stable binding of lipids to clusters of basic residues on the TM9-TM10 loop (Figures 1I and 1J). When the Ca2+-free and Ca2+-bound TMEM16F maps are compared at the same resolution, it appears that the changes of TMEM16F conformation induced by Ca2+ binding facilitate the binding of a greater number of lipids (Figures 1A and 1D). Considering that the channel and lipid scrambling activities of TMEM16F are Ca2+ dependent, having more stably associated lipids in Ca2+-bound condition may be related to these activities.
Figure 1. Ca2+-Dependent Conformation Changes and Lipid Binding to TMEM16F.

(A–C) Lipids with slanted orientations (model in magenta, superimposed on the sharpened density map in light gray; A) are associated with Ca2+-bound TMEM16F in digitonin (green) with lipids (magenta) and one Ca2+ ion (red sphere) in each monomer, overlaid on the electron density map (sharpened, in light gray; B). The Ca2+ ion is coordinated by N621 on TM6, E670 on TM7, and D703 on TM8 (C).
(D–F) Fewer lipids (model in magenta, superimposed on the sharpened electron density map in light gray; D) are associated with Ca2+-free TMEM16F in digitonin (yellow) with lipids (magenta) overlaid on the electron density map (sharpened, in light gray; E). TM6 remains close to TM7 and TM8 even without Ca2+ binding to the acidic residues on these transmembrane helices (F).
(G) Superimposition of ribbon diagrams of Ca2+-bound (green) and Ca2+-free (yellow) TMEM16F in digitonin, showing a bend of TM6 at G615 in Ca2+-bound, but not Ca2+-free, TMEM16F and R542, N621, and K706 with Ca2+-dependent placements.
(H) Superimposition of ribbon diagrams of Ca2+-bound (green) and Ca2+-free (yellow) TMEM16F in digitonin, showing different conformations of TM9-TM10 loop.
(I) A lipid with its headgroup coordinated by R813, K823, and H824 on TM9-TM10 loop of Ca2+-bound TMEM16F in digitonin (green).
(J) A lipid with its headgroup coordinated by R753 and H768 on TM9-TM10 loop of Ca2+-bound TMEM16F in digitonin (green).
See also Figures S1 and S2 and Table S1.
First, we test the functional roles of residues involved in Ca2+-induced conformational changes, such as N621 on the TM6 involved in Ca2+ binding (Figures 1C and S2A), R542 on the TM4-TM5 loop that contacts the TM2-TM3 loop only in Ca2+-free TMEM16F (Figures S2C and S2D), and K706 on TM8 that is in the proximity of E624 solely in the Ca2+-bound TMEM16F (Figures S2A and S2B). We used Ca2+ imaging to assay TMEM16F-dependent Ca2+ influx and live imaging of PS exposure to assay the lipid scrambling activity. We also examined giant plasma membrane vesicle (GPMV) generation, which requires both TMEM16F-dependent Ca2+ influx and lipid scrambling (Han et al., 2019). Time-lapse imaging of 500–1,000 cells with 10× magnification was performed to concurrently monitor GPMV formation and Ca2+ influx, and PS exposure was monitored in separate experiments involving time-lapse imaging of individual cells viewed with 603 magnification (Figures 2 and S3). All measurements were normalized by the expression levels (Figures S3G–S3I; Table S2). Control experiments revealed that, whereas TMEM16F is not required for the initial chemically induced Ca2+ release from internal stores, TMEM16F is essential for the subsequent Ca2+ influx (Figures S3A–S3F); HEK293 cells without heterologous expression of TMEM16F do not exhibit this Ca2+ influx, PS exposure, or GPMV formation (Han et al., 2019). Because the initiation of Ca2+ influx and PS exposure depends primarily on TMEM16F functions, we focused on the time of onset, which is the maximum of the second derivative of the curve (maximal acceleration; see STAR Methods). N621A and K706A mutations significantly reduced Ca2+ influx (Figure 2A) and GPMV formation (Figure 2C). N621A also suppressed PS exposure (Figure 2B). In contrast, R542A accelerated the onset of Ca2+ influx, PS exposure, and GPMV generation (Figures 2G–2I). These results support the notion that Ca2+ binding is important for both lipid scrambling and channel activities.
Figure 2. Functional Tests of Lipid-Binding Residues on TM9-TM10 Loop and Other Residues with Ca2+-Dependent Placements.

(A–F) Analysis of residues involved in Ca2+ binding is depicted (A)–(C), and (D)–(F) are for analysis of residues involved in lipid binding. Live imaging of TMEM16Fdependent Ca2+ influx (A and D), PS exposure (B and E), and GPMV generation (C and F) is shown. Time-lapse imaging of 500–1,000 cells with 10× magnification was performed to concurrently monitor GPMV formation and Ca2+ influx via Fluo-8 fluorescence. Time-lapse imaging of individual cells viewed with 603 magnification was performed to monitor PS exposure via pSIVA fluorescence. Data are represented as mean ± SEM.
(G–I) Scattered dot plots of time of onset of TMEM16F-dependent Ca2+ influx (G), PS exposure (H), and GPMV generation (I). Time of onset, which is the maximum of the second derivative of the curve (maximal acceleration), could not be determined for those time courses with a linear rather than sigmoidal rise. Data are represented as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Statistical significance of all mutants as compared to TMEM16F wild-type (WT) are determined by one-way ANOVA followed by Holm-Šídák multiple comparisons test.
Next, we tested the functional roles of two clusters of basic residues on the TM9-TM10 loop that bind lipids in Ca2+-bound TMEM16F (Figures 1I and 1J). Remarkably, the R753A/H768A double mutation specifically delayed the onset of PS exposure (Figures 2E and 2H) without affecting the onset of Ca2+ influx (Figures 2D and 2G), and the R813A/K823A/H824A triple mutation slowed Ca2+ influx, PS exposure, as well as GPMV generation (Figures 2D–2I). The finding that the lipid binding residues R753 and H768 specifically affect the onset of PS exposure, but not Ca2+ influx, indicates that they are functionally important for lipid scrambling rather than channel function.
PIP2 Facilitates Membrane Distortion and Thinning
We further reconstituted digitonin-solubilized TMEM16F into lipid nanodiscs for structure determinations (Figure S4; Table S1). We determined structures of Ca2+-bound TMEM16F in lipid nanodiscs without PIP2 to~ 7 Å and with supplement of PIP2. From the latter, we classified particles into two classes with distinct conformations (Figures 3, 4, and 5; Videos S1 and S2); whereas class 2 is slightly influenced by preferred orientation, both have better than 4-Å resolution (Figure S4). Major structural features, including TM helices and membrane surrounding the protein, are well resolved in all structures (Figures S4 and S5). Similar to what we observed in the digitonin structures, we found a single Ca2+ ion with similar arrangements of Ca2+-coordinating residues in the PIP2 supplemented nanodiscs (Figures S6A– S6C), at a location comparable to one of the densities for Ca2+ ions in TMEM16F in nanodiscs without PIP2 supplement (Figures S2E–S2J; Alvadia et al., 2019). Interestingly, comparison of structures after filtering all density maps to the same resolution of~7 Å revealed that, in both classes of TMEM16F structures in nanodiscs with PIP2 supplementation, the membrane is distorted and thinned by ~30% for class 1 and ~40% for class 2, respectively (Figures 3A, 3B, 4A, S6F, and S6G) near the site where TM6 is kinked at P628 with its lower part unwound and veering away (Figures 3D–3I). Many lipids found stably bound to the protein near the membrane distortion site are in slanted orientations; the slant angles vary with the protein conformation of these two classes of TMEM16F, however, we cannot be sure about the correspondence of the bound lipids in the two classes (Figure 4B).
