

Abstract
Glucocorticoid-induced osteoporosis (GIOP) is a frequent complication of systemic glucocorticoid (GC) therapy, is the most common form of secondary osteoporosis, and is associated with skeletal fragility and increased fracture risk. A soluble form of BMP receptor type 1A fusion protein (mBMPR1A-mFc) acts as an antagonist to endogenous BMPR1A and could increase bone mass in both ovariectomized and ovary-intact mice, but its effects in GIOP mice remained unclear. The aim of this study was to evaluate the effects of mBMPR1A-mFc on the skeleton in experimental models of GIOP. mBMPR1A-mFc treatment could increase the bone mineral density (BMD), trabecular bone volume, thickness, and number, and cortical thickness, and reduce the structure model index and trabecular separation in GIOP mice. mBMPR1A-mFc treatment could also prevent bone loss and enhance biomechanical strength in GIOP mice by promoting osteoblastic bone formation and inhibiting osteoclastic bone resorption. Mechanistic studies revealed that mBMPR1A-mFc treatment increased murine osteoblastogenesis by activating the Wnt/β-catenin signaling pathway while decreasing osteoclastogenesis by inhibiting the RANK/RANKL/osteoprotegerin (OPG) signaling pathway. These findings demonstrate that mBMPR1A-mFc treatment in GIOP mice improves bone mass, microarchitecture, and strength by enhancing osteoblastic bone formation and inhibiting osteoclastic bone resorption in GIOP mice and offers a promising novel alternative for the treatment of GIOP.
Keywords: Murine BMP receptor type 1A fusion protein (mBMPR1A-mFc), glucocorticoid-induced osteoporosis (GIOP), Wnt/β-catenin signaling, RANK/RANKL/OPG
Introduction
Currently, about 1% of the world’s population is under treatment with glucocorticoids (GCs) [1]. GC therapy is commonly used and effective for treating a variety of immune and inflammatory disorders, such as inflammatory bowel disease, asthma, rheumatoid arthritis, and chronic obstructive pulmonary disease, as well as organ transplants [2,3]. Unfortunately, GC therapy may have multiple detrimental effects on bones, which are incredibly complex and only partially understood [4,5].
The skeleton is a dynamic organ system that continuously undergoes remodeling, with osteoclasts resorbing old bone and osteoblasts forming new bone [6]. Patients under prolonged GC exposure consistently exhibit reduced osteoblastic bone formation and enhanced osteoclastic bone resorption [7-9]. Chronic GC treatment, which results in a significant reduction in bone mineral density (BMD) and in strength of cancellous and cortical bone, can increase the risk of atraumatic fractures by 30-50% [8,10,11]. Moreover, prolonged GC exposure can cause muscle weakness, body balance loss, and an increased risk of falls, resulting in an elevated risk of bone fractures [12]. Oral administration of as little as 2.5 mg prednisolone per day for more than 3 months already leads to a 1.5-fold increase in fracture risk, whereas the risk increases up to 5-fold for daily doses of 7.5 mg per day or higher [13].
Bone morphogenetic proteins (BMPs) are a member of the transforming growth factor-β (TGF-β) gene superfamily [14]. Previous studies have reported that BMP signaling plays a critical role in osteoblastogenesis [15]. The BMP signal is mediated through transmembrane serine/threonine kinase receptors such as BMP types I and II. Upon ligand binding, a highly conserved glycine- and serine-rich domain between the transmembrane and kinase domains in the type I receptor is phosphorylated and activated. Of the three type I receptors that recognize BMPs (BMPR1A, BMPR1B, and ACVR1), BMPR1A is the most effective receptor for transducing the canonical BMP ligands BMP2 [16] and BMP4 [17], which are abundantly expressed in bone [18].
Postnatal ablation of Bmpr1a expression has been used to investigate the effects of Bmpr1a disruption in osteoblasts [19]. Unexpectedly, mice with postnatal conditional Bmpr1a disruption exhibited a marked increase in bone mass [20], which is associated with reduced expression of the Wnt antagonists dickkopf-1 (Dkk1) and sclerostin (SOST) [21], as well as the receptor activator of the NF-κB ligand (RANKL) [19,20]. A recently developed soluble murine BMPR1A fusion protein (mBMPR1A-mFc) acts as an antagonist to endogenous BMPR1A. The mBMPR1A-mFc is a soluble fusion protein consisting of the extracellular domain of murine BMPR1A and the Fc portion of murine immunoglobulin G2a (IgG2a). Research has demonstrated that mBMPR1A-mFc binds to BMP2/4 specifically and with high affinity [22]. This, in turn, inhibits Dkk1 expression in osteoblasts to stimulate Wnt signaling and increase osteoblastic bone formation [23]. Additionally, mBMPR1A-mFc treatment clearly reduced RANKL expression in osteoblasts and decreased bone resorption. Interestingly, the treatment of ovariectomized and ovary-intact mice with mBMPR1A-mFc resulted in improved bone microarchitecture, increased bone mass, and increased strength, due to a reduction in bone resorption and an increase in bone formation [22]. All of these observations support the suggestion that inhibition of BMPR1A signaling with mBMPR1A-mFc may have a positive therapeutic effect for the treatment of pathological bone loss.
In the present study, we aimed to test the effects of mBMPR1A-mFc on GIOP in mice, and the potential cellular and molecular mechanisms by which mBMPR1A-mFc treatment exerts anabolic and antiresorptive effects in GIOP.
Material and methods
Overview of the study design
All animal procedures were approved by and performed in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) at Pizhou City Hospital, Xuzhou Medical University. Forty 12-week-old male C57Bl/6J mice were randomly divided into the following four groups (n=10 each): (i) CON-VEH, (ii) CON-mBMPR1A-mFc, (iii) DEX-VEH, and (iv) DEX-mBMPR1A-mFc. There was no significant difference in initial body weight between the four groups. Mice were exposed to DEX for 28 days [24]; 1 mg/kg DEX was injected subcutaneously, 5 days/week, once daily for four consecutive weeks [24]. mBMPR1A-mFc (4.5 mg/kg; Acceleron Pharma, USA) or vehicle was injected subcutaneously twice per week. Animals were housed at 23±1°C on a 12-hour/12-hour light/dark cycle and had ad libitum access to standard laboratory pellet rodent chow and fresh water. To trace in vivo mineralization, each mouse was intraperitoneally given 20 mg/kg calcein at 3 and 10 days before euthanasia.
Specimen harvest and preparation
Mice were euthanized via CO2 inhalation for sample collection. Fasting blood was drawn by an intracardiac needle to isolate serum. Femurs and tibiae were dissected, cleared of soft tissue, and weighed. The left femurs were wrapped in saline-soaked gauze and stored at -70°C until mechanical testing. The right femurs were used for micro-CT scanning and dynamic bone histomorphometric analysis. The left tibiae were crushed in liquid nitrogen under RNase-free conditions in order to extract total RNA for quantitative RT-PCR. The right femurs were prepared for histology and imaging in 4% paraformaldehyde (PFA) at 4°C.
Bone mass and microstructure assessment
The left femurs were scanned with a dual-energy X-ray absorptiometer (Hologic ODR 4500A) to measure total body, spine, and femur BMD (g/cm2). Bone specimens were then analyzed by micro-CT with a SkyScan 1072 scanner and associated analysis software (SkyScan, Belgium) [25]. The following 3D indices in the defined region of interest (ROI) were calculated automatically with the software: trabecular bone volume/total volume (BV/TV, %), trabecular number (Tb.N, 1/mm), trabecular thickness (Tb.Th, μm), cortical thickness (Ct.Th, mm), cortical volume (Ct.V, mm3), trabecular separation (Tb.Sp), and structure model index (SMI).
