

Abstract
Lung cancer is one of the most common human cancers and is the leading cause of cancer-related mortality. Previous studies have suggested that IL-22 might promote the survival of human lung cancer cells. However, the source of IL-22 and the regulatory mechanism of lung cancer cell proliferation remain unclear. In this study, we found that the expression of IL-22 was upregulated in non-small-cell lung cancer (NSCLC) tumor specimens, as revealed by RT-qPCR analysis. Furthermore, IL-22 was profoundly elevated in cell cultures of primary cancer-associated fibroblasts (CAFs) compared to the levels in cell cultures of normal fibroblasts. Moreover, treatment with the supernatant of CAF cell cultures significantly increased the proliferation, migration and invasion of A549 and H1650 cells but reduced apoptosis via the activation of PI3K-Akt-mTOR signaling, and the application of an anti-IL-22 antibody can partially block the effects induced by the CAF cell culture supernatant. Finally, we also identified a panel of critical genes with differential expression between A549 cells treated with and without IL-22. In summary, our results demonstrate a novel regulatory function of IL-22 secreted by CAFs in NSCLC and provide a potential therapeutic target for treating lung cancer.
Keywords: Interleukin-22, PI3K-Akt-mTOR, lung cancer, proliferation
Introduction
Lung cancer is one of the most common human cancers and is the leading cause of cancer-related mortality [1]. Among them, 85% of lung cancer patients suffered from non-small-cell lung cancer (NSCLC), which is often diagnosed at the advanced or metastatic stage [2]. Despite recent advances in targeted therapies, the 5-year overall survival rates of lung cancer patients still remain below 15% [3,4]. Although several studies have identified several immune-related cytokine biomarkers, including IL-1, IL-6, IL-13 and IL-12, in NSCLC, these biomarkers had relatively low sensitivity and specificity, especially for early diagnosis, prognosis evaluation, and prediction of recurrence. Therefore, novel molecular markers are urgently needed for clinical use.
Interleukin-22 (IL-22) is a recently identified IL-10 superfamily cytokine mainly activated by lymphocytes in chronically inflamed tissues, and its receptor consists of two chains: IL-22 receptor 1 (IL-22R1) and IL-10 receptor 2 (IL-10R2) [5]. Interestingly, IL-10R2 is expressed ubiquitously in various organs [6], whereas the expression of IL-22R1 is restricted to epithelial cells [7]. Therefore, IL-22 may have critical roles as a biological mediator from the immune system to epithelial cells. IL-22 likely exerts multiple functions under inflammatory conditions due to its regulatory roles in various genes involved in chemotaxis, proliferation, innate immunity and inflammation [8,9]. However, the role of IL-22 in carcinogenesis, especially NSCLC, remains obscure.
Cancerous tissue is a complex mix of tumor cells, stromal cells and extracellular matrix (ECM), all of which make up a disordered and aggressive niche in comparison with organized and homeostatic normal tissue. It has been well established that the tumor microenvironment plays an indispensable role in cancer development. Cancer-associated fibroblasts (CAFs) are involved in cancer initiation, growth, invasion, metastasis, therapeutic resistance and microenvironmental events, including angiogenesis/lymphangiogenesis, ECM remodeling, cancer-associated inflammation and metabolism reprogramming. In addition, CAFs are the predominant cellular components and play a central role in the tumor microenvironment [10]. At the molecular level, there is a sophisticated network underlying the interactions among CAFs, cancer cells and cytokines, accordingly targeting CAFs. This provides a novel strategy in cancer therapy.
In this study, we first attempted to highlight the critical role of IL-22 secreted by cancer-associated fibroblasts in lung cancer tumors. First, we validated the expression of IL-22 and IL-22 receptor (IL-22R) in lung cancer tissues collected from surgery, and the correlation of their expression and the advanced clinical pathology of NSCLC was analyzed. Furthermore, we quantified the IL-22 level in primary CAF cells as well as the NSCLC cell lines A549 and H1650. Moreover, we explored the potential role of IL-22 in regulating the proliferation, apoptosis, migration and invasion of lung cancer cells. Through transcriptome analysis, the activation of the critical signaling pathway PI3K-Akt-mTOR was examined. Finally, we identified a panel of critical genes with differential expression levels between CAFs alone and CAFs treated with IL-22. These results suggest a novel regulatory function of IL-22 secreted by CAFs in NSCLC and provide a potential therapeutic target for treating lung cancer.
Methods
Patients and clinical tissue samples
Forty pairs of lung cancer tissue samples and adjacent tissue samples were obtained from specimens surgically resected from NSCLC patients at Changzhou Tumor Hospital. The tissue specimens were immediately snap-frozen in liquid nitrogen at the time of surgery and stored at -80°C until processed. All the collected cases were diagnosed as NSCLC pathologically without any preoperative radiotherapy and/or chemotherapy. All patients provided informed consent. The present study was approved by the institutional review board of the hospital. The following information was collected from patients: age, sex, tumor size, tumor location, tumor invasion, lymph node metastasis, and tumor stage. The clinical information of the patients is shown in Table 1.
Table 1.