Figure 3. Membrane Distortion Depends on the TMEM16F Conformation.

(A) Ca2+-bound TMEM16F of class 2 (blue) in nanodiscs supplemented with PIP2 is associated with prominent distortion of the membrane (light gray, with border marked by dashed green lines) near TM3, TM4, TM6, and pre-TM1 elbow (red). Membrane distortion is quantified by measuring the membrane thickness, with the normalized values shown.
(B) Ca2+-bound TMEM16F of class 1 (orange) in nanodiscs supplemented with PIP2 is associated with moderate distortion of the membrane (light gray, with border marked by dashed green lines) near TM3, TM4, TM6, and pre-TM1 elbow (red). Membrane distortion is quantified by measuring the membrane thickness, with the normalized values shown.
(C) Ca2+-bound TMEM16F (yellow) in nanodiscs without PIP2 supplement is associated with minimal distortion of the membrane (light gray, with border marked by dashed green lines) as revealed by the membrane thickness measurements.
(D–F) Pairwise superimposition of density maps of TM3, TM4, TM6, and pre-TM1 elbow, showing the kink of TM6 at P628 (encircled with dotted line) of Ca2+-bound TMEM16F in nanodiscs supplemented with PIP2(D; orange for class 1;blue for class 2), but not in digitonin (E; green) or nanodiscs without PIP2 supplement (F; yellow).
(G–I) Pairwise superimposition of ribbon diagrams of TM3, TM4, TM6, and pre-TM1 elbow, Ca2+-bound TMEM16F in nanodiscs supplemented with PIP2 Class 1 (orange) vs Ca2+-bound TMEM16F in nanodiscs supplemented with PIP2 Class 2 (blue) (G) or Ca2+-bound TMEM16F in digitonin (green) (H), Ca2+-bound TMEM16F in nanodiscs supplemented with PIP2 Class 2 (blue) vs Ca2+-bound TMEM16F in digitonin (green) (I), showing the kink of TM6 at P628 (encircled with dotted line) of Ca2+-bound TMEM16F in nanodiscs supplemented with PIP2 (orange for class 1; blue for class 2), but not in digitonin (green).
See also Figures S2, S4, and S5, Table S1, and Videos S1 and S2.
Figure 4. TMEM16F-Conformation-Dependent Orientation of Bound Lipids and Functional Tests of Basic Residues Associated with Membrane Distortion.

(A) Transmembrane helices and loops in ribbon diagram for class 2 TMEM16F (blue) and bound lipids (magenta) shown with distorted membrane (gray). Membrane distortion is near TM3 and TM4 (red) and TM6 and pre-TM1 elbow (orange) with clusters of basic residues (encircled in dashed line) that are tested with mutagenesis.
(B) Superimposition of lipids bound to class 1 (gold) and class 2 (blue) TMEM16F in PIP2-supplemented nanodiscs. Dotted circles mark the lipid headgroups that interact with extracellular loops.
(C–E) Live imaging of TMEM16F-dependent Ca2+ influx (C), PS exposure (D), and GPMV generation (E). Time-lapse imaging of 500–1,000 cells with 10× magnification was performed to concurrently monitor GPMV formation and Ca2+ influx via Fluo-8 fluorescence. Time-lapse imaging of individual cells viewed with 603 magnification was performed to monitor PS exposure via pSIVA fluorescence. Data are represented as mean ± SEM.
(F–H) Scattered dot plots of time of onset of TMEM16F-dependent Ca2+ influx (F), PS exposure (G), and GPMV generation (H). Time of onset could not be determined for those time courses with a linear rather than sigmoidal rise. Data are represented as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Statistical significance of all mutants as compared to TMEM16F WT is determined by one-way ANOVA followed by Holm- Šídák multiple comparisons test.
Figure 5. PIP2-Dependent Lipid Binding Near Membrane Distortion and Functional Tests of Lipid-Binding Residues.

(A) Ca2+-bound TMEM16F (class 2) in PIP2 supplemented nanodiscs. Bound lipids are in cyan, and the one shown in (B) is in magenta.
(B) A phosphatidylserine (PS) (fatty acid tails in magenta and headgroup encircled with dashed line, superimposed on the sharpened electron density map in light gray) has its polar headgroup coordinated by R478 on TM3 and K590 and R592 on the TM5-TM6 loop. TMEM16Fs from class 1 (in orange, top panel) and class 2 (in blue, bottom panel) are shown with the same protein orientation.
(C–E) Live imaging of TMEM16F-dependent Ca2+ influx (C), PS exposure (D), and GPMV generation (E). Time-lapse imaging of 500–1,000 cells with 10× magnification was performed to concurrently monitor GPMV formation and Ca2+ influx via Fluo-8 fluorescence. Time-lapse imaging of individual cells viewed with 603 magnification was performed to monitor PS exposure via pSIVA fluorescence. Data are represented as mean ± SEM.
(F–H) Scattered dot plots of time of onset of TMEM16F-dependent Ca2+ influx (F), PS exposure (G), and GPMV generation (H). Time of onset could not be determined for those time courses with a linear rather than sigmoidal rise. Data are represented as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Statistical significance of all mutants as compared to TMEM16F WT is determined by one-way ANOVA followed by Holm- Šídák multiple comparisons test.
Comparisons between the two classes revealed that TMEM16F in class 1 is associated with less prominent membrane thinning (Figure 3B), bound to lipids that are less slanted (Figure 4B), and the TM6 is less kinked (Figures 3D and 3G). In contrast, in the structure without PIP2 supplementation, no membrane distortion is seen (Figures 3C and S6H) and TM6 is straight (Figures 3F, S2K, and S2M). Similarly, no distortion of micelle is seen in the digitonin structures with or without Ca2+, and TM6 does not display a kink at P628 (Figures 3E, 3H, 3I, S6D, and S6E). All comparisons of membrane or micelle thickness were made with all density maps low pass filtered to the same resolution of ~7 Å. These observations reveal that PIP2 is required for TMEM16F to adopt conformations with a kink of TM6, which in turn causes membrane thinning at TM3 and 4 and distortion at TM6 and pre-TM1.
Lipid Scrambling Is Related to Membrane Distortion
We then looked into how membrane distortion may affect lipid scrambling. The kink of TM6 creates a gap in the funnel-shaped opening facing the cytoplasmic side that is surrounded by TM3, TM4, and TM6. Instead of forming two parallel leaflets, lipids appear to be rearranged within the gap, thus causing thinning of membrane near TM3 and TM4 (Figures 3A and 3B). Furthermore, R636 and R639 at the lower end of TM6 now cluster with R271, H275, and R277 on the pre-TM1 elbow nearby (Figure 4A), which together appear to attract the lipid headgroups within the distorted bilayer, thus causing the membrane to curve slightly downward, creating a distortion in the membrane.