Mechanical testing
The left frozen femurs were thawed for 2 hours at room temperature and subjected to three-point bending (Bose ElectroForce 3200; Bose Corporation, USA) [26]. Force-displacement data were acquired at 30 Hz and used to determine maximum load (N), yield load (N), ultimate displacement (mm), yield displacement (mm), stiffness (N/mm), and energy absorption (N × mm).
Biochemistry assays
Serum levels of bone turnover markers (BTMs), i.e., bone-specific alkaline phosphatase (BALP), N-terminal propeptide of type I procollagen (P1NP), tartrate-resistant acid phosphatase 5b (TRAP-5b), C-terminal cross-linked telopeptides of type I collagen (CTX-I), RANKL, and OPG, were measured using respective mouse-specific enzyme-linked immunosorbent assays (R&D Systems, Inc., Minneapolis, MN, USA). All measurements were performed following the manufacturer’s instructions and all samples were assayed in duplicate within the same protocol.
Quantitative RT-PCR
Total RNA extraction and RT-PCR were performed [27]. Briefly, total RNA was isolated from left tibiae with Trizol reagent (Sigma, USA), following the manufacturer’s recommendations. The primer sequences used in this study were designed using an NCBI Bioinformatics database and purchased commercially. mRNA expression levels were normalized to the housekeeping gene GAPDH by using the ΔΔCt method. Data are expressed as fold change, where the ratio between the gene of interest and the housekeeping gene for control mice was set as 1. All PCRs were performed in triplicate; the primers are shown in Table 1.
Table 1.
qRT-PCR primer sequences
| Gene | Primer | Primer sequence (5’-3’) |
|---|---|---|
| Ahr | F | GGGACCTCGGGTGACAATAA |
| R | CCTCTGTCCTTTTCCAACCG | |
| Axin | F | CAGTGTGAAGGCCAATGGC |
| R | TGGGTTCTCGGAAAATGAGG | |
| Cyr6 | F | GTGAAGTGCGTCCTTGTGGA |
| R | TGCCCTTTTTTAGGCTGCTG | |
| Nkd2 | F | AATTTCAGTCCAAGCACGCC |
| R | CGGGACTCTCTCTCCTCTTGC | |
| Tagln | F | CAGCCCAGACACCGAAGCTA |
| R | AGGCTTGGTCGTTTGTGGAC | |
| TGFβ-3 | F | AGGCTTGGTCGTTTGTGGAC |
| R | AGGCTGATTGTGGCCAAGTT | |
| Thbs1 | F | GGACCGGGCTCAACTCTACA |
| R | AGCTCCGCGCTCTCCAT | |
| Twist1 | F | TCGACTTCCTGTACCAGGTCCT |
| R | CCATCTTGGAGTCCAGCTCG | |
| Wsp | F | ATGCCTGGCTGTGTACCAGC |
| R | CCTGCGAGAGTGAAGTTCGTG | |
| Lef 1 | F | CAGCTCCCCCATACTGTGAG |
| R | TGCTGTCTATATCCGCAGGAA | |
| TCF | F | CAGCTCCCCCATACTGTGAG |
| R | TGCTGTCTATATCCGCAGGAA | |
| Wnt3a | F | GGCTCCTCTCGGATACCTCT |
| R | ACAGAGAATGGGCTGAGTGC | |
| β-catenin | F | CCTAGCTGGTGGACTGCAGAA |
| R | CACCACTGGCCAGAATGATGA | |
| LRP5 | F | CACCATTGATTATGCCGACCAG |
| R | TGAGTCAGGCCAAACGGGTAG | |
| RANKL | F | GCAGCATCGCTCTGTTCCTGTA |
| R | CCTGCAGGAGTCAGGTAGTGTGTC | |
| OPG | F | CACACGAACTGCAGCACATT |
| R | TCCACCAAAACACTCAGCCA | |
| ALP | F | CCTAGACACAAGCACTAACACTA |
| R | GTCAGTCAGGTTGTTCCGATTC | |
| OCN | F | GCCTTCATGTCCAAGCAGGA |
| R | GCGCCGGAGTCTGTTCACTA | |
| RUNX2 | F | CTGTGGTTACCGTCATGGCC |
| R | GGAGCTCGGCGGAGTAGTTC | |
| Col-1 | F | CCCTACTCAGCCGTCTGTGC |
| R | GGGTTCGGGCTGATGTACC | |
| SPP1 | F | AGAGCGGTGAGTCTAAGGAGT |
| R | TGCCCTTTCCGTTGTTGTCC | |
| OPN | F | CACTCCAATCGTCCCTACAGT |
| R | CTGGAAACTCCTAGACTTTGACC | |
| Osterix | F | CATCTAACAGGAGGATTTTGGTTTG |
| R | AAGCCTTTGCCCACCTACTTTT | |
| TRAP | F | CACTCCCACCCTGAGATTTGT |
| R | CCCCAGAGACATGATGAAGTCA | |
| Oscar | F | GGTCCTCATCTGCTTG |
| R | TATCTGGTGGAGTCTGG | |
| CTSK | F | CTTCCAATACGTGCAGCAGA |
| R | TCTTCAGGGCTTTCTCGTTC | |
| GAPDH | F | AAATGGTGAAGGTCGGTGTGAAC |
| R | CAACAATCTCCACTTTGCCACTG |
Histomorphometry
The femoral diaphyseal region was decalcified in EDTA glycerol solution for 14 days at 4°C. Decalcified femurs were dehydrated, embedded in paraffin wax, and then cut into 5-μm-thick sections on a rotary microtome. The sections were stained with hematoxylin and eosin (H&E), or histochemically stained for total collagen and TRAP activity and with von Kossa stain and toluidine blue, following an earlier described protocol [28].
Double calcein labeling
Double calcein labeling was performed [29] by embedding undecalcified tibiae in LR white acrylic resin (London Resin Company, UK). Dynamic histomorphometry measurements were performed in 7-mm unstained bone sections by fluorescence microscopy. Mineral apposition rate (MAR, mm/day) and mineralizing surface per bone surface (MS/BS, %) were measured under ultraviolet light and used to calculate the bone formation rate with a surface referent (BFR/BS, mm3/mm2/day).
Immunohistochemical staining
The expression of SOST was visualized in paraffin-embedded femurs from different groups [30]. Goat anti-mouse SOST (R&D Systems, catalog no. AF1589, USA) was used.
Western blot analysis
Whole protein extracts from femurs were prepared as previously described [31]. Protein levels were determined by Western blot analysis. Membranes were probed with primary antibodies including Wnt3a, LRP5, SOST, β-catenin, RANKL, and OPG (Abcam, Cambridge, MA, USA) diluted 1:1000, overnight at 4°C, followed by incubation with corresponding secondary antibodies conjugated with horseradish peroxidase (Cell Signaling Technology) for 2 hours at room temperature. The proteins were detected using an enhanced chemiluminescence Western blotting detection kit (Amersham, Shanghai, China). Bands were quantified using ImageJ software (NIH).
Statistical analysis
All values were analyzed by one-way ANOVA followed by Tukey’s multiple comparison tests and are expressed as mean ± SD. Statistical analysis was carried out using SPSS version 16.0 (Chicago, IL, USA). Differences among groups were considered to be statistically significant at a P-value <0.05.