Clinical information of the patients
| Parameters | NO. | IL-22 | P value | IL-22R | p value |
|---|---|---|---|---|---|
| Age (years) | 0.824 | 0.742 | |||
| <40 | 16 | 1.461 ± 0.73 | 1.627 ± 0.74 | ||
| ≥40 | 24 | 1.569 ± 1.07 | 2.123 ± 0.88 | ||
| Tumor size (cm) | 0.015 | 0.048 | |||
| ≥5 | 14 | 3.615 ± 0.89 | 2.842 ± 1.01 | ||
| <5 | 26 | 1.899 ± 0.82 | 1.750 ± 0.97 | ||
| Histological grade | 0.738 | 0.685 | |||
| Differentiation | 19 | 2.311 ± 0.97 | 0.79 | ||
| Poor differentiation | 21 | 2.124 ± 1.12 | 1.868 ± 0.85 | ||
| Invasion depth | 0.139 | 0.417 | |||
| Tis-T2 | 25 | 1.785 ± 0.88 | 1.679 ± 0.82 | ||
| T3-T4 | 15 | 2.123 ± 0.89 | 1.821 ± 1.02 | ||
| Lymph node metastasis | 0.005** | 0.004** | |||
| N0 | 6 | 1.168 ± 0.65 | 1.612 ± 0.65 | ||
| N1-N3 | 34 | 3.080 ± 1.06 | 2.780 ± 0.98 | ||
| Distant metastasis | 0.003 | 0.038 | |||
| M0 | 12 | 1.768 ± 0.38 | 1.263 ± 0.60 | ||
| M1 | 28 | 2.866 ± 0.88 | 2.178 ± 1.21 |
P<0.01.
Immunohistochemistry
The tissue samples were perfused with PBS, collected and fixed with formalin. Then, the tissues were sliced into 3 μm sections and stained with hematoxylin and eosin (H&E). Later, the tissues were incubated with primary antibodies against IL-22, Akt, p-AKT, PI3K, p-PI3K mTOR and p-mTOR purchased from Abcam, USA. The immunoreactivity in sections was detected using a horseradish peroxidase-3,3’-diaminobenzidine kit (200x magnification, Invitrogen, USA). Five regions from three sections obtained were observed.
Detection of IL-22 in the CAFs
CAFs were isolated from the tumor tissues of NSCLC patients as previously described [10]. Elisa-Kit was purchased from Abcam (USA) to detect the expression level of IL-22 in the cell culture supernatant of CAFs. The operating procedure was followed by the Kit protocol.
Cell culture and treatment
Human NSCLC cell lines A549 and H1650 were cultivated in RPMI-1640 medium (Invitrogen, USA) supplemented with 10% FBS (fetal bovine serum, Invitrogen, USA) at 37°C in a humidified incubator (5% CO2). Cells were either treated with the supernatant from the CAFs or the supernatant from CAF+anti-IL-22 antibody (IL-22A) for 72 h.
Reverse transcription-PCR and quantitative real-time PCR
Total RNA was first extracted from cells or clinical tissue samples using TRIzol Reagent (Invitrogen) based on the manufacturer’s protocol. Total cDNA was reverse transcribed from isolated RNA using PrimeScript RT Master Mix (Takara, Dalian, China). Quantitative real-time PCR was performed to detect the expression levels of IL-22 and IL-22R using SYBR premix Ex Taq (Takara, Dalian, China) on an ABI Biosystems thermal cycler. The relative expression levels of target genes were normalized to those of GAPDH. The mRNA level was calculated with the 2-ΔΔCt method. The reactions were performed at 95°C for 30 seconds, followed by 40 cycles of 95°C for 5 seconds and 60°C for 30 seconds. The sequences of the primers were as follows: IL-22 forward, 5’-GCTAAGGAGGCTAGCTTG-3’ and reverse, 5’-CAGCAAATCCAGTTCTCC-3’; IL-22R forward, 5’-CTCCACAGCGGCATAGCCT-3’ and reverse, 5’-ACATGCAGCTTCCAGCTGG-3’; GAPDH forward, 5’-GGCTGCTTTTAACTCTGGTA-3’ and reverse, 5’-ATGCCAGTGAGCTTCCCGT-3’.
Cell proliferation analysis
After 72 h of treatment, the proliferation of A549 and H1650 cells was measured by a cell counting kit-8 (CCK-8) with the BrdU labeling method according to the manufacturer’s instructions. Briefly, the cells were washed with PBS buffer (pH 7.4), harvested by trypsinization and reseeded into a 96-well plate. Then, the plate was preincubated in a humidified incubator (37°C, 5% CO2). Ten microliters of the CCK-8 solution was added to each well of the plate, and the plate was incubated for 1 to 4 h. The absorbance was measured at 450 using a microplate reader.