Because these residues are strategically located at the site of membrane distortion, we further tested their functional roles. To differentiate the channel activity from the lipid scrambling, we assayed the former with calcium imaging and the latter with live imaging of PS exposure of cells stably transfected with wild-type or mutant TMEM16F. Importantly, the R636A/R639A double mutation accelerated the onset of PS exposure without affecting Ca2+ influx, and the R271A/H275A/R277A triple mutations accelerated the onset of Ca2+ influx, PS exposure, and GPMV generation (Figures 4C–4H; Table S2). Acceleration of lipid scrambling in the R636A/R639A double mutant could indicate that the rate of lipid scrambling is increased by weakening interactions of lipid polar headgroups with these basic residues at the location of membrane thinning, where the barrier for lipid scrambling is lower. The specific effect of neutralizing R636 and R639 on the onset of PS exposure, but not Ca2+ influx, implicates their involvement in lipid scrambling, in a process separable from channel function.
Interestingly, we observed one lipid (likely PS) with its polar headgroup stably coordinated by R478 on TM3 as well as K590 and R592 on the TM5-TM6 loop in both structures of TMEM16F in PIP2 supplemented nanodiscs (Figures 5A and 5B). Consistent to being at the thinnest site of membrane (Figure 5A), this is the most slanted lipid we observed in all of our structures (Figure 5B). Previous molecular dynamics studies of nhTMEM16 showed that lipids with their headgroups bound to a pair of conserved residues, E313 and R432 that correspond to R478 and E604 in TMEM16F, are being scrambled (Bethel and Grabe, 2016). We thus speculate that this lipid we observed in TMEM16F structures (Figure 5B) could have been trapped in a position of potential relevance to lipid scrambling. These intriguing observations prompted us to test the functional roles of the basic residues that contact this lipid. Whereas both R478A and the R478A/K590A double mutation delayed the onset of GPMV generation, the R478A/K590A/R592A triple mutation delayed the onset of PS exposure and abolished GPMV generation (Figures 5C–5H; Table S2), underscoring the functional importance of these lipid binding residues.
TMEM16F with Distorted Membrane Has an Intact Ion Permeation Pore
Resembling the TMEM16A pore (Dang et al., 2017; Paulino et al., 2017), the channel pore of TMEM16F has a funnel shape with its funnel opening toward the cytoplasmic side and is enclosed and surrounded by TM3–TM7 (Figures 6A–6C and S6I; Video S2), similar to the arrangements reported recently (Figures S2K– S2N; Alvadia et al., 2019). It remains intact in all of our structures with its hydrophilic interior enclosed hence not accessible by lipids from the surrounding membrane. Facing the channel pore is Q559 critical for ion selectivity of TMEM16F channels recorded from excised membrane patches (Alvadia et al., 2019; Yang et al., 2012; Ye et al., 2019). The pore appears to be closed in all structures (Figures 6D and S6J). This is expected because TMEM16F channel activation requires membrane depolarization even in the presence of PIP2 and elevated internal Ca2+ concentration (Yang et al., 2012; Ye et al., 2018). Comparisons among these structures show that Ca2+ binding and PIP2 supplementation lead to widening of the pore near the cytoplasmic side of the membrane (Figures 6A–6C and S6I). While maintaining an intact ion permeation pore, the kink of TM6 is the primary cause of membrane distortion. Therefore, in TMEM16F structures, membrane distortion does not require an open hydrophilic groove.
Figure 6. The TMEM16F Channel Pore and Functional Tests of Pore-Lining Residues.

(A–C) The solvent-accessible (mesh) pore of Ca2+-bound TMEM16F in PIP2-supplemented nanodiscs (orange for class 1; blue for class 2; A and B) or digitonin (green; C).
(D) Pore radius along the z axis (green for TMEM16F in digitonin; orange and blue for class 1 and class 2 of TMEM16F in PIP2 supplemented nanodiscs, respectively).
(E–G) Live imaging of TMEM16F-dependent Ca2+ influx (E), PS exposure (F), and GPMV generation (G). Time-lapse imaging of 500–1,000 cells with 10× magnification was performed to concurrently monitor GPMV formation and Ca2+ influx via Fluo-8 fluorescence. Time-lapse imaging of individual cells viewed with 603 magnification was performed to monitor PS exposure via pSIVA fluorescence. Data are represented as mean ± SEM.
(H–J) Scattered dot plots of time of onset of TMEM16F-dependent Ca2+ influx (H), PS exposure (I), and GPMV generation (J). Time of onset could not be determined for those time courses with a linear rather than sigmoidal rise. Data are represented as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Statistical significance of all mutants as compared to TMEM16F WT is determined by one-way ANOVA followed by Holm-Šídák multiple comparisons test.
(K) Normalized currents fit to the Hill equation. The Hill coefficient is 2, and the EC50 values of E529A and E604 (24.0 ± 2.3 μM and 20.1 ± 2.2 μM, respectively) are significantly increased compared to the EC50 of wild-type control (7.4 ± 1.9 mM; p < 0.0001; corrected Dunnett test following one-way ANOVA). EC50 of K530A (6.5 ± 2.9 μM) is not significantly different from WT value (p = 0.99).
See also Figures S2, S3, and S6, Table S2, and Videos S1 and S2.
In the case of the nhTMEM16 and afTMEM16 fungal scramblases, this equivalent pore is not intact but opens to the membrane (Brunner et al., 2014; Falzone et al., 2019; Kalienkova et al., 2019), and the TM6 helix is straight. Therefore, unlike TMEM16F structures, the membrane distortion in nhTMEM16 and afTMEM16 structures is likely caused by the open hydrophilic groove.
To explore the functional involvement of pore-lining residues, we compared the effects of alanine substitutions of E529 and K530 on TM4 and E604 on TM6 (Figures 6E–6J and S3; Table S2). E529A delayed the onset of Ca2+ influx, PS exposure, and GPMV generation, and K530A had a milder effect on these processes and E604A had stronger effects on PS exposure and GPMV formation. Consistent with the strong effects on Ca2+ influx, E529A and E604A mutations reduced TMEM16F channel activity by causing a right shift of its Ca2+ sensitivity for channel activation (Figure 6K), indicating that these mutations may have altered the relative stability of the open and closed conformations of the channel. The reduction of Ca2+ influx by mutations of these pore-lining residues could indirectly impact the Ca2+-activated scramblase activity as well as GPMV generation that requires both TMEM16F-dependent Ca2+ influx and lipid scrambling (Han et al., 2019).
DISCUSSION
Members of TMEM16 family share similar overall structural architecture but with diverse and distinct functionalities (Falzone et al., 2018; Pedemonte and Galietta, 2014). Although TMEM16A and TMEM16B are Ca2+-activated chloride channels (Ferrera et al., 2010; Huang et al., 2012), most members of this family have lipid scramblase activities (Falzone et al., 2018; Suzuki et al., 2013). Among them, TMEM16F is a bona fide ion channel and lipid scramblase (Alvadia et al., 2019; Watanabe et al., 2018; Yang et al., 2012), with important physiological functions in many different cell types (Batti et al., 2016; Bricogne et al., 2019; Ehlen et al., 2013; Fujii et al., 2015; Hu et al., 2016; Mattheij et al., 2016; Ousingsawat et al., 2015a, 2015b; Yang et al., 2012). Therefore, an intriguing question is how a protein carries out such dual functionality of channel and scramblase.
