

Abstract
Chronic myelomonocytic leukemia (CMML) can be complicated by autoimmune features associated with rheumatologic disorders, which have been commonly reported by more researches currently. The intrinsic correlation between CMML and autoimmune diseases can create significant pitfalls in differential diagnosis. CMML occasionally presents with clinical and histopathological manifestations that are similar to those of Immunoglobulin G4-related disease (IgG4-RD), a newly recognized systemic autoimmune disorder. Both CMML and IgG4-RD can have significant overlaps due to the common nature of these systemic disorders, especially when atypical clinical phenotypes are present. It is significant for physicians to accurately distinguish CMML and IgG4-RD because the therapy modalities could differ extremely between the two entities. Here we present a unique case of a 70-year-old female who had a condition that mimicked the onset of IgG4-RD not only in terms of clinical manifestations but also in serology and histopathology analyses. Following a series of rigorous examinations, this patient was ultimately diagnosed as having CMML. Herein, we discuss the aspects of IgG4-RD’s differential diagnosis and the need for careful comparison of clinical and laboratory features as well as consideration of the pathogenesis of both IgG4-RD and CMML. We also stress a concept that the presence of autoimmune conditions cannot be the sole basis to exclude diagnosis of CMML, as these disorders can appear concomitantly.
Keywords: Chronic myelomonocytic leukemia (CCML), IgG4-related disease (IgG4-RD), malignancy, autoimmune diseases (AIDs)
Introduction
Chronic myelomonocytic leukemia (CMML) is a clonal hematopoietic stem cell neoplasm characterized by overlapping features of myelodysplastic syndrome (MDS) and myeloproliferative neoplasm (MPN). CMML has an inherent tendency to transform into acute myeloid leukemia with a relatively poor prognosis [1,2]. Patients with CMML can present with a variety of manifestations including constitutional symptoms, splenomegaly, bleeding, infection and lesions even in the presence of multifarious autoimmune and inflammatory processes [3]. CMML was previously considered to be a subtype of MDS that comprises a heterogeneous group of clonal stem cell neoplasms in the past, yet it has already been differed from MDS due to unique genetic and prognostic traits nowadays [4]. Despite their differences, the two conditions do share some similarities and can coexist with autoimmune disorders. To our knowledge, CMML and MDS are frequently concomitant with some autoimmune diseases (AIDs) such as polyarthritis, psoriasis and vasculitis. CMML and MDS have an AIDs complication rate of 7% and 20%, respectively [5,6]. Features of MDS/CMML-associated AIDs are typically nonspecific and varied, which make their recognition difficult.
IgG4-RD is a rarely benign immune-mediated disease that often mimics many malignant, inflammatory and infectious disorders. Multiple organs are typically involved in IgG4-RD, although until recently the specific etiology of the disease was unknown. It was recently recognized as a unified autoimmune disease and histopathological examination remains the gold standard for diagnosis [7]. Considering the connection between CMML and AIDs, some malignant and autoimmune processes such as multi-organ lesions and arthralgia may be easily misinterpreted as manifestations of IgG4-RD. Thus, early differential diagnosis of IgG4-RD or CMML can be challenging for clinicians, particularly because the two conditions have overlapping histopathological and serological features as well as clinical manifestations.
Here, we describe an unusual case in which the patient had symptoms that mimicked those associated with IgG4-RD onset. The patient exhibited elevated serum IgG4 levels and histopathological analysis showed infiltration of IgG4-positive lymphoplasmacytic cells. The patient was eventually diagnosed with CMML based on examination of bone marrow, detection of mutations in relevant genes and flow cytometry analyses.