Western blot analysis
Total proteins were isolated from A549 and H1650 cells using a protease inhibitor cocktail. Protein concentrations were determined by a BCA Protein Assay Kit (Pierce, USA). The appropriate amount of proteins were resolved by 10% SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) gel, transferred onto a polyvinylidene fluoride (PVDF) membrane, blocked in 5% nonfat dry milk in Tris-buffered saline (pH 7.4) containing 0.05% Tween 20, and blotted with antibodies against Akt, p-AKT, PI3K, p-PI3K mTOR and p-mTOR. β-actin served as the internal control. Then, the cells were incubated with HRP and secondary antibodies. The relative expression of the protein was evaluated with the gray value ratio of each protein and β-actin.
RNA isolation, cDNA library preparation, and RNA-seq analysis
TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) was used to extract the total RNA from the obtained cells according to the manufacturer’s instructions. Total RNA quality was assessed by NanoDrop 2000 spectrophotometry (Thermo Fisher, Waltham, MA, USA). Only samples with a total RNA concentration of .1 mg/mL and a 260 nm/280 nm optical density ratio between 1.8 and 2.0 were submitted for double-stranded cDNA synthesis. The rRNA was removed, the mRNA was enriched using magnetic beads with oligo (dT) and the mRNA randomly fragmented using fragmentation buffer. The mRNA was used as a template to synthesize the first-strand cDNA using 1st Strand Enzyme Mix (Vazyme). The second-strand cDNA was synthesized using 2nd Strand Marking Buffer and 2nd Strand/End Repair Enzyme Mix (Vazyme). The products were purified by VAHTSTM DNA Clean Beads, and then the ends of the double-stranded products were repaired and A-tailed. An adapter was ligated to the A-tailed products using ligation mix. Suitably sized fragments were selected using VAHTSTM DNA Clean Beads to construct the cDNA library by PCR. RNA sequencing was performed using Illumina HiSeq XTen (Vazyme Biotech Co., Ltd).
The quantitative analysis of gene expression was completed by Cufflinks based on RPKM (reads per kb per million reads). Differentially expressed genes (DEGs) were identified with adjusted P<0.05 for multiple tests by Benjamini-Hochberg Method [11] for controlling the false discovery rate. Cluster analysis was performed on differentially expressed genes using the software cluster and Java Treeview. Gene functional annotations were based on a genome database and mapped by GO terms. GO enrichment analysis was carried out using WEGO. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were identified according to p-values and adjusted q-values using a BLAST search against the KEGG database and were then mapped onto KEGG pathways [12].
Statistics
All values were expressed as the means ± SDs. The data for cell invasion were analyzed using an unpaired two-tailed t-test. Chi-squared analyses were performed to determine the correlation between various pathological parameters, and Fisher’s exact test was also performed when necessary. They all use SPSS-19 software. P<0.05 was considered to indicate statistical significance.
Results
IL-22 and IL-22R were upregulated in lung cancer tissue
First, we compared the expression of IL-22 and IL-22R in tissue samples of patients with NSCLC and adjacent tissue using RT-qPCR. As shown in Figure 1, the expression of both IL-22 and IL-22R was significantly increased in the tissue samples of patients with NSCLC compared with that in the adjacent tissue (Figure 1A and 1B). Immunohistochemistry was performed to examine the expression levels of IL-22 and IL-22R in NSCLC tissues and adjacent normal tissues. The results showed that the expression levels of IL-22 and IL-22R in NSCLC tissues were increased compared with those in adjacent normal tissues (Figure 1C). Furthermore, we isolated CAFs from the NSCLC tissue samples and cultured them for 72 h. Compared with that of normal fibroblasts, the expression of IL-22 was significantly upregulated in the cell culture supernatant of CAFs (Figure 1D). These results indicated that IL-22 and IL-22R were upregulated in lung cancer tissue and IL-22 enriched in cell culture supernatant of CAFs.
Figure 1.
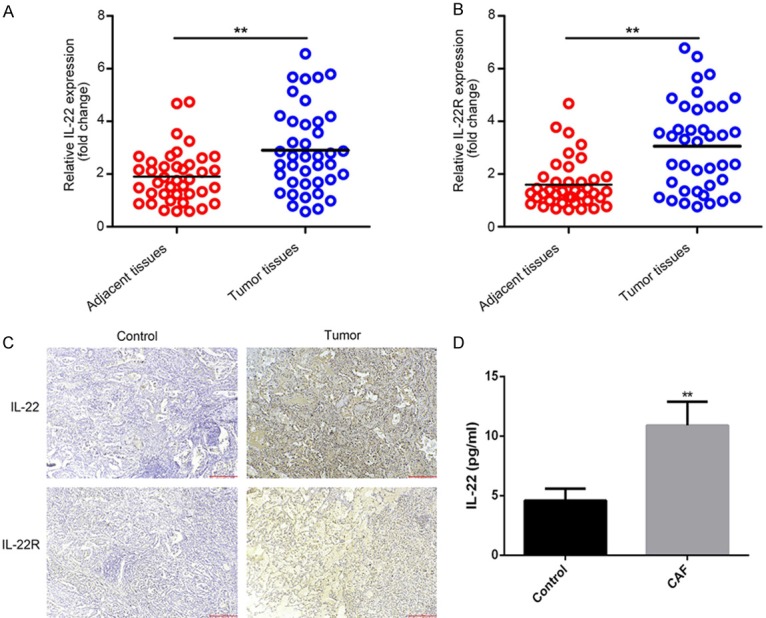
Comparison of the levels of IL-22 and IL-22R in tissue samples of patients with NSCLC and the adjacent tissues. A: RT-qPCR analysis of the expression of IL-22 in tissue samples of NSCLC patients. B: RT-qPCR analysis of the expression of IL-22R in tissue samples of NSCLC patients. C: IHC was performed to examine the expression levels of IL-22 and IL-22R in NSCLC tissues and adjacent normal tissues. D: Elisa-Kit analyze the expression of IL-22 in cell culture supernatant of CAFs.