In contrast to the current view, in which lipid scrambling requires an open groove facing the membrane interior (Bethel and Grabe, 2016; Falzone et al., 2018; Whitlock and Hartzell, 2016; Yu et al., 2015), the structural features of TMEM16F presented in this study suggest a model of how the same protein can fulfil dual functionality of channel and scramblase while maintaining an intact ion permeation pore (Figure 7). In the presence of Ca2+ but without PIP2, the configuration of ion permeation pore of TMEM16F resembles that of TMEM16A, formed by TM3–7 with a funnel shape opening toward the cytoplasmic side. Supplementing PIP2 to TMEM16F causes a swing of the lower part of TM6 and subtle rearrangements of other pore-forming helices, thereby causing membrane distortion and thinning (Figures 7B and 7C). Such changes widen the funnel opening further but without changing the ion permeation pore (Figure 7A), which remains fully intact as in the structure of TMEM16A. The widened funnel opening, however, causes membrane distortion and thinning that is similar to that found in the fungal scramblase structures (Falzone et al., 2019; Kalienkova et al., 2019). Neutralization of R753/H768 or R636/R639, which are evolutionarily conserved basic residues (Figure S7), specifically affects the latency for PS exposure without affecting the onset for Ca2+ rise (Figures 2 and 4), indicating that these two pairs of residues are crucial for lipid scrambling, but not ion permeation, and there is a connection between membrane distortion observed in the structure and lipid scrambling. Therefore, even without an open groove, membrane distortion and thinning appears sufficient to facilitate lipid scrambling.
Figure 7. Model for Lipid Scrambling.

(A) Schematics showing the half-open subunit cavity of the fungal scramblases nhTMEM16 and afTMEM16 (left) and the intact enclosed channel pore ofTMEM16F, which has dual function of channel and scramblase, in the presence (right) or absence (middle) of PIP2 that causes changes of TMEM16F conformation. The cross section of dimeric TMEM16F in nanodiscs with or without PIP2 supplement is very similar.
(B) Membrane distortion is associated with fungal scramblases with half-open subunit cavity (left). Whereas TMEM16F in nanodiscs without PIP2 supplementation is not associated with membrane distortion, PIP2-induced conformation changes of TMEM16F causes membrane distortion near the enclosed channel pore (right).
(C) In addition to the two lipids stably bound to clusters of basic residues in the TM9-TM10 loop of Ca2+-bound TMEM16F, a lipid (likely PS) is bound to basic residues on TM4 and the TM4-TM5 loop of Ca2+-bound TMEM16F in PIP2-supplemented nanodiscs. PIP2 induces displacements of TM3, TM4, and TM6, which displays a kink to bring clusters of basic residues on TM6 and the pre-TM1 elbow together, thereby causing membrane distortion. Alanine substitutions of a subset of these basic residues involved in lipid binding and membrane distortion specifically affect PS exposure, indicating that lipid scrambling may proceed in a pathway separable from ion permeation.
Our model differs from the three models recently proposed by Alvadia and colleagues, namely the “out-of-the-groove mechanism” for lipids to move outside of the protein-enclosed pore but without tight contact with the protein and two alternative scenarios invoking a conformation resembling the fungal TMEM16 scramblases either for both ions and lipids to permeate the same open cavity conformation or for the lipid-conductive open cavity to alternate with the ion-conductive protein-surrounded pore (Alvadia et al., 2019). Our model for the out-ofthe-groove lipid scrambling (Figure 7) is based on PIP2-dependent protein conformation changes that are critical for lipid distortion and thinning (Figure 3), and the specific functional dependence of lipid scrambling, but not Ca2+ influx, on basic residues that either coordinate the polar headgroup of a bound lipid or reside near membrane distortion and thinning (Figures 2 and 4).
We did not resolve densities in our structures that can be interpreted as bound PIP2. However, our earlier studies showed that the N-terminal segment bearing a cluster of positively charged residues is crucial for PIP2 modulation of TMEM16F channel activity (Ye et al., 2018). In our structures, this cluster is presumably located very close to the TM6-TM7 loop below P628 that swings away upon PIP2 binding (Figures S6K and S6L). These observations are consistent with the notion that PIP2 plays a crucial role in allowing TMEM16F to adopt conformations with a kink of TM6, thus bringing positively charged residues on TM6 to the proximity of positively charged residues in the pre-TM1 elbow (Figure 4B), causing membrane distortions and thinning.
In summary, our findings reveal that TMEM16F maintains an intact ion permeation channel while adopting a conformation that causes membrane distortion and facilitates lipid scrambling.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yifan Cheng (ycheng@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Full length mouse TMEM16F was fused to a 3C consensus sequence, a Strep-tag II peptide, and a GFP moiety at its C terminus, and expressed in HEK293 GnTi- cells obtained by ATCC using the BacMam system as described previously (Dang et al., 2017).
METHOD DETAILS
Protein purification
Protein purification and sample processing were carried out at 4°C. For nanodisc-reconstituted preparations, approximately 10 g of cell pellet (from about 1 L of culture) was lysed by stirring for 40 min in 100 mL hypotonic buffer containing 20 mM HEPES (pH 7.5) supplemented with 0.1 mg/ml DNase, 1 × complete protease inhibitor cocktail (Roche) and 1 mM phenylmethylsulfonyl (PMSF). The membrane fraction was collected by centrifugation at 30,000 g for 30 min, and then homogenized with a Dounce homogenizer in extraction buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, and 10 mM EGTA (PH 7.5) supplemented with 0.1 mg/ml DNase, 1 × complete protease inhibitor cocktail, and 1 mM PMSF. Protein was extracted in 100 mL extraction buffer plus 0.5% n-dodecyl-b-d-maltopyranoside (DDM) and 0.1% cholesteryl hemisuccinate (CHS) with gentle stirring for 2 h. The insoluble fraction was removed by centrifugation at 30,000 g for 30 min. The recombinant protein was affinity purified with an anti-GFP nanobody immobilized on CNBr-activated Sepharose resin (GE Healthcare) in wash buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, 10 mM EGTA, 0.06% Digitonin supplemented with a 0.1 mg/ml lipid mixture containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoylsn-glycero-3-phosphoethanolamine (POPE) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS) at a ratio of 3:1:1. The purified protein was recovered by incubation with 1.5 CV wash buffer containing 0.5 mM dithiothreitol (DTT) and 50 mg 3C protease for 4 h. To reconstitute the protein in nanodiscs, the lipid was prepared as described previously (Dang et al., 2017) in ddH2O. After purification, the protein sample was mixed with MSP2N2 and soy PC (Avanti) at a molar ratio of 1:4:100 of TMEM16F monomer:MSP2N2:lipids (The ratio of SoyPC:POPC:POPE:POPS is about 6:3:1:1.) The mixture was allowed to equilibrate overnight and Bio-beads SM2 (Bio-Rad) were added to the mixture three times within 24 h to gradually remove detergents from the system. PIP2 was applied to the sample for 15 min. Afterward, the sample was filtered through a 0.45 mm filter, and the reconstituted protein was separated on a Superdex-200 column in column buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, and 1 mM EGTA. PIP2 was added to nanodiscs with incorporated protein (4 PIP2 per TMEM16F monomer; 1 PIP2 per 50 lipid molecules). PIP2 was added to protein in nanodiscs for a period of 15 min of incubation before injection onto the column for gel filtration to isolate TMEM16F reconstituted in nanodiscs with PIP2 supplement. The peak fraction was collected and concentrated to 0.6–0.7 mg/ml using a 100 kDa MWCO Amicon Ultra filter device (Millipore). 2 mM CaCl2 was added to the protein sample right before freezing the grids. The lipid compositions used for protein purification and nanodisc reconstitutions are chosen based on previous studies (Dang et al., 2017; Jin et al., 2017; Whicher and MacKinnon, 2016).