Results
mBMPR1A-mFc treatment reverses DEX-induced osteopenia in mice
We first examined whether mBMPR1A-mFc treatment could increase bone mass in GIOP mice. Total, spinal, and femoral BMD, measured by DXA, all declined significantly in the DEX-VEH group (Figure 1A-C). mBMPR1A-mFc treatment increased the BMD in CON-mBMPR1A-mFc mice and prevented BMD loss in the DEX-mBMPR1A-mFc group (Figure 1A-C). BMD levels in DEX-mBMPR1A-mFc mice were comparable to those of CON-VEH animals (Figure 1A-C). The micro-CT images clearly indicated improved microarchitectural properties after mBMPR1A-mFc treatment (Figure 1D, 1E). Low values for BV/TV (Figure 1F), Tb.N (Figure 1G), Tb.Th (Figure 1H), Ct.Th (Figure 1I), and Ct.V (Figure 1J) and high values for SMI (Figure 1K) and Tb.Sp (Figure 1L) were observed in the DEX-VEH group. However, these microarchitectural parameters improved after mBMPR1A-mFc treatment, as demonstrated by significant increases in BV/TV, Tb.N, Tb.Th, Ct.Th, and Ct.V and a marked reduction in SMI and Tb.Sp (Figure 1F-L). These results show that mBMPR1A-mFc treatment reverses DEX-induced osteopenia.
Figure 1.

mBMPR1A-mFc treatment protects against DEX-induced bone loss in mice. (A-C) The BMD of total, spinal, and femoral were measured by DXA. (D, E) Representative reconstructed μCT images of femur cancellous bone (3D) and cortical bone (2D). (F) The trabecular bone volume (BV/TV), (G) trabecular number (Tb.N), (H) trabecular thickness (Tb.Th), (I) cortical thickness (Ct.Th), (J) cortical bone volume (Ct.V), (K) structural model index (SMI) and (L) trabecular space (Tb.Sp) from μCT analysis. Values are expressed as mean ± SD, n=10 per group. ns, not statistically significant; *, P<0.05; **, P<0.01; ***, P<0.001, versus the indicated group.
mBMPR1A-mFc treatment increases femoral bone strength
To examine whether mBMPR1A-mFc increased bone strength, three-point bending of the left femoral diaphysis was performed. DEX treatment resulted in lower maximum load (Figure 2A), lower energy absorption (Figure 2B), less stiffness (Figure 2C), less ultimate displacement (Figure 2D), less yield displacement (Figure 2E), and lower yield load (Figure 2F) compared with CON-VEH mice. CON-mBMPR1A-mFc mice exhibited enhanced bone strength, with a higher maximum load, greater energy absorption, more stiffness, greater ultimate displacement, increased yield displacement, and higher yield load when compared with DEX-VEH mice (Figure 2A-F). Taken together, these results suggest that mBMPR1A-mFc treatment enhances skeletal properties in adult mice and prevents skeletal deterioration in GIOP mice.
Figure 2.
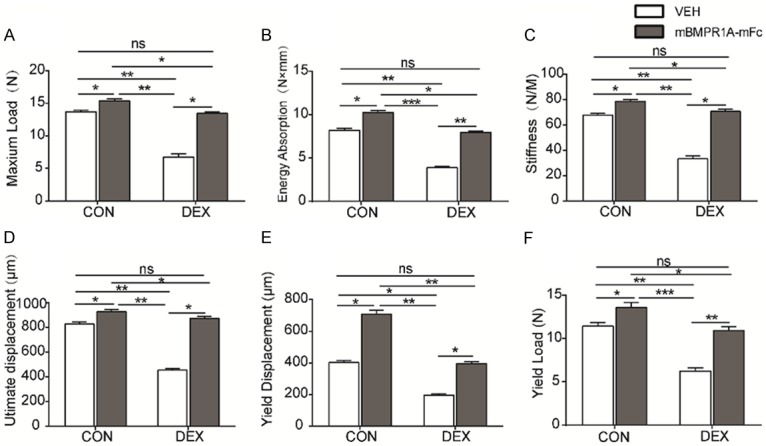
mBMPR1A-mFc treatment protects against DEX-induced reduction of mechanical properties in mice. The (A) maximum load, (B) energy absorption, (C) stiffness, (D) ultimate displacement, (E) yield displacement, and (F) yield load through three-point bending test. Values are expressed as mean ± SD, n=10 per group. ns, not statistically significant; *, P<0.05; **, P<0.01; ***, P<0.001, versus the indicated group.
mBMPR1A-mFc treatment increases osteoblastic bone formation
Static histomorphometric analysis (400× magnification) (Figure 3A-D) of trabecular bone in the distal femora following DEX treatment showed lower BV/TV (Figure 3F), lower total collagen-positive area/tissue area (Figure 3G), lower von Kossa-positive area/tissue area (Figure 3H), a lower number of OBs per bone surface (N.Ob/BS) (Figure 3I), and a lower percentage of bone surface covered by OBs (Ob.S/BS) (Figure 3J) compared with VEH-treated mice. However, dynamic histomorphometric analysis (Figure 3E) revealed that all bone formation indices, i.e., mineral apposition rate (MAR) (Figure 3K), bone formation rate per bone surface (BFR/BS) (Figure 3L), and mineralizing surface per bone surface (MS/BS) (Figure 3M), were decreased on the endocortical surface in DEX-VEH mice when compared to CON-VEH mice, consistent with the decreased ALP (Figure 3N) and P1NP (Figure 3O) levels observed in DEX-VEH mice. To understand the molecular mechanisms responsible for the significant decrease in osteoblastogenesis, mRNA levels of ALP, OCN, Runx2, COL-1, SPP1, OPN, and Osterix were assessed by RT-PCR (Figure 3P). Consistent with previous reports, DEX treatment significantly downregulated the mRNA levels of each of the osteogenic genes analyzed. mBMPR1A-mFc treatment increased the expression levels of ALP, OCN, Runx2, COL-1, SPP1, OPN, and Osterix in DEX-treated mice, which confirms that mBMPR1A-mFc increases osteoblast differentiation (Figure 3P).
Figure 3.