Il-22 promoted the proliferation of NSCLC cells in vitro
Moreover, we treated cells with the cell culture supernatant of CAFs, and it was observed that compared with untreated cells, the treatment of the cell culture supernatant of CAFs induced a significant increase in proliferation (P<0.01; Figure 2) and decreased the apoptosis of A549 and H1650 cells (Figure 3), while the addition of the IL-22 antibody partially blocked the effects induced by the cell culture supernatant of CAFs.
Figure 2.
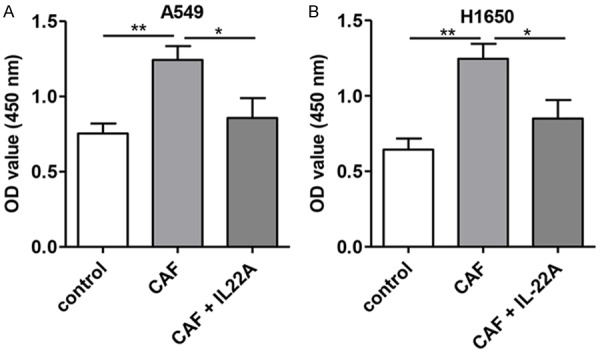
Effect of IL-22 on the proliferation of NSCLC cells in vitro. A: Compared with that of the control group, the proliferation rate of A549 cells was significantly increased after CAF treatment, which was significantly decreased in the CAF+IL-22A group. B: The proliferation rate of H1650 was significantly higher than that in the control group, which was significantly reversed after treatment with IL-22A. *P<0.05 vs. CAF group. **P<0.01 vs. CAF group.
Figure 3.

Effect of IL-22 on the apoptosis of NSCLC cells in vitro. (A) Compared with that in the control group, the apoptosis rate of A549 was significantly decreased in the CAF group, which was significantly augmented in the CAF+IL-22A group. (B) Quantitative analysis of (A). (C) The proliferation rate of H1650 was significantly lower than that in the control group, which was significantly reversed after treatment with IL-22A. (D) Quantitative analysis of (C). *P<0.05 vs. CAF group. **P<0.01 vs. CAF group.
IL-22 promoted the migration and invasion of NSCLC cells in vitro
Furthermore, we also examined the effects of the cell culture supernatant of CAFs on the migration and invasion ability of A549 and H1650 cells. As shown in Figures 4 and 5, treatment with the cell culture supernatants of CAFs induced a significant increase in the migration and invasion abilities of A549 and H1650 cells, while the addition of the IL-22 antibody partially blocked the effects induced by the cell culture supernatant of CAFs.
Figure 4.
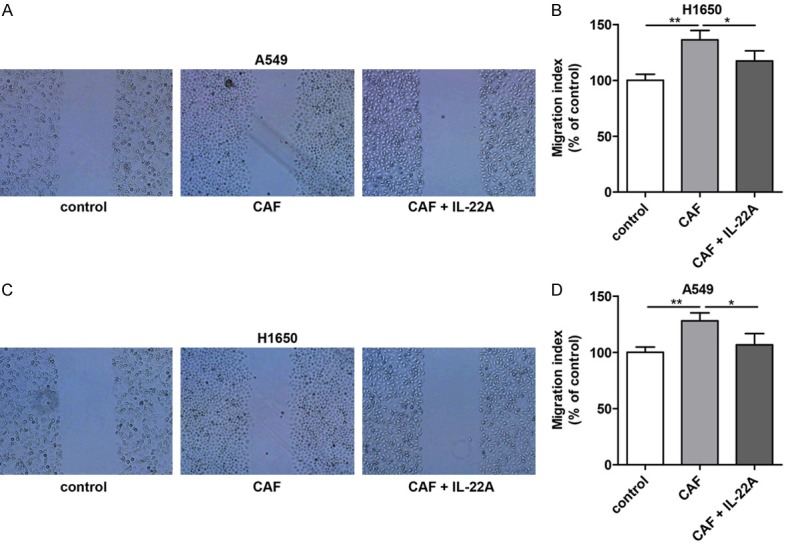
Effect of IL-22 on the migration of NSCLC cells in vitro. (A) Compared with that in the control group, the migration of A549 cells was significantly increased after CAF treatment, which was significantly decreased in the CAF+IL-22A group. (B) Quantitative analysis of (A). (C) The migration of H1650 cells in the CAF group was significantly higher than that in the control group, which was significantly reversed after treatment with IL-22A. (D) Quantitative analysis of (C). *P<0.05 vs. CAF group. **P<0.01 vs. CAF group.