Similar purification procedures were used to prepare digitonin solubilized TMEM16F except that 0.5 mM CaCl2 was used in all buffers instead of EGTA and 1% digitonin was in the extraction buffer. After digestion by 3C protease, the protein was collected and immediately separated on a Superdex-200 column in column buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, 0.5 mM CaCl2 and 0.06% digitonin. The peak fractions were collected and concentrated to 5 mg/ml via a 100-kDa Amicon Ultra filter device.
Electron microscopy
For cryo-EM, 2.5 mL of purified mTMEM16F was applied to holey carbon grids (Quantifoil 400 mesh Cu R1.2/1.3) glow-discharged for 30 s. After 30 s incubation on the grids at 4°C under 100% humidity, grids were blotted with Whatman #4 filter paper for 6–8 s and plunge-frozen in liquid ethane cooled by liquid nitrogen using a FEI Mark IV Vitrobot. Grids were transferred to an FEI Titan Krios electron microscope equipped with a field emission electron source and operated at 300 kV at the UCSF Cryo-EM facility.
Images were recorded using SerialEM (Mastronarde, 2005) in super-resolution mode using a K2 summit direct electron detector (Gatan) at a calibrated magnification of 22,500X, yielding a physical pixel size of 1.059 Å (0.5295 Å super resolution pixel size). Dose rate, total dose and defocus range used for data collection are summarized in Table S1.
Data processing
For cryo-EM data, drift correction was performed using MotionCor2 (Zheng et al., 2017) and images were binned 2×2 by Fourier cropping to a pixel size of 1.059 Å The contrast transfer function (CTF) was estimated using CTFFIND4 (Rohou and Grigorieff, 2015) using motion-corrected sums without dose-weighting. Motion-corrected sums with dose-weighting were used for all other image processing. RELION (Scheres, 2012) was used for 2D classification, 3D classification, and refinement procedures. We used the inbuilt local resolution by RELION for local resolution estimation.
All mTMEM16F data were processed with similar strategy. In summary, motion corrected sums were imported into cisTEM (Grant et al., 2018) for CTF estimation and particle picking. The refinement package including all picked particles were exported and used for further processing with RELION. All particles were binned in groups of four to pixel size of 4.236 for 3D classification. Particles from good classes without binning were further sorted by 2D classification and another round of 3D classification. 3D auto- refinement with a C2 symmetry was processed to get a density map, which was subjected to ‘post-processing’ in RELION to generate the final map for model building. Detailed information of data processing can be found in Figure S5 Directional FSC (dFSC) was calculated using similar approach as reported previously (Dang et al., 2017) with an improved script. Local resolution estimates were calculated with unsharpened raw density maps using ResMap (Kucukelbir et al., 2014). The initial reference used for data processing was the TMEM16A density map for the Ca2+-bound TMEM16F in digitonin dataset. For other datasets, the density map of TMEM16F in digitonin was used as initial reference.
All five density maps were low pass filtered to 6.7 Å before measuring the thickness of the membrane or micelle (Figures S6D–S6H). The distance between the extracellular and intracellular edges of membrane was measured in Chimera. We picked three regions, the thinnest region, the thickest region, and a region distant from the protein thus corresponding to the average thickness of the membrane in the Ca2+ bound TMEM16F nanodisc Class 2 density map. After aligning all five maps, the same three regions were measured in each density map. We show the absolute thickness for these three regions in Figures S6D–S6H. We also normalized the thickness of the thinnest region and the thickest region of the membrane to that of the region corresponding to the average thickness of the membrane in each map, as shown in Figure 3.
Model building
Ab initio model building was carried out in COOT (Emsley et al., 2010) and PHENIX. Initial model was generated with SWISS-MODEL (Waterhouse et al., 2018) on the basis of sequence alignment of TMEM16F with cryo-EM structure of TMEM16A. The model was refined in real space with Phenix.real_space_refine (Adams et al., 2010) and followed by further manual adjustment in COOT. This process was repeated until Ramachandran validation was satisfied. Structural validation was carried out by MolProbity and EMRinger in PHENIX. We used the difference map to locate the Ca2+ ion bound to TMEM16F in digitonin or nanodiscs.
Caver (Pavelka et al., 2016) was used to calculate the pore profile shown in Figures 6 and S6. The narrowest point is defined as the location of the pore with the smallest radius. Difference map was generated based on refined structure and density map using PHENIX with the command line option “phenix.real_space_diff_map.” The program PyMol (The PyMOL Molecular Graphics System, 2002). and UCSF Chimera (Pettersen et al., 2004) were used for figure preparation.
Electrophysiology
Coverslips with cells were transferred to a recording chamber on a Nikon-TE2000 Inverted Scope (Nikon) and transfection was confirmed with fluorescent microscopy. Patch borosilicate pipets (Sutter Instrument, Novato, CA, USA) were pulled from a Sutter P-97 puller with resistances of 2–3 MΩ for inside-out patch recordings. Solutions were puffed to the excised patch using VC3–8xP pressurized perfusion system (ALA Science, Farmingdale, NY, USA). Bath solution contained 145 mM NaCl, 10 mM HEPES, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, pH 7.2 with NaOH. Pipette solution contained 150 mM NaCl, 10 mM HEPES, 1 mM CaCl2. The membrane patch for inside-out recording was excised to in Ca2+-free solution to prevent desensitization (Ye et al., 2018): 150 mM NaCl, 10 mM HEPES, 2 mM EGTA. For solutions with Ca2+ < 100 μM, Ca2+ was added to solutions containing 2 mM HEDTA, and the final concentration was confirmed with Oregon Green BAPTA-5N (Thermo Fisher). The osmolality of each solution was adjusted to 290~310 mOsm/kg. Data were acquired using a Multiclamp 700B amplifier controlled by Clampex 10.2 via Digidata 1440A (Axon Instruments, Sunnyvale, CA, USA). All experiments were performed at room temperature (22–24°C). All data were analyzed using pClamp10 (Molecular Devices, Sunnyvale, CA, USA), OriginLab (OriginLab Corporation, Northampton, MA, USA), and Graphpad Prism. For the measurement of Ca2+-sensitivity, every trace was fit with the Hill equation to generate its respective EC50 and H (Hill coefficient). The curves in the figures display the averaged current magnitudes normalized to their respective maximal values (I/Imax %). Significant differences for EC50s of Ca2+-response curves were determined with Kruskal-Wallis tests followed by Dunn’s multiple comparison tests. In all cases, data represent mean ± SEM.