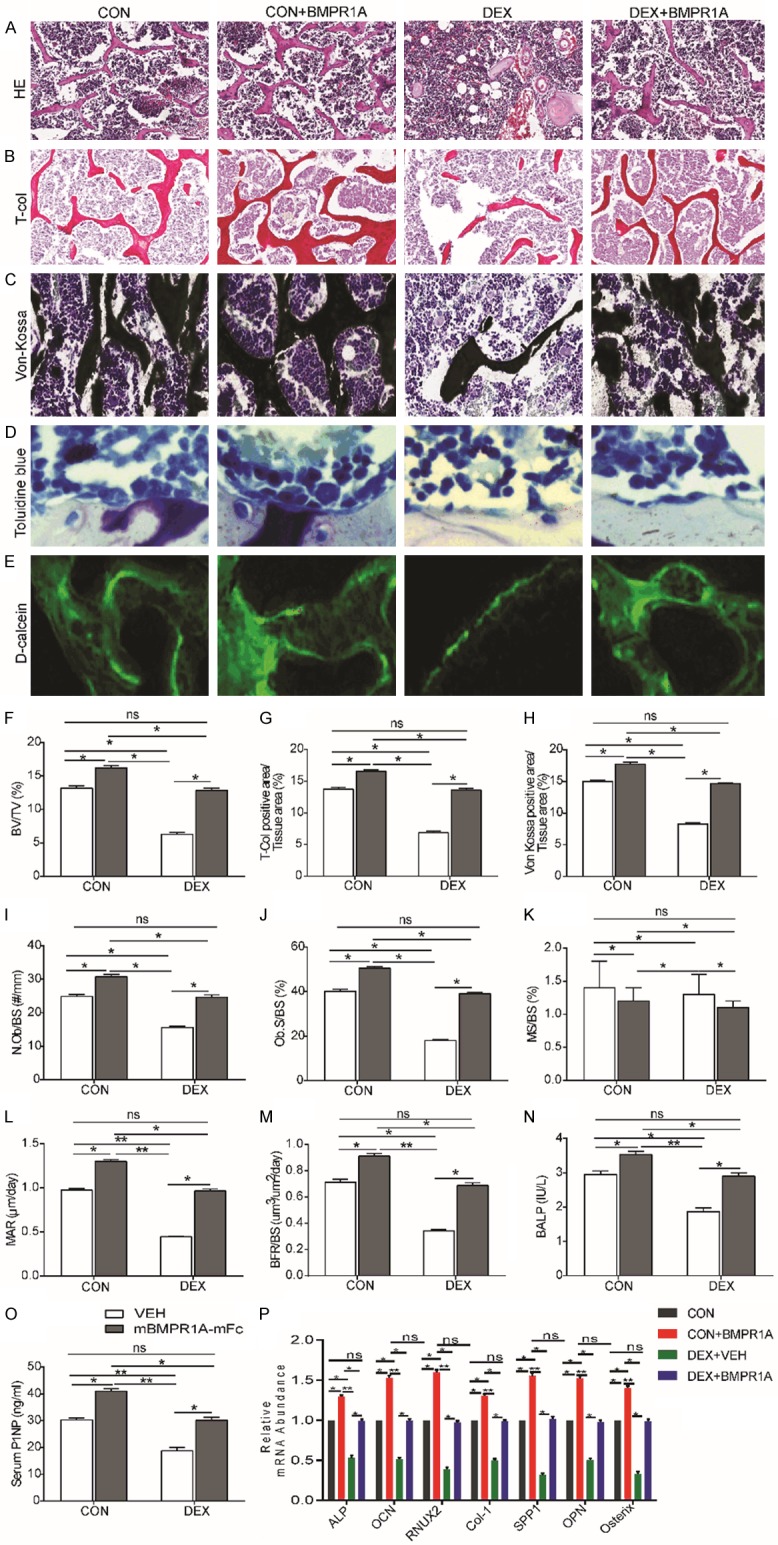
mBMPR1A-mFc treatment protects against the DEX-induced reduction of osteoblastic bone formation in mice. The histomorphometric analysis of distal femur by (A) hematoxylin & eosin (H&E), 400×, (B) total collagen, 400×, (C) von kossa, 400×, (D) toluidine blue, 400×, and (E) fluorescent calcein labeling staining, 400×. Histomorphometric analysis of (F) trabecular bone volume (BV/TV), (G) Total-Col positive area/Tissue area, (H) von kossa positive area/Tissue area, (I) Number of OBs per bone surface (N.Ob/BS), (J) Percentage of bone surfaces covered by OBs (Ob.S/BS), (K) percentage of bone surfaces covered by mineralized surfaces (MS/BS), (L) Mineral apposition rate (MAR), (M) Bone formation rate/bone surface (BFR/BS). Serum bone formation markers (N) BALP and (O) PINP were measured by ELISA. (P) The expression of osteoblast-specific genes, including ALP, OCN, Runx2, COL-1, SPP1, OPN and Osterix was examined by qRT-PCR in femurs. Values are expressed as mean ± SD, n=10 per group. ns, not statistically significant; *, P<0.05; **, P<0.01; ***, P<0.001, versus the indicated group.
mBMPR1A-mFc treatment protects against DEX-induced osteoclastic bone resorption
Static histomorphometric analysis (400× magnification) (Figure 4A) of the trabecular bone of the distal femora revealed a significant decrease in the number of osteoclasts per bone surface (N.Oc/BS) (Figure 4B), a decrease in eroded bone surface (ES/BS) (Figure 4C), and a decrease in bone surface occupied by osteoclasts (Oc.S/BS) (Figure 4D) in the DEX-mBMPR1A-mFc group compared with the VEH-treated group. The decrease in osteoclast number was associated with a reduction in TRAP-5b (Figure 4E) and CTX-1 (Figure 4F) serum levels in mBMPR1A-mFc-treated mice compared with VEH-treated animals. The qRT-PCR results indicate that expression levels of genes related to osteoclastogenesis, such as Acp5 (which encodes TRAP), osteoclast-associated receptor (Oscar), and Cathepsin K (Ctsk), were also significantly reduced after mBMPR1A-mFc treatment (Figure 4G). These data suggest that osteoclast numbers and resorption were significantly decreased, leading to increased bone mass after mBMPR1A-mFc treatment.
Figure 4.
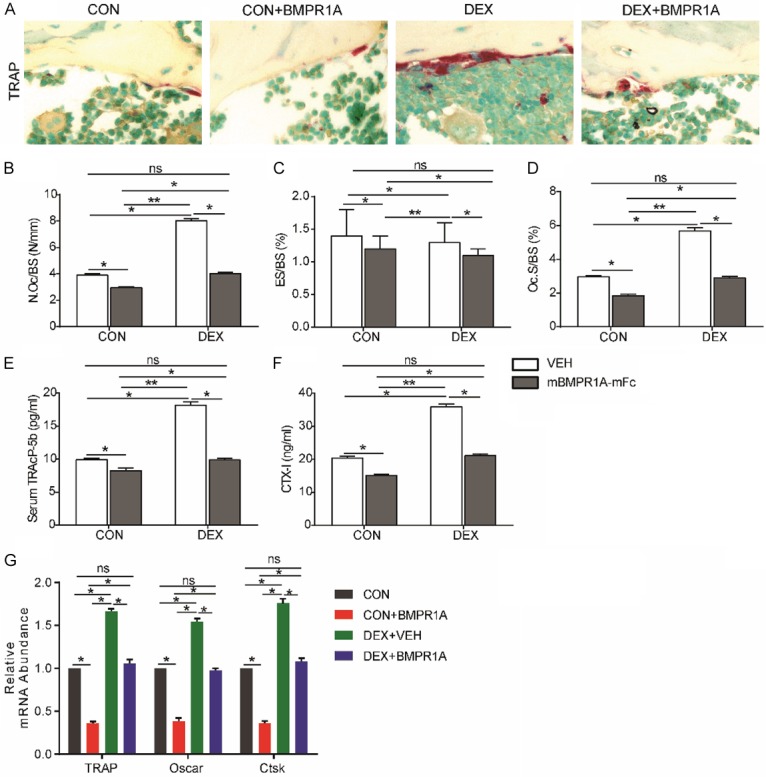
mBMPR1A-mFc treatment protects against the DEX-induced osteoclastic bone resorption in mice. (A) Sections of the metaphyseal regions of the distal femurs were performed TRAP staining, 400×. (B) The osteoclast number/bone surface (N.Oc/BS), (C) eroded surface/bone surface (ES/BS) and (D) osteoclast surface/bone surface (Oc.S/BS) were analyzed. Serum bone resorption markers (E) TRAP5b and (F) CTX were measured by ELISA. (G) RT-PCR of bone tissue extracts for the expression of TRAP, Oscar and CTSK. Values are expressed as mean ± SD, n=10 per group. ns, not statistically significant; *, P<0.05; **, P<0.01; ***, P<0.001, versus the indicated group.