Case report
A 70 year-old Chinese woman who presented with a 10-year history of arthralgia and splenomegaly was admitted to hospital for “acute left abdominal pain for two days”. Further investigation of the patient’s medical history showed that she had a history of skin allergies without other underlying disease. Laboratory examination revealed an increased monocyte proportion of 26.2% and mild anemia. Abdominal computed tomography (CT) indicated an enlarged spleen. Other laboratory work-ups including coagulation tests, liver enzymes and renal function were within normal ranges. However, the patient’s abdominal pain did not resolve and she subsequently developed a continuous decrease in hemoglobin in spite of management with proper treatments. She was initially diagnosed with perisplenitis with suspicion of splenic tumors and underwent “laparoscopic splenectomy and pericardial devascularization”. Unfortunately, the patient continued to complain of hypogastric pain after the operation. The abdominal CT indicated spontaneous hemorrhage in the left kidney and a left nephrectomy was performed after embolization of the renal artery had a poor hemostatic effect. Biopsy specimens of the spleen and kidney were sent to another hospital for pathological examination, which revealed infiltrate comprised of a large number of histocytes (highlighted by CD68) (Figure 1A and 1B) as well as proliferation of plasma cells and tumor-like cells without pathological mitoses in the spleen (Figure 2A and 2B). Focal chronic inflammation, hemorrhage and necrosis were seen in the kidney samples. Immunochemistry staining of spleen specimens showed lymphoplasmacytic infiltration rich in IgG4+ plasma cells with >10 IgG4+ cells per high power filed (Figure 3A and 3B) and a IgG4:IgG ration of 34.5% (Figure 3C and 3D); no diagnostic evidence of malignancy was identified. Meanwhile, the patient had elevated serum IgG4 immunoglobulin levels (2.390 g/L; cutoff value for IgG4-RD >1.35 g/L). Based on these histopathology and serology outcomes, an initial diagnosis of IgG4-RD was established.
Figure 1.
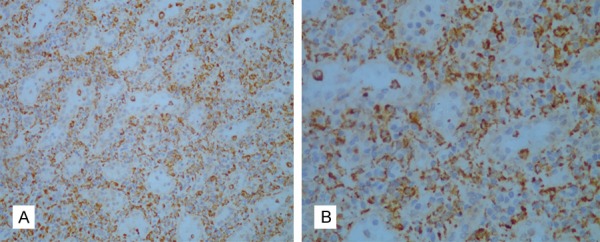
Immunostaining for CD68 in a spleen biopsy sample. Photomicrograph of the spleen showed the presence of many CD68+ cells in a spleen biopsy sample (20× and 40× magnification in A and B, respectively).
Figure 2.
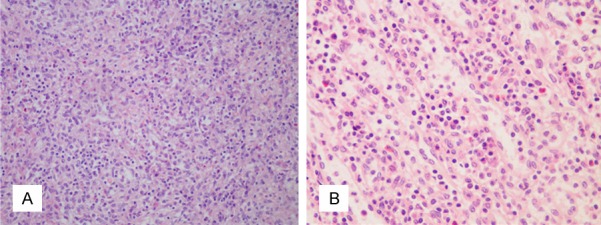
Histopathology of spleen tissue stained with H&E. Photomicrograph of the spleen showed tumor-like hyperplasia of splenic histocytes without evident pathological mitoses. The spleen tissue is extensively infiltrated by inflammatory cells, mainly plasma cells and lymphocytes (20× and 40× magnification in A and B, respectively).
Figure 3.
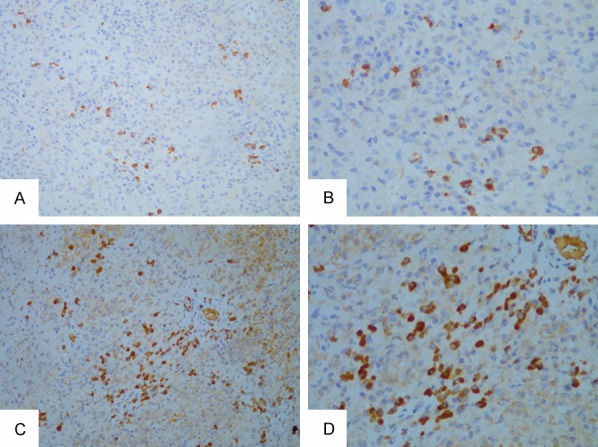
Immunostaining for IgG4 and IgG of spleen tissue. Staining for IgG4 with diffuse infiltrates of IgG4+ plasma cells and >10 positive cells per high powered filed (20× and 40× magnification in A and B, respectively). Immunostaining for IgG shows a dense infiltration of IgG+ cells in the spleen biopsy sample. In the IgG-stained section the ratio of IgG4 to IgG-positive plasma cells is up to 34.5% (20× and 40× magnification in C and D, respectively).