Giant plasma membrane vesicle (GPMV) quantification and calcium rise measurement
Cultured cells were incubated with with NucBlue Live Cell Stain (Thermo Fisher) and 1 μM of the calcium reporter dye Fluo-8 AM (AAT Bioquest) for 15 min, washed twice in Dulbecco’s phosphate buffered saline (DPBS), and treated with 25 mM paraformaldehyde (PFA)/2 mM dithiothreitol (DTT) in HEPES-buffered modified Tyrode’s buffer (10 mM HEPES, 143 mM NaCl, 4 mM NaCl, 10 mM glucose, 2 mM CaCl2) for live cell imaging on a Nikon-TE2000 inverted microscope (Nikon Instruments, Melville, NY, USA) equipped with a thermostat chamber. Images for both brightfield and Fluo-8 were acquired in parallel once every minute starting 10 min posttreatment for 50 min for a total of 1 hour treatment time. Quantification of the number of GPMVs in each frame were collected using custom software (Han et al., 2019). Images of nuclear staining were acquired after each experiment and used to quantify the number of cells in each field for normalization. Statistical significance for GPMV quantification was determined by one-way ANOVA followed by Holm-Šídák multiple comparisons test. Data are represented as mean ± SEM for curves, and as mean ± stdev for scattered dot plots. To calculate the values for Vmax, time of onset, and duration, we fit the GPMV quantification data with the Weibull growth model using Graph Pad Prism 7: Y = YM −(YM −Y0) ∗exp( −1 ∗ ((k ∗ X)g) Vmax is defined as the maximum of the first derivative of this curve, and time of onset is the maximum of the second derivative (indicating the maximal acceleration of vesiculation).
GPMVs are counted using custom Python code that employs a combination of a Canny edge detection algorithm (Canny, 1986; van der Walt et al., 2014) and a custom designed convolutional neural net program (Han et al., 2019). The Canny edge detection was combined with a modified Hough transform to detect circles in each image. The convolutional neural net was trained on 32 X 32 pixel images of centered vesicles. Over 1400 images were selected manually to train the neural net in our recent study (Han et al., 2019). To increase the specificity of detection, a particular vesicle was only counted if both the edge algorithm and neural net detected the vesicle.
Lipid scrambling assay
Stable HEK293 cell lines expressing wild-type or mutant mTMEM16F were plated in glass bottom dishes coated with 1X poly-L-lysine (Sigma) and 1:2000 matrigel (Sigma) at least two hours prior to imaging. Cells were incubated with NucBlue Live Cell Stain for 10 min, washed twice in HEPES-buffered modified Tyrode’s buffer (10 mM HEPES, 143 mM NaCl, 4 mM KCl, 10 mM glucose, 2 mM CaCl2). Image acquisition began once PFA/DTT/CaCl2 and 1:100 pSIVA (BioRad, Hercules, CA) was added to a final concentration of 25 mM/ 2 mM/2 mM and terminated after 1 hour. Image analysis was carried out on each individual cell using the Nikon Elements Software (Han et al., 2019). To calculate the values for Vmax and time of onset, we fit the pSIVA imaging data with the Weibull growth model using Graph Pad Prism 7: Y = YM −(YM −Y0) ∗exp( −1 ∗ ((k ∗ X)g) Vmax is defined as the maximum of the first derivative of this curve, and time of onset is the maximum of the second derivative.
Microplate reader calcium rise measurement
Cultured HEK293 cells were pre-loaded with 5 μM Fluo-5N (Thermo) and NucBlue Live Cell Stain (Invitrogen) for 15 min and washed twice in DPBS and incubated in calcium-free GPMV buffer. 25 mM PFA/2 mM DTT in calcium-free GPMV buffer is added 3 min after data acquisition begins to visualize PFA-induced calcium release from internal store. Finally, 2 mM CaCl2 (final concentration) is added 10 min after PFA. Fluorescence is continuously recorded using a Synergy H4 plate reader (BioTek, Winooski, VT) reading with a minimal interval of 1 read/11 s for calcium release and once every min for 60 min for calcium influx.
QUATIFICATION AND STATISTICAL ANALYSIS
Statistics and Graphs were generated by using pClamp10, GraphPad Prism 7 or Origin 8 software. Data are represented as mean ± SEM for curves, and as mean ± stdev for scattered dot plots. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Statistical significance of all mutants as compared to TMEM16F wild-type (WT) was determined by one-way ANOVA followed by Holm- Šídák multiple comparisons test.
DATA AND CODE AVAILABILITY
The accession numbers of cryo-EM density maps of TMEM16F reported in this paper are EMD: 20244, 20245, 20246, and 20247. The accession numbers of particle image stacks after motion correction related to TMEM16F reported in this paper are EMPIAR: 10278 (TMEM16F in Digitonin with calcium bound), EMPIAR: 10279 (TMEM16F in Digitonin without calcium bound), EMPIAR: 10280 (TMEM16F in Nanodisc). The accession numbers of atomic coordinates for TMEM16F reported in this paper are PDB: 6P46, 6P47, 6P48, and 6P49. All electrophysiological data generated or analyzed during this study are included in the published article. All other data are available from the corresponding authors upon reasonable request.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM medium | Corning cellgro | 10–013-CV |
| Mammalian cell culture medium | Expression System | 98–001-01 |
| SF-900 III SFM medium | GIBCO | 12658–027 |
| Penicillin/Streptomycin | Corning | 30–002-CI |
| Fetal bovine serum | AXENIA BIOLOGIX | F001 |
| n-Dodecyl-b-D-Maltopyranoside (DDM) | Anatrace | D310 |
| Cholesteryl hemisuccinate | Anatrace | CH210 |
| Digitonin | AppliChem | A1905 |
| Protease inhibitor cocktail | Roche | 18970600 |
| DNase | Roche | 10104159001 |
| Lipofectamine 2000 transfection reagent | Invitrogen | 11668019 |
| Critical Commercial Assays | ||
| CNBR-activated Sepharose beads | GE Healthcare | 17–0430-01 |
| Superdex 200, 10/300 GL | GE Healthcare | 17–5175-01 |
| Deposited Data | ||
| TMEM16F in Digitonin with calcium bound | This paper | EMPIAR-10278 |
| TMEM16F in Digitonin without calcium bound | This paper | EMPIAR-10279 |
| TMEM16F in Nanodisc | This paper | EMPIAR-10280 |
| Cryo-EM structure of TMEM16F in digitonin with calcium bound | This paper | PDB: 6P46 and EMD: 20244 |
| Cryo-EM structure of TMEM16F in digitonin without calcium | This paper | PDB: 6P47 and EMD: 20245 |
| Cryo-EM structure of calcium-bound TMEM16F in nanodisc with supplement of PIP2 in Cl1 | This paper | PDB: 6P48 and EMD: 20246 |
| Cryo-EM structure of calcium-bound TMEM16F in nanodisc with supplement of PIP2 in Cl2 | This paper | PDB: 6P49 and EMD: 20247 |
| Experimental Models: Cell Lines | ||
| xSf9 | ATCC | CRL-1711 |
| HEK293S GnTI | ATCC | CRL-3022 |
| Software and Algorithms | ||
| RELION | Scheres, 2012 | http://www2.mrc-lmb.cam.ac.uk/relion |
| UCSF Chimera | Pettersen et al., 2004 | http://www.cgl.ucsf.edu/chimera |
| PHENIX | Adams et al., 2010 | https://www.phenix-online.org |
| COOT | Emsley et al., 2010 | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Caver | Pavelka et al., 2016 | https://www.caver.cz/ |
| PyMOL | Molecular Graphics System, Version 1.8 Schrodinger, LLC | https://pymol.org/2/ |
| cisTEM | Grant et al., 2018 | https://cistem.org/ |
| Other | ||
| Quantifoil R1.2/1.3 400 mesh Cu holey carbon grids | SPI supplies | 4240C-XA |
Highlights.