mBMPR1A-mFc treatment alters Wnt/β-catenin signaling
Because osteoblast differentiation is induced by Wnt activation, we investigated the effects of DEX and BMPR1A treatment on Wnt signaling. The percentage of SOST-positive osteocytes, as detected by SOST immunostaining, was increased 2-fold in DEX-VEH mice compared with CON-VEH mice (400× magnification) (Figure 5A, 5B), which is in agreement with the femoral SOST mRNA levels (Figure 5C). The qRT-PCR results indicate that genes related to Wnt signaling, such as Wnt3a, LRP5, and β-catenin, were also significantly upregulated after mBMPR1A-mFc treatment (Figure 5D). Furthermore, we measured the femoral mRNA expression levels of genes specifically upregulated by Wnt signaling, i.e., aryl-hydrocarbon receptor (Ahr), axin2, cysteine-rich protein 61 (Cyr61), naked cuticle 2 homolog (Nkd2), transgelin (tagline), transforming growth factor β3 (TGFβ3), thrombospondin 1 (Thbs1), Twist gene homolog 1 (Twist1), Wnt1-inducible signaling pathway protein 1 (Wisp1), T-cell factor (TCF), and lymphoid enhancer factor-1 (Lef-1) (Figure 5E). These genes were chosen because they are sensitive markers of Wnt activation [32].The protein expression of Wnt3a, LRP5, β-catenin and SOST in each group were also assessed by Western blot analysis (Figure 5F). The results reveal that the levels of protein expression and mRNA for the genes related to Wnt signaling and the genes specifically upregulated by Wnt signaling were all dramatically decreased by DEX treatment compared with the CON-VEH group (Figure 5D-J). Moreover, treatment with mBMPR1A-mFc caused a significant increase in the levels of protein expression of Wnt3a, LRP5, β-catenin, and SOST and the mRNA levels of the genes related to Wnt signaling and the genes upregulated by Wnt signaling in the DEX-mBMPR1A-mFc and CON-mBMPR1A-mFc groups (Figure 5D-J), suggesting that mBMPR1A-mFc stimulates bone formation by activating Wnt signaling.
Figure 5.

mBMPR1A-mFc treatment alters Wnt/β-catenin signaling. A. The longitudinal sections of femoral bone stained with an anti-SOST antibody, 400×. B. The percentage of SOST-positive osteocytes were quantified. C. Sost gene expression was quantified by qPCR in femur. D. RT-PCR of bone tissue extracts for the expression of Wnt3a, LRP5 and β-catenin. E. RT-PCR analysis of mRNA expression of genes specifically upregulated by Wnt signaling in flushed femurs. F. Protein expression of Wnt3a, LRP5, β-catenin and SOST in each group, as assessed by Western blot analysis. G-J. Densitometry results of Wnt3a, LRP5, β-catenin and SOST protein expression in each group. Values are expressed as mean ± SD, n=10 per group. ns, not statistically significant; *, P<0.05; **, P<0.01; ***, P<0.001, versus the indicated group.
mBMPR1A-mFc treatment alters RANKL/OPG signaling
To elucidate the molecular mechanisms underlying the suppression of osteoclastogenesis, we examined the effects of mBMPR1A-mFc on RANKL and OPG expression in the femur. mBMPR1A-mFc treatment caused a dramatic decrease in the mRNA expression levels of RANKL and a modest increase in the mRNA levels of OPG (Figure 6C). The ratio of RANKL/OPG, a key index for osteoclastic activity, was reduced by mBMPR1A-mFc treatment (Figure 6C). Consistent with our PCR results, RANKL serum levels were decreased (Figure 6A) while serum OPG levels were increased (Figure 6B) after mBMPR1A-mFc treatment compared with VEH-treated mice. Furthermore, we measured the protein expression of RANKL and OPG in the femur in each group by Western blot analysis (Figure 6D). The RANKL protein expression levels were reduced (Figure 6E) while the OPG protein expression levels were increased (Figure 6F) after mBMPR1A-mFc treatment compared with VEH-treated mice.
Figure 6.
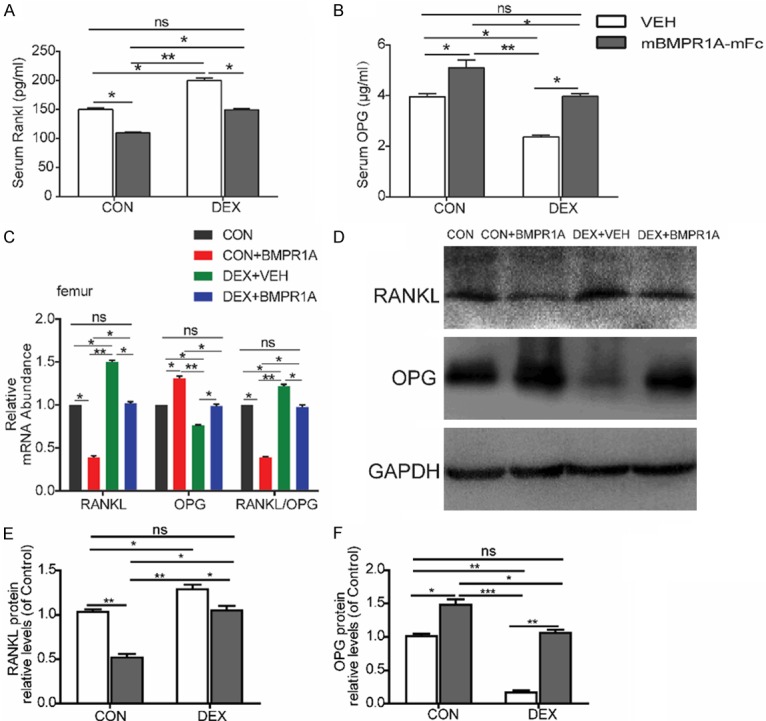
mBMPR1A-mFc treatment alters RANKL/OPG signaling. A, B. Serum Rankl and OPG were measured by ELISA. C. qPCR analysis of mRNA expression of of RANKL, OPG, and RANKL/OPG ratio in femurs. D. Protein expression of RANKL and OPG in each group, as assessed by Western blot analysis. E, F. Densitometry results of RANKL and OPG protein expression in each group. Values are expressed as mean ± SD, n=10 per group. ns, not statistically significant; *, P<0.05; **, P<0.01; ***, P<0.001, versus the indicated group.
Discussion
Glucocorticoid-mediated osteoporosis is the most common form of secondary osteoporosis and is associated with substantial morbidity and mortality, a reduction in quality of life, and a large socioeconomic impact [8,11,33]. Given the importance of GCs as a common therapy for rheumatologic diseases, asthma, and immunosuppression, new ways to prevent and treat GIOP are urgently needed. The goal of our study was to evaluate the role of mBMPR1A-mFc treatment on GIOP. The current study demonstrates that mBMPR1A-mFc treatment prevents the deterioration of femoral bone microarchitecture, mass, and mechanical properties of GIOP mice by increasing bone formation and suppressing bone resorption. The effects of mBMPR1A-mFc treatment on the femur appear to be associated with the regulation of skeletal gene expression levels of Wnt3a/β-catenin and RANKL/RANK/OPG signaling pathways.
Inhibition of mineral resorption with bisphosphonates is the current standard treatment strategy for GIOP [34]. Bisphosphonates prevent GC-induced bone loss in animal models and human GIOP patients. However, bisphosphonates also reduce bone formation even further [35,36]. Treatment with anti-RANKL antibodies induces even more remarkable reductions in bone formation compared to treatment with bisphosphonates in patients receiving GCs [37]. Therefore, there is a clinical need to identify new agents for the treatment of GIOP, particularly agents that promote bone formation and simultaneously inhibit bone resorption.
Laboratory mice are often used for establishing animal models of GIOP [38-45]. The C57BL/6J mouse is a reliable and reproducible model for GIOP, as demonstrated by significant decreases in BMD, reductions in osteoblastic bone formation, and increases in osteoclastic bone absorption [43,44]. In this study, supraphysiologic DEX (1 mg/kg/day, 5 days/week for 4 weeks) was given to 12-week-old male C57BL/6J mice by subcutaneous injection to reproduce previous findings [24]. The femur, the part most sensitive to atypical fractures in approximately 40% of European women treated with GCs [46,47], was chosen to assess the effects of mBMPR1A-mFc on GIOP. As expected, DEX disrupted bone homeostasis, reduced bone mass, decreased bone mechanical strength, and increased cortical and trabecular bone damage in GIOP mice. Interestingly, we found that mBMPR1A-mFc increases bone mass in the GIOP model. mBMPR1A-mFc treatment completely reversed DEX-induced bone loss and restored both trabecular bone volume, number, and thickness and cortical thickness. The increase in BMD, improvement in bone microarchitecture, increase in bone formation rate, and decrease in osteoclast-positive surface observed in mBMPR1A-mFc-treated animals are consistent with our previous research [22]. In the current study, we demonstrated that the adverse effects of GC treatment were reversed by the inhibition of BMP2/4 signaling via treatment with mBMPR1A-mFc, which led to increased bone mass and improved bone microarchitecture by increasing the bone formation rate and decreasing the osteoclast-positive surface.