A positron-emission CT using 13mCi F18 fluorodeoxyglucose showed multiple lymph nodes spread over the body including the bilateral cervix and, mediastinum as well as, hilar, distal esophageal, para-abdominal aortic, mesenteric and inguinal regions with some slightly hypermetabolic lymph nodes, which were considered to be suggestive of an AID. Furthermore, diffused hypermetabolism was observed in bone marrow throughout the body that was possibly caused by reactive hyperplasia in hematopoietic tissue. No malignancies were noted in the CT scan. Together these assessments raised concern about possible benign AIDs. Based on the patient’s medical history of frequent skin allergies and arthralgia that are associated with autoimmune features, a diagnosis of IgG4-RD seemed to fit well as a unified diagnosis for the tumefactive lesions seen in the spleen and kidney, as well as the positive serology and histopathology findings. Both the clinical manifestations and histopathology findings were judged to be consistent with IgG4-RD. Thus, the patient was diagnosed with IgG4-RD and received glucocorticoid therapy.
The patient experienced a recurrent hemorrhage of the right kidney during the glucocorticoid treatment and her condition appeared to worsen with the presence of progressive renal function deterioration. The creatinine level rose from 72 μmol/L at baseline to 348.8 μmol/L (reference value: 49-90 μmol/L) over several weeks. Consequently, continuous renal replacement therapy (CRRT) was initiated. The absence of autoimmune features including favorable response to glucocorticoids and history of sustained monocytosis and splenomegaly, both of which were incompatible with IgG4-RD but highly consistent with CMML, led the patient’s clinicians to reconsider the previous diagnosis. A subsequent bone marrow (BM) biopsy showed granulocytic/monocytic hyperplasia with the number of segmented granulocytes exceeding that for stab granulocytes and a decreasing proportion of erythrocytic lineages that lacked dysplastic blasts (Figure 4). Peripheral blood smears exhibited a significant increase in monocytes of up to 29% without promonocytes, a normal proportion of neutrophils that had increased and thickened granules, and irregular erythrocytes (Figure 5). Immunostaining for neutrophil alkaline phosphatase (N-ALP), Periodic Acid-Schiff (PAS) and iron in the bone marrow demonstrates that staining for N-ALP was positive. PAS and iron staining are within normal limits (Figure 6A). Immunostaining for CD41 in the bone marrow showed normal megakaryocytes and the absence of micromegakaryocytes (Figure 6B). Flow cytometry analysis of the monocyte subsets distribution displayed: i) an increased proportion of CD14+ monocytes of up to 25.32% (Figure 7A) and ii) a proportion of classical monocytes (CD14+CD16-), referred to MO1, of up to 90.6% (Figure 7B), which was close to the cutoff value of 94% that was validated in prior reports to distinguish CMML from reactive monocytes [8-10]. Continuous monitoring of routine blood tests yielded a persistently absolute monocyte count of ≥1×109/L with >10% of leukocytes, mild thrombocytopenia and moderate anemia, whereas the leukocyte and neutrophil counts were within normal ranges. Molecular genetics further revealed that the mutation frequency of CBL is up to 97.1% in this patient, followed in decreasing order by SF3A1, ARID1A and TPMT at 53.2%, 47.1% and 6.6%, respectively (Table 1). Taking the aforementioned results together, a diagnosis of CMML-0 was made according to the 2016 World Health Organization criteria [1]. Upon diagnosis, a front line chemotherapy regimens for CMML was immediately prescribed for her. This patient experienced several fevers and infections but with a good condition in protopathy that had achieved clinical remission and her CMML-associated constitutional symptoms improved significantly over a 7-month follow up period.
Figure 4.