Figure 5.
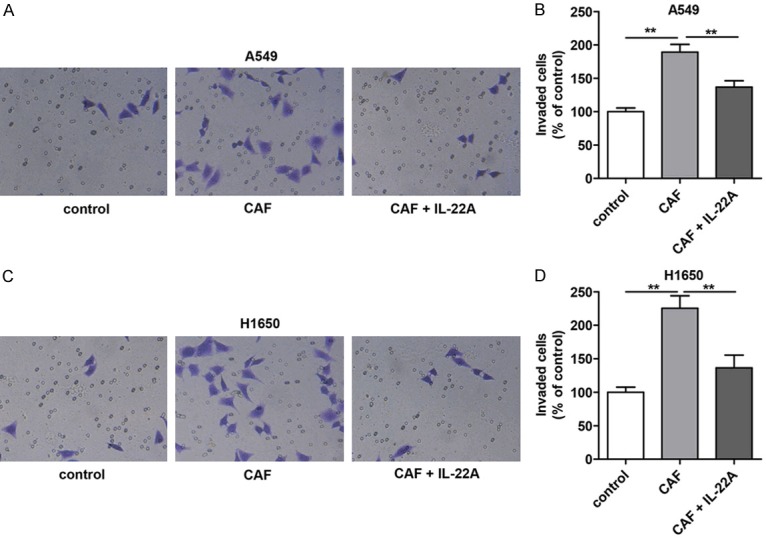
Effect of IL-22 on the invasion of NSCLC cells in vitro. (A) Compared with that in the control group, the migration of A549 cells was significantly increased after CAF treatment, which was significantly decreased in the CAF+IL-22A group. (B) Quantitative analysis of (A). (C) The migration of H1650 cells in the CAF group was significantly higher than that in the control group, which was significantly reversed after treatment with IL-22A. (D) Quantitative analysis of (C). *P<0.05 vs. CAF group. **P<0.01 vs. CAF group.
Il-22 regulated the expression of the PI3K-AKT-mTOR pathway in A549 and H1650 cells in vitro
Next, we performed Western blot analysis to investigate the effect of the cell culture supernatant of CAFs on the expression of PI3K-AKT-mTOR in A549 and H1650 cells in vitro. The treatment of the cell culture supernatant of CAFs induced a significant increase in the expression and phosphorylation of PI3K, AKT and mTOR (Figure 6), while the addition of the IL-22 antibody partially blocked the activation of the PI3K/AKT/mTOR signaling pathway induced the cell culture supernatant of CAFs.
Figure 6.
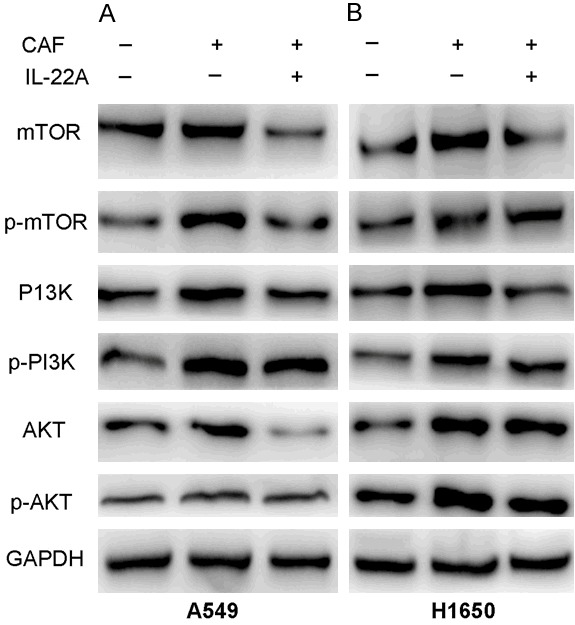
Effect of IL-22 on the expression of PI3K/AKT/mTOR in NSCLC cells in vitro. A: The protein levels of p-mTOR and p-PI3K/AKT in A549 cells treated with CAF were significantly increased compared with those in the control group, which were reversed after treatment with IL-22A. B: The protein levels of p-mTOR and p-PI3K/AKT in H1650 cells treated with CAF were significantly increased compared with those the control group, which were reversed after treatment with IL-22A.
IL-22 altered gene expression profiling of A549 cells
To profile the differentially expressed genes for A549 cells treated with or without IL-22, genome-wide RNA sequencing was conducted between three independent experiments. Hierarchical cluster analysis was applied for the changed gene with a fold-change >1.5 (P<0.05, FDR<30%) or a fold-change <0.667. As shown in Figure 7, we found that 2158 genes were significantly altered when treated with IL-22 compared to those in the control. Of those, 1375 genes were upregulated, while 783 genes were downregulated. To understand the important biological process affected by IL-22 cocultured with A549 cells, a gene ontology (GO) analysis was performed and revealed that IL-22 was involved in the regulation of small molecule metabolic processes, response to oxygen-containing compounds, response to organic substances, the regulation of locomotion, cell proliferation, motility, cell migration, cellular responses to chemical stimuli, and the positive regulation of cellular processes and biological processes. Moreover, in terms of cellular components, we found that the most abundant genes were focused on protein binding and the binding of cofactors, coenzymes and other molecules, suggesting that IL-22 affected the interaction of proteins and biological molecules. In addition, for molecular function, the most enriched GOs are vesicle, extracellular organelle and exosome, cytosol, cytoplasmic part and cytoplasm (Figure 8). The differentially expressed mRNAs in A549 cells treated with or without IL-22 were further subjected to pathway analyses. Twenty pathways were significantly enriched (P<0.05), including the p53 signaling pathway, metabolic pathways and peroxisome pathway (Figure 9), suggesting that IL-22 was able to regulate global signaling pathways regarding apoptotic and metabolic regulatory functions.