The RANKL/RANK/OPG signaling pathway plays a key role in the regulation of bone remodeling by modulating osteoclast development and activation [48]. Both RANKL and OPG are secreted predominantly by osteoblasts, which regulates the differentiation of osteoclasts [49]. RANKL binds to RANK on the cell surface of osteoclasts or osteoclast precursors to induce differentiation, activation, and survival of osteoclasts; OPG, a decoy receptor for RANKL, inhibits osteoclastogenesis [48]. The ratio of RANKL/OPG can be used to assess bone remodeling. In agreement with a previous study [50], RANKL mRNA expression was upregulated after DEX treatment in mice. Furthermore, the RANKL/OPG ratio was higher in the DEX-VEH group than in the CON-VEH group, which is in agreement with other studies [45]. Moreover, we found that the RANKL mRNA expression was markedly downregulated by mBMPR1A-mFc in mice, which is consistent with another study [51]. These findings indicate that mBMPR1A-mFc regulates the RANKL/RANK/OPG signaling pathway to reduce bone resorption, resulting in improved lumbar vertebral trabecular bone mass, increased mechanical strength, and a reduction in bone resorption marker serum levels in GIOP mice.
In the current study, we found that mBMPR1A-mFc treatment in mice not only inhibits DEX-induced bone loss but also results in significantly greater bone mass and strength compared to CON-VEH mice, confirming our previous results that mBMPR1A-mFc treatment results in a profound increase in mouse bone mass [22]. The increased bone mass was associated with greater trabecular number and width, larger cortical thickness, and lower trabecular separation [22]. This finding suggests that the anabolic effect of mBMPR1A-mFc treatment was retained despite continuous DEX administration.
Previous studies have revealed that canonical Wnt signaling plays a critical role in the regulation of bone mass, bone remodeling, and bone homeostasis [52]. Wnt binds to frizzled receptors and their low-density lipoprotein co-receptor receptor-related proteins (LRP5/6) to stabilize cytosolic β-catenin, which then enters the nucleus and initiates the transcription of Wnt target genes [53]. In concordance with previous studies [54], the femoral mRNA expression levels of Wnt3a, LRP5, and β-catenin were downregulated in the DEX-VEH group when compared with the CON-VEH group. Interestingly, the expression levels of Wnt3a, LRP5, and β-catenin were all markedly upregulated after mBMPR1A-mFc treatment, demonstrating the potential activation of the Wnt3a/β-catenin signaling pathway by mBMPR1A-mFc, which is consistent with our previous results [23]. Also consistent with previous results [55,56], we demonstrated that mBMPR1A-mFc inhibits the expression of the soluble Wnt antagonist SOST. SOST has been shown to be a negative regulator of Wnt signaling and osteoblast differentiation [34,57]. Previous studies reported that the Wnt-signaling antagonist SOST is upregulated following GC treatment [58,59]. Significantly increased levels of SOST following GC treatment may contribute to skeletal deterioration by enhancing osteoclastogenesis by increasing levels of the pro-resorptive cytokine RANKL and decreasing levels of the RANKL decoy receptor OPG [60]. mBMP1RA-mFc treatment inhibits SOST expression and activates canonical Wnt signaling [22], which could account for the increase in osteoblast numbers and bone formation, which in turn prevents skeletal deterioration associated with DEX treatment. Thus, our study indicates that mBMPR1A-mFc upregulates the Wnt3a/β-catenin signaling pathway, resulting in increased serum levels of bone formation markers, higher femoral trabecular bone mass, and improved mechanical properties in GIOP mice.
The main limitation of our current study is that we studied only male mice at a single time point, with a single mBMPR1A-mFc dosing regimen. Therefore, it remains to be confirmed whether the osteoprotective effects of mBMPR1A-mFc will continue with longer treatment. It also remains to be investigated whether more frequent dosing or higher doses will enhance the osteoprotective effects. Additional research is required to investigate the optimal dose and further elucidate the functional mechanisms underlying its protective effects.
In summary, mBMPR1A-mFc treatment results in increases in bone mass, structure, and strength. Furthermore, mBMPR1A-mFc treatment can reverse GC-induced bone loss through enhancing osteoblastic bone formation by activating Wnt/β-catenin signaling and suppressing osteoclastic bone resorption by inhibiting the RANK/RANKL/OPG pathway. These results suggest that mBMPR1A-mFc may be a promising therapeutic agent for the treatment of GC-associated secondary osteoporosis.
Acknowledgements
This study was supported by the Research Funding Project of Xuzhou Medical University (Grant No. 2018KJ23).
Disclosure of conflict of interest
None.
References
- 1.Overman RA, Toliver JC, Yeh JY, Gourlay ML, Deal CL. United States adults meeting 2010 American college of rheumatology criteria for treatment and prevention of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken) 2014;66:1644–1652. doi: 10.1002/acr.22346. [DOI] [PubMed] [Google Scholar]
- 2.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids - new mechanisms for old drugs. N Engl J Med. 2005;353:1711–1723. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 3.Jin HO, Seo SK, Woo SH, Kim ES, Lee HC, Yoo DH, An S, Choe TB, Lee SJ, Hong SI, Rhee CH, Kim JI, Park IC. Activating transcription factor 4 and CCAAT/enhancer-binding protein-beta negatively regulate the mammalian target of rapamycin via Redd1 expression in response to oxidative and endoplasmic reticulum stress. Free Radic Biol Med. 2009;46:1158–1167. doi: 10.1016/j.freeradbiomed.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 4.Manolagas SC. Steroids and osteoporosis: the quest for mechanisms. J Clin Invest. 2013;123:1919–1921. doi: 10.1172/JCI68062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seibel MJ, Cooper MS, Zhou H. Glucocorticoid-induced osteoporosis: mechanisms, management, and future perspectives. Lancet Diabetes Endocrinol. 2013;1:59–70. doi: 10.1016/S2213-8587(13)70045-7. [DOI] [PubMed] [Google Scholar]
- 6.Parfitt AM. Osteonal and hemi-osteonal remodeling: the spatial and temporal framework for signal traffic in adult human bone. J Cell Biochem. 1994;55:273–286. doi: 10.1002/jcb.240550303. [DOI] [PubMed] [Google Scholar]
- 7.Reid IR. Glucocorticoid osteoporosis--mechanisms and management. Eur J Endocrinol. 1997;137:209–217. doi: 10.1530/eje.0.1370209. [DOI] [PubMed] [Google Scholar]