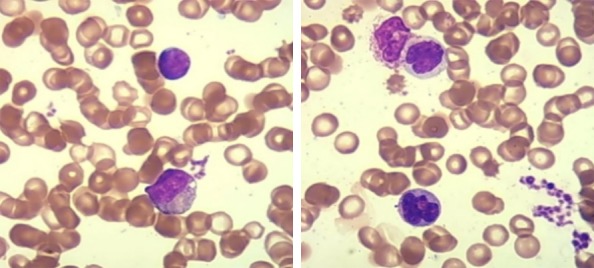
Morphological characteristics in bone marrow smears. Bone marrow aspirates show an increased proportion of granulocytes and monocytes without abnormalities in morphology as well as evidence of immature monocytes, decreased proportion of mature erythrocytes with enlargement of a light-stained central area in erythrocytes and a decreased proportion of lymphocytes (Wright-Giemsa, 1,000× original magnification).
Figure 5.
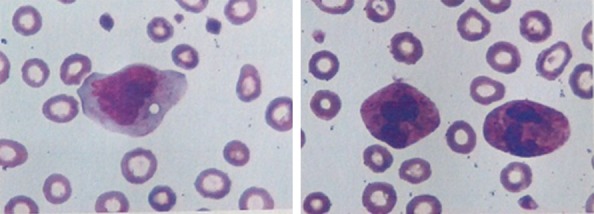
Morphological characteristics in peripheral blood smear. Peripheral blood smear showing significantly increased frequency of monocytes (up to 29%), which were mainly mature. A normal proportion of neutrophil having increased and thickened granules in the cytoplasm and irregular erythroids with an enlarged central light-stained area were found (Wright-Giemsa, 400× original magnification).
Figure 6.
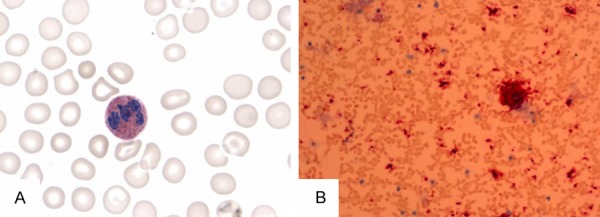
Immunostaining for neutrophil alkaline phosphatase (N-ALP), Periodic Acid-Schiff (PAS) and iron in the bone marrow. Staining for N-ALP demonstrates that the positive ratio and positive index for N-ALP is 96.00% and 236.00, respectively (reference range: 66.28±22.75% and 103.28±69.63, respectively). PAS and iron staining are within normal limits (200× magnification in A). Immunostaining for CD41 in the bone marrow showed 15 normal megakaryocytes in the whole film and the absence of micromegakaryocytes (20× magnification in B).
Figure 7.
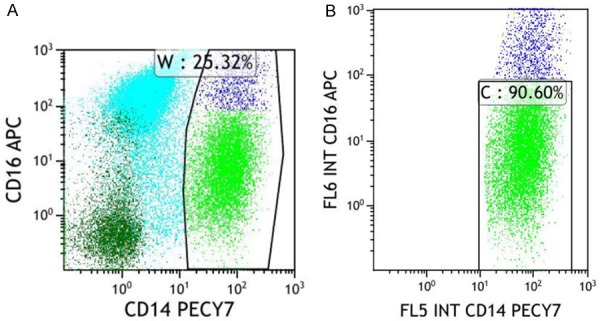
Sorting PBMCs to obtain CD14+ and CD14+CD16- monocyte subsets of this patient. Flow cytometry panel of CD16 and CD14 expression on gated monocytes revealed that the proportion of CD14+ monocytes is 25.32% in (A) and the ratio of CD14+CD16- monocytes to CD14+ monocytes is 90.60% in (B).
Table 1.
Gene mutations associated with hematological disorders detected in this patient
| Mutated gene | Transcript ID | Mutation location | Nucleotide change | Amino acid change | dbSNP | Mutation frequency (%)† |
|---|---|---|---|---|---|---|
| CBL* | NM-005188 | Exon 8 | c.1151G>A | p.C384Y | -- | 97.1 |
| TPMT# | NM-000367 | Exon 9 | c.659T>C | p.V200A | -- | 6.6 |
| SF3A1# | NM-005877 | Exon 4 | c.423C>G | p.I141M | -- | 53.2 |
| ARID1A# | NM-006015 | Exon 16 | c.3999-4001dupGCA | p.Q1334dupQ | rs570962335 | 47.1 |
The mutations location is identified as being associated with CMML in specific studies, or is definitively to mediated functional changes of disease-related proteins.