Ca2+ binding enhances lipid association with TMEM16F calcium-activated scramblase
TMEM16F in nanodiscs supplemented with PIP2 is stably bound to a PS-like lipid
PIP2 supplement leads to membrane distortion that varies with TMEM16F conformation
Lipid binding residues outside of the protein enclosed pore affect lipid scrambling
ACKNOWLEDGMENTS
We thank D. Bulkley, A. Myasnikov, and M. Braunfeld at UCSF cryo-EM facility for help with data acquisition. We also thank Z. Yu and his colleagues at the HHMI Janelia Cryo-EM Facility for help with data acquisition and colleagues in our laboratories for discussion. This work was supported by grants from the NIH (R01GM098672, S10OD0020054, and S10OD021741 to Y.C.; R01NS069229 to L.Y.J.; and R35NS097229 to Y.N.J.). S.D. is supported by a Human Frontier Science Program (HFSP) Postdoctoral Fellowship. T.W.H. is supported by the Jane Coffin Childs Memorial Fund for Medical Research. L.Y.J., Y.N.J., and Y.C. are Investigators with the Howard Hughes Medical Institute.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.06.023.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvadia C, Lim NK, Clerico Mosina V, Oostergetel GT, Dutzler R, and Paulino C (2019). Cryo-EM structures and functional characterization of the murine lipid scramblase TMEM16F. eLife 8, e44365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batti L, Sundukova M, Murana E, Pimpinella S, De Castro Reis F, Pagani F, Wang H, Pellegrino E, Perlas E, Di Angelantonio S, et al. (2016). TMEM16F regulates spinal microglial function in neuropathic pain states. Cell Rep. 15, 2608–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethel NP, and Grabe M (2016). Atomistic insight into lipid translocation by a TMEM16 scramblase. Proc. Natl. Acad. Sci. USA 113, 14049–14054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisseau P, Bene MC, Besnard T, Pachchek S, Giraud M, Talarmain P, Robillard N, Gourlaouen MA, Bezieau S, and Fouassier M (2018). A new mutation of ANO6 in two familial cases of Scott syndrome. Br. J. Haematol 180, 750–752. [DOI] [PubMed] [Google Scholar]
- Bricogne C, Fine M, Pereira PM, Sung J, Tijani M, Wang Y, Henriques R, Collins MK, and Hilgemann DW (2019). TMEM16F activation by Ca2+ triggers plasma membrane expansion and directs PD-1 trafficking. Sci. Rep 9, 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner JD, Lim NK, Schenck S, Duerst A, and Dutzler R (2014). X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature 516, 207–212. [DOI] [PubMed] [Google Scholar]
- Canny J (1986). A computational approach to edge detection. IEEE Trans. Pattern Anal. Mach. Intell 8, 679–698. [PubMed] [Google Scholar]
- Dang S, Feng S, Tien J, Peters CJ, Bulkley D, Lolicato M, Zhao J, Zuberbühler K, Ye W, Qi L, et al. (2017). Cryo-EM structures of the TMEM16A calcium-activated chloride channel. Nature 552, 426–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlen HW, Chinenkova M, Moser M, Munter HM, Krause Y, Gross S, Brachvogel B, Wuelling M, Kornak U, and Vortkamp A (2013). Inactivation of anoctamin-6/Tmem16f, a regulator of phosphatidylserine scrambling in osteoblasts, leads to decreased mineral deposition in skeletal tissues. J. Bone Miner. Res 28, 246–259. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzone ME, Malvezzi M, Lee BC, and Accardi A (2018). Known structures and unknown mechanisms of TMEM16 scramblases and channels. J. Gen. Physiol 150, 933–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzone ME, Rheinberger J, Lee BC, Peyear T, Sasset L, Raczkowski AM, Eng ET, Di Lorenzo A, Andersen OS, Nimigean CM, and Accardi A (2019). Structural basis of Ca2+-dependent activation and lipid transport by a TMEM16 scramblase. eLife 8, e43229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrera L, Caputo A, and Galietta LJ (2010). TMEM16A protein: a new identity for Ca(2+)-dependent Cl− channels. Physiology (Bethesda) 25, 357–363. [DOI] [PubMed] [Google Scholar]
- Fujii T, Sakata A, Nishimura S, Eto K, and Nagata S (2015). TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc. Natl. Acad. Sci. USA 112, 12800–12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, Rohou A, and Grigorieff N (2018). cisTEM, user-friendly software for single-particle image processing. eLife 7, e35383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb S, Poulsen KA, Juul CA, Kyed T, Klausen TK, Larsen EH, and Hoffmann EK (2013). TMEM16F (Anoctamin 6), an anion channel of delayed Ca(2+) activation. J. Gen. Physiol 141, 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- György B, Szabó TG, Pásztói M, Pál Z, Misják P, Aradi B, Lászloó V, Pállinger E, Pap E, Kittel A, et al. (2011). Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell. Mol. Life Sci 68, 2667–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han TW, Ye W, Bethel NP, Zubia M, Kim A, Li KH, Burlingame AL, Grabe M, Jan YN, and Jan LY (2019). Chemically induced vesiculation as a platform for studying TMEM16F activity. Proc. Natl. Acad. Sci. USA 116, 1309–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Headland SE, Jones HR, Norling LV, Kim A, Souza PR, Corsiero E, Gil CD, Nerviani A, Dell’Accio F, Pitzalis C, et al. (2015). Neutrophilderived microvesicles enter cartilage and protect the joint in inflammatory arthritis. Sci. Transl. Med 7, 315ra190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Kim JH, He K, Wan Q, Kim J, Flach M, Kirchhausen T, Vortkamp A, and Winau F (2016). Scramblase TMEM16F terminates T cell receptor signaling to restrict T cell exhaustion. J. Exp. Med 213, 2759–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Wong X, and Jan LY (2012). International Union of Basic and Clinical Pharmacology. LXXXV: calcium-activated chloride channels. Pharmacol. Rev 64, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Yu K, Hartzell HC, and Tajkhorshid E (2017). Lipids and ions traverse the membrane by the same physical pathway in the nhTMEM16 scramblase. eLife 6, e28671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Bulkley D, Guo Y, Zhang W, Guo Z, Huynh W, Wu S, Meltzer S, Cheng T, Jan LY, et al. (2017). Electron cryo-microscopy structure of the mechanotransduction channel NOMPC. Nature 547, 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalienkova V, Clerico Mosina V, Bryner L, Oostergetel GT, Dutzler R, and Paulino C (2019). Stepwise activation mechanism of the scramblase nhTMEM16 revealed by cryo-EM. eLife 8, e44364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukelbir A, Sigworth FJ, and Tagare HD (2014). Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BC, Khelashvili G, Falzone M, Menon AK, Weinstein H, and Accardi A (2018). Gating mechanism of the extracellular entry to the lipid pathway in a TMEM16 scramblase. Nat. Commun 9, 3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvezzi M, Andra KK, Pandey K, Lee BC, Falzone ME, Brown A, Iqbal R, Menon AK, and Accardi A (2018). Out-of-the-groove transport of lipids by TMEM16 and GPCR scramblases. Proc. Natl. Acad. Sci. USA 115, E7033–E7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Mattheij NJ, Braun A, van Kruchten R, Castoldi E, Pircher J, Baaten CC, Wülling M, Kuijpers MJ, Köhler R, Poole AW, et al. (2016). Survival protein anoctamin-6 controls multiple platelet responses including phospholipid scrambling, swelling, and protein cleavage. FASEB J. 30, 727–737. [DOI] [PubMed] [Google Scholar]