Figure 7.
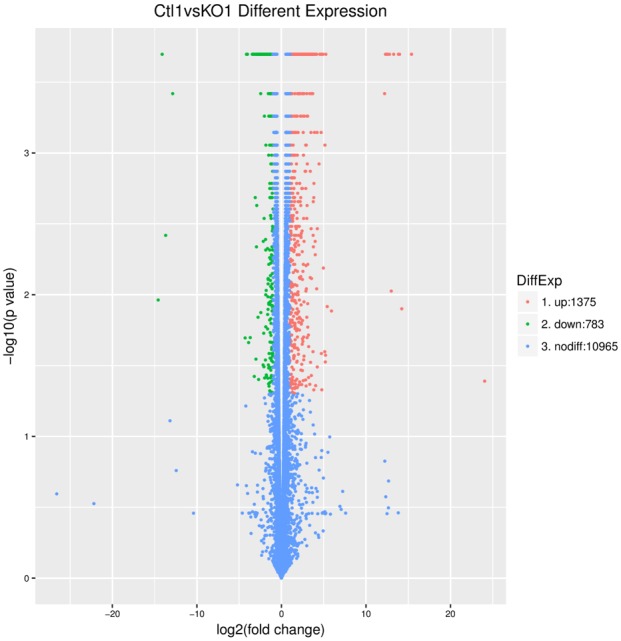
Differentially expressed genes for A549 cells treated with IL-22 or without treatment. Hierarchical cluster analysis was applied for the changed gene with a fold-change >1.5 (P<0.05, FDR<30%) or a fold-change <0.667. A total of 1375 genes were upregulated after treatment with IL-22, while 783 genes were downregulated.
Figure 8.
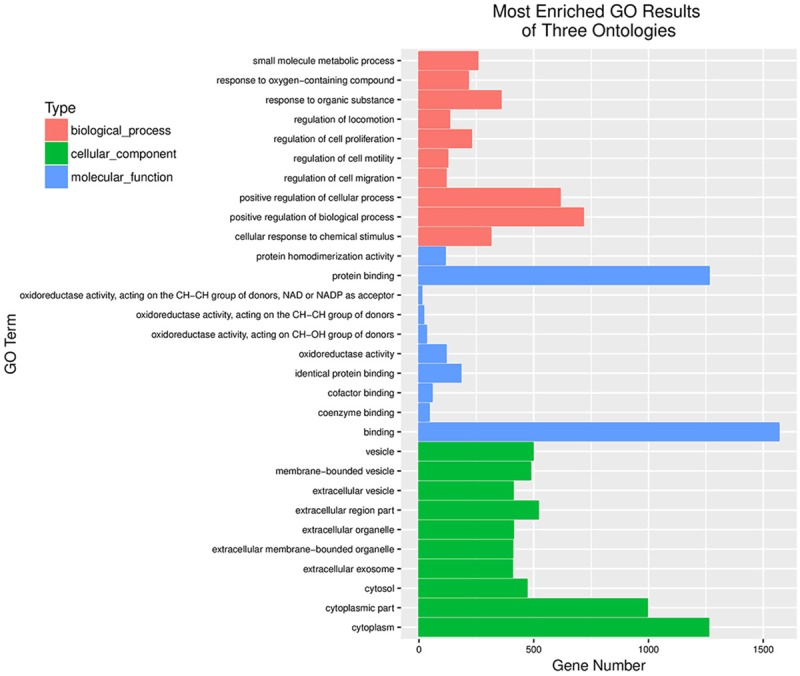
Results of GO analysis. The regulation of IL-22 was prominent in small molecule metabolic processes, responses to organic substances, cell proliferation, cell migration, cellular responses to chemical stimuli, the positive regulation of cellular processes and biological processes. IL-22 affected the interaction of proteins and biological molecules. The most enriched GOs are vesicle, extracellular organelle and exosome, cytosol, cytoplasmic part and cytoplasm.
Figure 9.