The mutation location has not yet been associated with CMML in published reports, but is supported by at least two databases.
dbSNP: database of single Nucleotide Polymorphisms;
Reported frequency of the mutation in this patient.
Discussion
This case highlights that both CMML and IgG4-RD are systemic protean diseases that can affect almost any organ or system in the body. Although CMML is a malignant and IgG4-RD is a benign autoimmune disorder, the two diseases share certain similarities in both clinical manifestations and pathophysiology. CMML is, a clonal myeloid neoplasm that could share a wide spectrum infiltration of multiple organs, especially the spleen. Multiple studies have reported that CMML patients occasionally present with concomitant or pre-existing autoimmune disorders or systemic inflammatory syndromes (SIS), such as arthritis, psoriasis, systemic vasculitis, chronic inflammatory demyelinating polyneuropathy and other rare AIDs [11,12]. Therefore, the presence of AIDs should not be the exclusion criteria for CMML, since these conditions can present concomitantly, although, the specific mechanisms involved in interactions between CMML and AIDs remain unclear. Notably, flow cytometry analysis of this patient revealed an aberrant elevation in the infraction of CD14+ monocytes (25.32%) in peripheral blood. Interestingly, Tomohiro et al. reported that activation of NOD-2 in CD14+ monocytes induced IgG4 production by B cells in a BAFF-dependent and T cell-independent manner [13]. Based on these findings from earlier studies, we presumed that CMML patients along with an increased proportion of CD14+ monocytes could be accompanied by elevated serum levels of IgG4 immunoglobulin, which was the case for this patient. The elevated serum IgG4 levels in this patient may be explained by this reason. Such a potential similarity between CMML and IgG4-RD contributes to great challenges and dilemma for reaching to the accurate diagnosis of CMML in view of results of serological analysis.
IgG4-RD is newly recognized as a systemic autoimmune disorder that often mimics malignant, infectious and inflammatory disorders. Patients with IgG4-RD are prone to form tumefactive lesions that involve multiple organs in common sites such as the parotid gland, salivary gland and pancreas, but the lung, kidney and retroperitoneal tissue can also be affected [7,14]. Representative features of IgG4-RD histopathology comprise of: dense lymphoplasmacytic infiltration (IgG4+ plasma cells) with or without elevated serum IgG4 levels, fibrosis (often presenting in a storiform pattern), and obliterative phlebitis [15]. Although, CMML and IgG4-RD can be discriminated upon careful and extensive examinations including histopathology, gene detection and flow cytometry, the pathology of these two disorders share some similarities. The inflammatory and autoimmune profile of CMML patients may involve a high proportion of IgG4+ cells among plasma cells owing to the abovementioned correlation between CMML and AIDs. Even though the combination of serological results and histopathological features provides strongly supportive evidence for a diagnosis of IgG4-RD, careful correlation with clinical presentations is often required to contribute to a definitive diagnosis.
Hence, when arriving at a conclusion based on interpretation of pathology results alone, terms such as ‘definitive’ should be avoided in favor of terms such as ‘histologically suggestive of IgG4-RD’. Given the underlying relationship between CMML and IgG4-RD, distinguishing the histopathological features of the two diseases is challenging. A widely recognized clinical hallmark of IgG4-RD is its dramatic response to a glucocorticoid regimen, but this patient showed no response to steroids and her clinical condition instead deteriorated. The previous diagnosis IgG4-RD was overturned upon failure of glucocorticoid therapy. This outcome emphasizes the importance of repeated bone marrow biopsy and flow cytometry for patients exhibiting atypical monocytosis in order to obtain an accurate diagnosis as early as possible.