- 8.Weinstein RS. Glucocorticoid-induced bone disease. N Engl J Med. 2011;365:62–70. doi: 10.1056/NEJMcp1012926. [DOI] [PubMed] [Google Scholar]
- 9.Dempster DW. Bone histomorphometry in glucocorticoid-induced osteoporosis. J Bone Miner Res. 1989;4:137–141. doi: 10.1002/jbmr.5650040202. [DOI] [PubMed] [Google Scholar]
- 10.Weinstein RS. Glucocorticoid-induced osteoporosis. Rev Endocr Metab Disord. 2001;2:65–73. doi: 10.1023/a:1010007108155. [DOI] [PubMed] [Google Scholar]
- 11.Mazziotti G, Angeli A, Bilezikian JP, Canalis E, Giustina A. Glucocorticoid-induced osteoporosis: an update. Trends Endocrinol Metab. 2006;17:144–149. doi: 10.1016/j.tem.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 12.Rizzoli R, Adachi JD, Cooper C, Dere W, Devogelaer JP, Diez-Perez A, Kanis JA, Laslop A, Mitlak B, Papapoulos S, Ralston S, Reiter S, Werhya G, Reginster JY. Management of glucocorticoid-induced osteoporosis. Calcif Tissue Int. 2012;91:225–243. doi: 10.1007/s00223-012-9630-5. [DOI] [PubMed] [Google Scholar]
- 13.Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Use of oral corticosteroids and risk of fractures. J Bone Miner Res. 2000;15:993–1000. doi: 10.1359/jbmr.2000.15.6.993. [DOI] [PubMed] [Google Scholar]
- 14.Massague J. Receptors for the TGF-beta family. Cell. 1992;69:1067–1070. doi: 10.1016/0092-8674(92)90627-o. [DOI] [PubMed] [Google Scholar]
- 15.Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. Growth Factors. 2004;22:233–241. doi: 10.1080/08977190412331279890. [DOI] [PubMed] [Google Scholar]
- 16.Keller S, Nickel J, Zhang JL, Sebald W, Mueller TD. Molecular recognition of BMP-2 and BMP receptor IA. Nat Struct Mol Biol. 2004;11:481–488. doi: 10.1038/nsmb756. [DOI] [PubMed] [Google Scholar]
- 17.Hatta T, Konishi H, Katoh E, Natsume T, Ueno N, Kobayashi Y, Yamazaki T. Identification of the ligand-binding site of the BMP type IA receptor for BMP-4. Biopolymers. 2000;55:399–406. doi: 10.1002/1097-0282(2000)55:5<399::AID-BIP1014>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 18.Wozney JM. The bone morphogenetic protein family and osteogenesis. Mol Reprod Dev. 1992;32:160–167. doi: 10.1002/mrd.1080320212. [DOI] [PubMed] [Google Scholar]
- 19.Mishina Y, Starbuck MW, Gentile MA, Fukuda T, Kasparcova V, Seedor JG, Hanks MC, Amling M, Pinero GJ, Harada S, Behringer RR. Bone morphogenetic protein type IA receptor signaling regulates postnatal osteoblast function and bone remodeling. J Biol Chem. 2004;279:27560–27566. doi: 10.1074/jbc.M404222200. [DOI] [PubMed] [Google Scholar]
- 20.Kamiya N, Ye L, Kobayashi T, Lucas DJ, Mochida Y, Yamauchi M, Kronenberg HM, Feng JQ, Mishina Y. Disruption of BMP signaling in osteoblasts through type IA receptor (BMPRIA) increases bone mass. J Bone Miner Res. 2008;23:2007–2017. doi: 10.1359/JBMR.080809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamiya N, Kobayashi T, Mochida Y, Yu PB, Yamauchi M, Kronenberg HM, Mishina Y. Wnt inhibitors Dkk1 and Sost are downstream targets of BMP signaling through the type IA receptor (BMPRIA) in osteoblasts. J Bone Miner Res. 2010;25:200–210. doi: 10.1359/jbmr.090806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baud’huin M, Solban N, Cornwall-Brady M, Sako D, Kawamoto Y, Liharska K, Lath D, Bouxsein ML, Underwood KW, Ucran J, Kumar R, Pobre E, Grinberg A, Seehra J, Canalis E, Pearsall RS, Croucher PI. A soluble bone morphogenetic protein type IA receptor increases bone mass and bone strength. Proc Natl Acad Sci U S A. 2012;109:12207–12212. doi: 10.1073/pnas.1204929109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ko FC, Van Vliet M, Ellman R, Grasso D, Brooks DJ, Spatz JM, Conlon C, Aguirre JI, Wronski TJ, Bouxsein ML. Treatment with a soluble bone morphogenetic protein type 1A receptor (BMPR1A) fusion protein increases bone mass and bone formation in mice subjected to hindlimb unloading. JBMR Plus. 2017;1:66–72. doi: 10.1002/jbm4.10012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi J, Wang L, Zhang HY, Jie Q, Li XJ, Shi QY, Huang Q, Gao B, Han YH, Guo K, Liu J, Yang L, Luo ZJ. Glucocorticoids: dose-related effects on osteoclast formation and function via reactive oxygen species and autophagy. Bone. 2015;79:222–232. doi: 10.1016/j.bone.2015.06.014. [DOI] [PubMed] [Google Scholar]
- 25.Xue Y, Karaplis AC, Hendy GN, Goltzman D, Miao D. Genetic models show that parathyroid hormone and 1,25-dihydroxyvitamin D3 play distinct and synergistic roles in postnatal mineral ion homeostasis and skeletal development. Hum Mol Genet. 2005;14:1515–1528. doi: 10.1093/hmg/ddi160. [DOI] [PubMed] [Google Scholar]
- 26.Jing D, Luo E, Cai J, Tong S, Zhai M, Shen G, Wang X, Luo Z. Mechanical vibration mitigates the decrease of bone quantity and bone quality of leptin receptor-deficient Db/Db mice by promoting bone formation and inhibiting bone resorption. J Bone Miner Res. 2016;31:1713–1724. doi: 10.1002/jbmr.2837. [DOI] [PubMed] [Google Scholar]
- 27.Xue YB, Zhang ZL, Karaplis AC, Hendy GN, Goltzman D, Miao DS. Exogenous PTH-related protein and PTH improve mineral and skeletal status in 25-hydroxyvitamin D-1 alpha-hydroxylase and PTH double knockout mice. J Bone Miner Res. 2005;20:1766–1777. doi: 10.1359/JBMR.050608. [DOI] [PubMed] [Google Scholar]
- 28.Miao DS, Scutt A. Recruitment, augmentation and apoptosis of rat osteoclasts in 1,25-(OH)(2)D-3 response to short-term treatment with 1,25-dihydroxyvitamin D-3 in vivo. BMC Musculoskelet Disord. 2002;3:16. doi: 10.1186/1471-2474-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miao D, He B, Lanske B, Bai XY, Tong XK, Hendy GN, Goltzman D, Karaplis AC. Skeletal abnormalities in Pth-null mice are influenced by dietary calcium. Endocrinology. 2004;145:2046–2053. doi: 10.1210/en.2003-1097. [DOI] [PubMed] [Google Scholar]
- 30.Taut AD, Jin Q, Chung JH, Galindo-Moreno P, Yi ES, Sugai JV, Ke HZ, Liu M, Giannobile WV. Sclerostin antibody stimulates bone regeneration after experimental periodontitis. J Bone Miner Res. 2013;28:2347–2356. doi: 10.1002/jbmr.1984. [DOI] [PubMed] [Google Scholar]
- 31.Geng Q, Gao H, Yang R, Guo K, Miao D. Pyrroloquinoline quinone prevents estrogen deficiency-induced osteoporosis by inhibiting oxidative stress and osteocyte senescence. Int J Biol Sci. 2019;15:58–68. doi: 10.7150/ijbs.25783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson A, Vayssiere B, Garcia T, Newell W, Baron R, Roman-Roman S, Rawadi G. Gene array analysis of Wnt-regulated genes in C3H10T1/2 cells. Bone. 2005;36:585–598. doi: 10.1016/j.bone.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 33.Kawai M, Moedder UI, Khosla S, Rosen CJ. Emerging therapeutic opportunities for skeletal restoration. Nat Rev Drug Discov. 2011;10:141–156. doi: 10.1038/nrd3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tian E, Zhan F, Walker R, Rasmussen E, Ma Y, Barlogie B, Shaughnessy JD Jr. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med. 2003;349:2483–2494. doi: 10.1056/NEJMoa030847. [DOI] [PubMed] [Google Scholar]