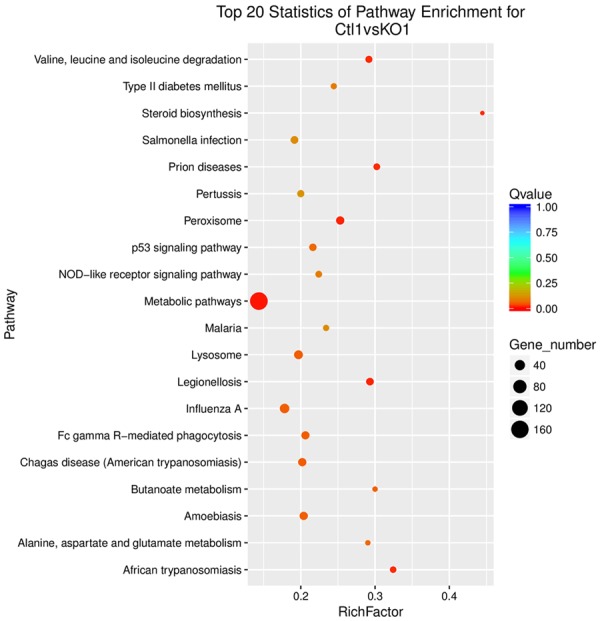
Results of pathway analysis. Twenty pathways were significantly enriched (P<0.05), including the p53 signaling pathway, metabolic pathways and peroxisome pathway. IL-22 was able to regulate global signaling pathways regarding apoptotic and metabolic regulatory functions. *P<0.05 vs. cells not treated with IL-22. **P<0.01 vs. cells not treated with IL-22.
Discussion
IL-22 has been shown to be critical in the progression of inflammatory disease, but its biological function in carcinogenesis remains obscure. In the present study, we first identified that the expression of IL-22 and the IL-22 receptor (IL-22R) in clinical lung cancer tissues, demonstrated by RT-qPCR, was positively correlated with the tumor size and metastasis of NSCLC. Notably, we noticed a strong expression of IL-22 consistently in stromal cells at the invasive front of the tumor, suggesting that IL-22 signaling may play a critical role in lung cancer cell invasion. A significantly elevated level of IL-22 was secreted by primary CAFs compared to that secreted by normal fibroblast cells, which was consistent in the NSCLC cell lines A549 and H1650 cocultured with CAFs. Moreover, we found that IL-22 was able to regulate the proliferation, apoptosis, migration and invasion of lung cancer cells via the activation of the critical signaling pathway PI3K-akt-mTOR, facilitating the progression of NSCLC.
In NSCLC, increasing evidence has revealed the importance of the local microenvironment in carcinoma initiation and progression. Activated stromal cells might be essential for tumor initiation, the progression of ductal carcinoma in situ to an invasive type, and metastasis processes [13,14]. Approximately 80% of fibroblasts acquire an aggressive phenotype and contribute to cancer progression, including tumor growth, invasion and metastasis, directly or indirectly through paracrine activation by various growth factors and cytokines. Notably, the activation of these pro-carcinogenic fibroblasts, termed CAFs, is not reversible, similar to the activation of related myofibroblasts. In our study, we first provide evidence that the increased level of IL-22 in NSCLC was secreted by CAFs, further leading to the activation of the PI3K-Akt-mTOR signaling pathway, ultimately resulting in cell proliferation, apoptosis, migration, and invasion in lung cancer.
Indeed, consistent with our current findings, previous studies have suggested that IL-22 may possibly promote cell proliferation and anti-apoptosis via the activation of various pathways, such as STAT3, MAPK, and/or NF-κB signaling in different types of cells [15,16]. The autocrine production of IL-22 has been suggested to contribute to human lung cancer cell survival, migration and resistance to chemotherapy through the upregulation of anti-apoptotic proteins, including Bcl-2, and via IL-22R1/STAT3 [9,17]. Furthermore, IL-22 promotes epithelial cell transformation and breast tumorigenesis via MAP3K8 activation [18]. Interestingly, in gastric cancer, CAFs have been shown to produce IL-22 to further support the invasion of gastric cancer cells, possibly via STAT3 and ERK signaling [19]. Genetic evidence has also suggested that a specific genetic polymorphism in IL-22 (rs2227484 genotype) was associated with a 2.263-fold increased risk for advanced NSCLC [20]. The corresponding high level of plasma IL-22 might contribute to the development of NSCLC. In addition, elevated IL-22 was also shown to promote survival and resistance to chemotherapy in human lung cancer cells via elevated anti-apoptotic proteins [9].
In the current study, genome-wide RNA sequencing was performed to further understand the global genetic changes associated with IL-22 treatment. Interestingly, we found that 2158 genes were significantly changed between cells treated with IL-22 and cells treated without IL-22. GO analysis revealed that IL-22 treatment was able to regulate several major biological events, including metabolic processes. Glucose metabolism, a metabolic process, in tumor cells significantly differs from that in normal cells. On one hand, it was considered that tumor cells principally take up high levels of glucose and secrete lactate under aerobic conditions, while normal cells catabolize glucose by mitochondrial oxidative phosphorylation. On the other hand, in a metastatic lymph node of breast cancer patients, immunohistochemical analysis revealed that metastatic breast cancer cells are positively stained by a glycolytic marker [21]. Thus, mCAFs likely contribute to metabolic reprogramming of the tumor microenvironment via cytokine secretion, such as IL-22.