CMML and IgG4-RD are two completely different entities not only in pathogenesis but also in terms of treatment and prognosis. Making an early differential diagnosis for the two disorders is critical for selecting appropriate therapeutic approaches to ensure optimum prognosis and avoid potentially correlated complications. Treatment of CMML with regular chemotherapy and allogeneic hematopoietic stem cell transplant (HSCT) can achieve clinical remission [16]. Meanwhile, for IgG4-RD patients glucocorticoids are commonly recognized as the cornerstone of treatment with or without use of steroid-sparing immunosuppressive agents and biological agents (e.g., B cell depletion with rituximab) depending on disease severity and corticosteroid response [7,17]. In the case presented here, there is an intense requirement for physicians to review the histopathological examination results repeatedly and carried out a series of hematological examinations including bone marrow biopsy and peripheral blood smears for dynamic monitoring. It is of note that IgG4-RD should be judged preferentially as an exclusive diagnosis due to its rarity, and a definitive diagnosis of IgG4-RD can be made by excluding a variety of mimickers, such as malignancies, infections, and other immune-mediated disorders. When clinicians encounter a possible diagnosis of IgG4-RD that is based merely on an appropriate interpretation of histopathology results, they should take into account the possibility that a hematological neoplasm may be responsible for sustained monocytosis despite persistently normal WBC levels.
Conclusion
CMML can occasionally show analogous properties with IgG4-RD in clinical, serological and even histopathological examinations. As such, to distinguish these two diseases precisely, physicians must carry out comprehensive analyses of the pathological results and consider the various clinical scenarios. Furthermore, the findings in our study suggested that CMML can concomitantly present with certain autoimmune features, which is consistent with previous reports. The effects of intrinsic immune pathogenesis on the correlation between CMML and AIDs will require additional examination to determine an applicable diagnostic strategy to accurately distinguish these two disorders.
Acknowledgements
We thank the patient and her family for allowing us to describe her case in detail. This work was supported by National Natural Science Foundation of China (No. 81771754 to L.L. Dong), Tongji Hospital Clinical Research Flagship Program (No. 2019CR206 to L.L. Dong) and Science Foundation of Tongji Hospital (No. 2018A09 to Y.X. Chen).
Disclosure of conflict of interest
None.
References
- 1.Arber DA, Orazi A, Hasserjian R. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;128:462–463. doi: 10.1182/blood-2016-06-721662. [DOI] [PubMed] [Google Scholar]
- 2.Loghavi S, Khoury JD. Recent updates on chronic myelomonocytic leukemia. Curr Hematol Malig Rep. 2018;13:446–454. doi: 10.1007/s11899-018-0475-5. [DOI] [PubMed] [Google Scholar]
- 3.McCullough KB, Patnaik MM. Chronic myelomonocytic leukemia: a genetic and clinical update. Curr Hematol Malig Rep. 2015;10:292–302. doi: 10.1007/s11899-015-0271-4. [DOI] [PubMed] [Google Scholar]
- 4.Gangat N, Patnaik MM, Tefferi A. Myelodysplastic syndromes: contemporary review and how we treat. Am J Hematol. 2016;91:76–89. doi: 10.1002/ajh.24253. [DOI] [PubMed] [Google Scholar]
- 5.Braun T, Fenaux P. Myelodysplastic syndromes (MDS) and autoimmune disorders (AD): cause or consequence? Best Pract Res Clin Haematol. 2013;26:327–336. doi: 10.1016/j.beha.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 6.Al Ustwani O, Ford LA, Sait SJ, Block AM, Barcos M, Vigil CE, Griffiths EA, Thompson JE, Wang ES, Ambrus J Jr, Wetzler M. Myelodysplastic syndromes and autoimmune diseases--case series and review of literature. Leuk Res. 2013;37:894–899. doi: 10.1016/j.leukres.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet. 2015;385:1460–1471. doi: 10.1016/S0140-6736(14)60720-0. [DOI] [PubMed] [Google Scholar]