- 35.Saag KG, Emkey R, Schnitzer TJ, Brown JP, Hawkins F, Goemaere S, Thamsborg G, Liberman UA, Delmas PD, Malice MP, Czachur M, Daifotis AG. Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. Glucocorticoid-induced osteoporosis intervention study group. N Engl J Med. 1998;339:292–299. doi: 10.1056/NEJM199807303390502. [DOI] [PubMed] [Google Scholar]
- 36.Saag KG, Shane E, Boonen S, Marin F, Donley DW, Taylor KA, Dalsky GP, Marcus R. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med. 2007;357:2028–2039. doi: 10.1056/NEJMoa071408. [DOI] [PubMed] [Google Scholar]
- 37.Mok CC, Ho LY, Ma KM. Switching of oral bisphosphonates to denosumab in chronic glucocorticoid users: a 12-month randomized controlled trial. Bone. 2015;75:222–228. doi: 10.1016/j.bone.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 38.Ko JY, Chuang PC, Ke HJ, Chen YS, Sun YC, Wang FS. MicroRNA-29a mitigates glucocorticoid induction of bone loss and fatty marrow by rescuing Runx2 acetylation. Bone. 2015;81:80–88. doi: 10.1016/j.bone.2015.06.022. [DOI] [PubMed] [Google Scholar]
- 39.Karunaratne A, Xi L, Bentley L, Sykes D, Boyde A, Esapa CT, Terrill NJ, Brown SDM, Cox RD, Thakker RV, Gupta HS. Multiscale alterations in bone matrix quality increased fragility in steroid induced osteoporosis. Bone. 2016;84:15–24. doi: 10.1016/j.bone.2015.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dai W, Jiang L, Lay YA, Chen H, Jin G, Zhang H, Kot A, Ritchie RO, Lane NE, Yao W. Prevention of glucocorticoid induced bone changes with beta-ecdysone. Bone. 2015;74:48–57. doi: 10.1016/j.bone.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu K, Jing Y, Zhang W, Fu X, Zhao H, Zhou X, Tao Y, Yang H, Zhang Y, Zen K, Zhang C, Li D, Shi Q. Silencing miR-106b accelerates osteogenesis of mesenchymal stem cells and rescues against glucocorticoid-induced osteoporosis by targeting BMP2. Bone. 2017;97:130–138. doi: 10.1016/j.bone.2017.01.014. [DOI] [PubMed] [Google Scholar]
- 42.Sato AY, Cregor M, Delgado-Calle J, Condon KW, Allen MR, Peacock M, Plotkin LI, Bellido T. Protection from glucocorticoid-induced osteoporosis by anti-catabolic signaling in the absence of sost/sclerostin. J Bone Miner Res. 2016;31:1791–1802. doi: 10.1002/jbmr.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Brien CA, Jia D, Plotkin LI, Bellido T, Powers CC, Stewart SA, Manolagas SC, Weinstein RS. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology. 2004;145:1835–1841. doi: 10.1210/en.2003-0990. [DOI] [PubMed] [Google Scholar]
- 44.Sato AY, Tu X, McAndrews KA, Plotkin LI, Bellido T. Prevention of glucocorticoid induced-apoptosis of osteoblasts and osteocytes by protecting against endoplasmic reticulum (ER) stress in vitro and in vivo in female mice. Bone. 2015;73:60–68. doi: 10.1016/j.bone.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thiele S, Ziegler N, Tsourdi E, De Bosscher K, Tuckermann JP, Hofbauer LC, Rauner M. Selective glucocorticoid receptor modulation maintains bone mineral density in mice. J Bone Miner Res. 2012;27:2242–2250. doi: 10.1002/jbmr.1688. [DOI] [PubMed] [Google Scholar]
- 46.de Vries F, Pouwels S, Lammers JW, Leufkens HG, Bracke M, Cooper C, van Staa TP. Use of inhaled and oral glucocorticoids, severity of inflammatory disease and risk of hip/femur fracture: a population-based case-control study. J Intern Med. 2007;261:170–177. doi: 10.1111/j.1365-2796.2006.01754.x. [DOI] [PubMed] [Google Scholar]
- 47.Giusti A, Hamdy NAT, Papapoulos SE. Atypical fractures of the femur and bisphosphonate therapy A systematic review of case/case series studies. Bone. 2010;47:169–180. doi: 10.1016/j.bone.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 48.Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 49.Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takuma A, Kaneda T, Sato T, Ninomiya S, Kumegawa M, Hakeda Y. Dexamethasone enhances osteoclast formation synergistically with transforming growth factor-beta by stimulating the priming of osteoclast progenitors for differentiation into osteoclasts. J Biol Chem. 2003;278:44667–44674. doi: 10.1074/jbc.M300213200. [DOI] [PubMed] [Google Scholar]
- 51.Pearsall RS, Canalis E, Cornwall-Brady M, Underwood KW, Haigis B, Ucran J, Kumar R, Pobre E, Grinberg A, Werner ED, Glatt V, Stadmeyer L, Smith D, Seehra J, Bouxsein ML. A soluble activin type IIA receptor induces bone formation and improves skeletal integrity. Proc Natl Acad Sci U S A. 2008;105:7082–7087. doi: 10.1073/pnas.0711263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006;116:1202–1209. doi: 10.1172/JCI28551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rossini M, Gatti D, Adami S. Involvement of WNT/beta-catenin signaling in the treatment of osteoporosis. Calcif Tissue Int. 2013;93:121–132. doi: 10.1007/s00223-013-9749-z. [DOI] [PubMed] [Google Scholar]
- 54.Lui PP, Lee YW, Wong YM, Zhang X, Dai K, Rolf CG. Expression of Wnt pathway mediators in metaplasic tissue in animal model and clinical samples of tendinopathy. Rheumatology (Oxford) 2013;52:1609–1618. doi: 10.1093/rheumatology/ket214. [DOI] [PubMed] [Google Scholar]
- 55.Mao B, Wu W, Li Y, Hoppe D, Stannek P, Glinka A, Niehrs C. LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature. 2001;411:321–325. doi: 10.1038/35077108. [DOI] [PubMed] [Google Scholar]
- 56.Brott BK, Sokol SY. Regulation of Wnt/LRP signaling by distinct domains of Dickkopf proteins. Mol Cell Biol. 2002;22:6100–6110. doi: 10.1128/MCB.22.17.6100-6110.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fujita K, Janz S. Attenuation of WNT signaling by DKK-1 and -2 regulates BMP2-induced osteoblast differentiation and expression of OPG, RANKL and M-CSF. Mol Cancer. 2007;6:71. doi: 10.1186/1476-4598-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gifre L, Ruiz-Gaspa S, Monegal A, Nomdedeu B, Filella X, Guanabens N, Peris P. Effect of glucocorticoid treatment on Wnt signalling antagonists (sclerostin and Dkk-1) and their relationship with bone turnover. Bone. 2013;57:272–276. doi: 10.1016/j.bone.2013.08.016. [DOI] [PubMed] [Google Scholar]
- 59.Belaya ZE, Grebennikova TA, Melnichenko GA, Nikitin AG, Solodovnikov AG, Brovkina OI, Grigoriev AU, Rozhinskaya LY, Dedov II. Effects of endogenous hypercortisolism on bone mRNA and microRNA expression in humans. Osteoporos Int. 2018;29:211–221. doi: 10.1007/s00198-017-4241-7. [DOI] [PubMed] [Google Scholar]
- 60.Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19:179–192. doi: 10.1038/nm.3074. [DOI] [PubMed] [Google Scholar]