- Nguyen DM, Chen LS, Yu WP, and Chen TY (2019). Comparison of ion transport determinants between a TMEM16 chloride channel and phospholipid scramblase. J. Gen. Physiol 151, 518–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousingsawat J, Wanitchakool P, Kmit A, Romao AM, Jantarajit W, Schreiber R, and Kunzelmann K (2015a). Anoctamin 6 mediates effects essential for innate immunity downstream of P2X7 receptors in macrophages. Nat. Commun 6, 6245. [DOI] [PubMed] [Google Scholar]
- Ousingsawat J, Wanitchakool P, Schreiber R, Wuelling M, Vortkamp A, and Kunzelmann K (2015b). Anoctamin-6 controls bone mineralization by activating the calcium transporter NCX1. J. Biol. Chem 290, 6270–6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulino C, Kalienkova V, Lam AKM, Neldner Y, and Dutzler R (2017). Activation mechanism of the calcium-activated chloride channel TMEM16A revealed by cryo-EM. Nature 552, 421–425. [DOI] [PubMed] [Google Scholar]
- Pavelka A, Sebestova E, Kozlikova B, Brezovsky J, Sochor J, and Damborsky J (2016). CAVER: algorithms for analyzing dynamics of tunnels in macromolecules. IEEE/ACM Trans. Comput. Biol. Bioinform 13, 505–517. [DOI] [PubMed] [Google Scholar]
- Pedemonte N, and Galietta LJ (2014). Structure and function of TMEM16 proteins (anoctamins). Physiol. Rev 94, 419–459. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pomorski T, and Menon AK (2006). Lipid flippases and their biological functions. Cell. Mol. Life Sci 63, 2908–2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, and Stoorvogel W (2013). Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol 200, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Iehara T, Sato K, Fujii T, Sakai H, and Okada Y (2013). TMEM16F is a component of a Ca2+-activated Cl- channel but not a volume-sensitive outwardly rectifying Cl- channel. Am. J. Physiol. Cell Physiol 304, C748–C759. [DOI] [PubMed] [Google Scholar]
- Sims PJ, Wiedmer T, Esmon CT, Weiss HJ, and Shattil SJ (1989). Assembly of the platelet prothrombinase complex is linked to vesiculation of the platelet plasma membrane. Studies in Scott syndrome: an isolated defect in platelet procoagulant activity. J. Biol. Chem 264, 17049–17057. [PubMed] [Google Scholar]
- Suzuki J, Umeda M, Sims PJ, and Nagata S (2010). Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468, 834–838. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Fujii T, Imao T, Ishihara K, Kuba H, and Nagata S (2013). Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J. Biol. Chem 288, 13305–13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Walt S, Schönberger JL, Nunez-Iglesias J, Boulogne F, Warner JD, Yager N, Gouillart E, and Yu T; scikit-image contributors (2014). scikit-image: image processing in Python. PeerJ 2, e453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe R, Sakuragi T, Noji H, and Nagata S (2018). Single-molecule analysis of phospholipid scrambling by TMEM16F. Proc. Natl. Acad. Sci. USA 115, 3066–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. (2018). SWISSMODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46 (W1), W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whicher JR, and MacKinnon R (2016). Structure of the voltage-gated K+ channel Eag1 reveals an alternative voltage sensing mechanism. Science 353, 664–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock JM, and Hartzell HC (2016). A Pore Idea: the ion conduction pathway of TMEM16/ANO proteins is composed partly of lipid. Pflugers Arch. 468, 455–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock JM, and Hartzell HC (2017). Anoctamins/TMEM16 proteins: chloride channels flirting with lipids and extracellular vesicles. Annu. Rev. Physiol 79, 119–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf P (1967). The nature and significance of platelet products in human plasma. Br. J. Haematol 13, 269–288. [DOI] [PubMed] [Google Scholar]
- Yang H, Kim A, David T, Palmer D, Jin T, Tien J, Huang F, Cheng T, Coughlin SR, Jan YN, and Jan LY (2012). TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 151, 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye W, Han TW, Nassar LM, Zubia M, Jan YN, and Jan LY (2018). Phosphatidylinositol-(4, 5)-bisphosphate regulates calcium gating of smallconductance cation channel TMEM16F. Proc. Natl. Acad. Sci. USA 115, E1667–E1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye W, Han TW, He M, Jan YN, and Jan LY (2019). Dynamic change of electrostatic field in TMEM16F permeatioin pathway shifts its ion selectivity. bioRxiv. 10.1101/515569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Whitlock JM, Lee K, Ortlund EA, Cui YY, and Hartzell HC (2015). Identification of a lipid scrambling domain in ANO6/TMEM16F. eLife 4, e06901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwaal RF, Comfurius P, and Bevers EM (2004). Scott syndrome, a bleeding disorder caused by defective scrambling of membrane phospholipids. Biochim. Biophys. Acta 1636, 119–128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers of cryo-EM density maps of TMEM16F reported in this paper are EMD: 20244, 20245, 20246, and 20247. The accession numbers of particle image stacks after motion correction related to TMEM16F reported in this paper are EMPIAR: 10278 (TMEM16F in Digitonin with calcium bound), EMPIAR: 10279 (TMEM16F in Digitonin without calcium bound), EMPIAR: 10280 (TMEM16F in Nanodisc). The accession numbers of atomic coordinates for TMEM16F reported in this paper are PDB: 6P46, 6P47, 6P48, and 6P49. All electrophysiological data generated or analyzed during this study are included in the published article. All other data are available from the corresponding authors upon reasonable request.