- 8.Selimoglu-Buet D, Wagner-Ballon O, Saada V, Bardet V, Itzykson R, Bencheikh L, Morabito M, Met E, Debord C, Benayoun E, Nloga AM, Fenaux P, Braun T, Willekens C, Quesnel B, Ades L, Fontenay M, Rameau P, Droin N, Koscielny S, Solary E. Characteristic repartition of monocyte subsets as a diagnostic signature of chronic myelomonocytic leukemia. Blood. 2015;125:3618–3626. doi: 10.1182/blood-2015-01-620781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Talati C, Zhang L, Shaheen G, Kuykendall A, Ball M, Zhang Q, Lancet JE, Zuckerman KS, List AF, Komrokji R, Moscinski L, Padron E. Monocyte subset analysis accurately distinguishes CMML from MDS and is associated with a favorable MDS prognosis. Blood. 2017;129:1881–1883. doi: 10.1182/blood-2016-12-753210. [DOI] [PubMed] [Google Scholar]
- 10.Patnaik MM, Timm MM, Vallapureddy R, Lasho TL, Ketterling RP, Gangat N, Shi M, Tefferi A, Solary E, Reichard KK, Jevremovic D. Flow cytometry based monocyte subset analysis accurately distinguishes chronic myelomonocytic leukemia from myeloproliferative neoplasms with associated monocytosis. Blood Cancer J. 2017;7:e584. doi: 10.1038/bcj.2017.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zahid MF, Barraco D, Lasho TL, Finke C, Ketterling RP, Gangat N, Hanson CA, Tefferi A, Patnaik MM. Spectrum of autoimmune diseases and systemic inflammatory syndromes in patients with chronic myelomonocytic leukemia. Leuk Lymphoma. 2017;58:1488–1493. doi: 10.1080/10428194.2016.1243681. [DOI] [PubMed] [Google Scholar]
- 12.Peker D, Padron E, Bennett JM, Zhang X, Horna P, Epling-Burnette PK, Lancet JE, Pinilla-Ibarz J, Moscinski L, List AF, Komrokji RS, Zhang L. A close association of autoimmune-mediated processes and autoimmune disorders with chronic myelomonocytic leukemia: observation from a single institution. Acta Haematol. 2015;133:249–256. doi: 10.1159/000365877. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe T, Yamashita K, Fujikawa S, Sakurai T, Kudo M, Shiokawa M, Kodama Y, Uchida K, Okazaki K, Chiba T. Involvement of activation of toll-like receptors and nucleotide-binding oligomerization domain-like receptors in enhanced IgG4 responses in autoimmune pancreatitis. Arthritis Rheum. 2012;64:914–924. doi: 10.1002/art.33386. [DOI] [PubMed] [Google Scholar]
- 14.Wolfson AR, Hamilos DL. Recent advances in understanding and managing IgG4-related disease. F1000Research. 2017:6. doi: 10.12688/f1000research.9399.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, Kloeppel G, Heathcote JG, Khosroshahi A, Ferry JA, Aalberse RC, Bloch DB, Brugge WR, Bateman AC, Carruthers MN, Chari ST, Cheuk W, Cornell LD, Fernandez-Del Castillo C, Forcione DG, Hamilos DL, Kamisawa T, Kasashima S, Kawa S, Kawano M, Lauwers GY, Masaki Y, Nakanuma Y, Notohara K, Okazaki K, Ryu JK, Saeki T, Sahani DV, Smyrk TC, Stone JR, Takahira M, Webster GJ, Yamamoto M, Zamboni G, Umehara H, Stone JH. Consensus statement on the pathology of IgG4-related disease. Modern Pathol. 2012;25:1181–1192. doi: 10.1038/modpathol.2012.72. [DOI] [PubMed] [Google Scholar]
- 16.Hunter AM, Zhang L, Padron E. Current management and recent advances in the treatment of chronic myelomonocytic leukemia. Curr Treat Options Oncol. 2018;19:67. doi: 10.1007/s11864-018-0581-6. [DOI] [PubMed] [Google Scholar]
- 17.Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, Chari ST, Della-Torre E, Frulloni L, Goto H, Hart PA, Kamisawa T, Kawa S, Kawano M, Kim MH, Kodama Y, Kubota K, Lerch MM, Löhr M, Masaki Y, Matsui S, Mimori T, Nakamura S, Nakazawa T, Ohara H, Okazaki K, Ryu JH, Saeki T, Schleinitz N, Shimatsu A, Shimosegawa T, Takahashi H, Takahira M, Tanaka A, Topazian M, Umehara H, Webster GJ, Witzig TE, Yamamoto M, Zhang W, Chiba T, Stone JH Second International Symposium on IgG4-Related Disease. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheumatol. 2015;67:1688–99. doi: 10.1002/art.39132. [DOI] [PubMed] [Google Scholar]