CAFs could be a novel prospective therapeutic target in cancer immunotherapy. As a crucial component within the cancer microenvironment, CAFs are activated at the early stages and widely contribute to tumor initiation, growth, invasion, metastasis, and therapy resistance via growth factors, cytokines and other biological molecules. Current CAF-targeted cellular therapies have undergone preclinical evaluation and clinical trials [22], including the inhibition of CAF activation, blockage of CAF-epithelial interaction, inhibition of CAF-related angiogenesis, and destruction of metabolism uncoupling between CAFs and cancer cells. For example, the inhibition of IL-22 secretion from CAFs in NSCLC could be a promising therapeutic strategy.
In summary, we have demonstrated the elevated expression of IL-22 in NSCLC tumor specimens, which was correlated with the advanced clinicopathological stages of patients. Furthermore, IL-22 was profoundly elevated in the coculture of primary CAFs and lung cancer cells. Moreover, IL-22 treatment induced increased the proliferation and migration of A549 and H1650 cells but reduced apoptosis, possibly via the activation of PI3K-Akt-mTOR signaling. Finally, IL-22 regulates a globally altered gene expression profile in lung cancer cells, including metabolic signaling pathways, cell proliferation and migration. Together, these results suggest that IL-22 produced by CAFs promotes lung cancer invasion via the PI3K-Akt-mTOR signaling pathway.
Acknowledgements
This work was supported by Social Development Program of Jiangsu Province (BE2018643), the Scientific Program of Shandong Provincial Department of Education (J18KA254), 333 project of Jiangsu scientific research program (BRA2018168), and the Scientific Research Program of Changzhou (CE20165024; 2016C2BJ007; ZD201602; CE20175025; CE20185045; CJ20180029).
Disclosure of conflict of interest
None.
References
- 1.IARC. World cancer report 2014. Geneva: WHO; 2014. [Google Scholar]
- 2.Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83:584–594. doi: 10.4065/83.5.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.NIOH. SEER cancer statistics review, 1975-2011. National Cancer Institute, Bethesda. 2011 [Google Scholar]
- 4.Reck M, Popat S, Reinmuth N, De Ruysscher D, Kerr KM, Peters S ESMO Guidelines Working Group. Metastatic non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23:vii56–vii64. doi: 10.1093/annonc/mds226. [DOI] [PubMed] [Google Scholar]
- 5.Lim C, Savan R. The role of the IL-22/IL-22R1 axis in cancer. Cytokine Growth Factor Rev. 2014;25:257–271. doi: 10.1016/j.cytogfr.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 6.Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. 2011;12:383–390. doi: 10.1038/ni.2025. [DOI] [PubMed] [Google Scholar]
- 7.Zenewicz LA, Flavell RA. Recent advances in IL-22 biology. Int Immunol. 2011;23:159–163. doi: 10.1093/intimm/dxr001. [DOI] [PubMed] [Google Scholar]
- 8.Nagalakshmi ML, Rascle A, Zurawski S, Menon S, de Waal Malefyt R. Interleukin-22 activates STAT3 and induces IL-10 by colon epithelial cells. Int Immunopharmacol. 2004;4:679–691. doi: 10.1016/j.intimp.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 9.Zhang W, Chen Y, Wei H, Zheng C, Sun R, Zhang J, Tian Z. Antiapoptotic activity of autocrine interleukin-22 and therapeutic effects of interleukin-22-small interfering RNA on human lung cancer xenografts. Clin Cancer Res. 2008;14:6432–6439. doi: 10.1158/1078-0432.CCR-07-4401. [DOI] [PubMed] [Google Scholar]
- 10.Luo H, Tu G, Liu Z, Liu M. Cancer-associated fibroblasts: a multifaceted driver of breast cancer progression. Cancer Lett. 2015;361:155–163. doi: 10.1016/j.canlet.2015.02.018. [DOI] [PubMed] [Google Scholar]
- 11.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practicle and powerful approach to multiple testing. J R Stat Soc Ser B (Methodol) 1995;57:289–300. [Google Scholar]
- 12.Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pontiggia O, Sampayo R, Raffo D, Motter A, Xu R, Bissell MJ, Joffé EB, Simian M. The tumor microenvironment modulates tamoxifen resistance in breast cancer: a role for soluble stromal factors and fibronectin through beta1 integrin. Breast Cancer Res Treat. 2012;133:459–471. doi: 10.1007/s10549-011-1766-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu M, Yao J, Carroll DK, Weremowicz S, Chen H, Carrasco D, Richardson A, Violette S, Nikolskaya T, Nikolsky Y, Bauerlein EL, Hahn WC, Gelman RS, Allred C, Bissell MJ, Schnitt S, Polyak K. Regulation of in situ to invasive breast carcinoma transition. Cancer Cell. 2008;13:394–406. doi: 10.1016/j.ccr.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wen Z, Liao Q, Zhao J, Hu Y, You L, Lu Z, Jia C, Wei Y, Zhao Y. High expression of interleukin-22 and its receptor predicts poor prognosis in pancreatic ductal adenocarcinoma. Ann Surg Oncol. 2014;21:125–132. doi: 10.1245/s10434-013-3322-x. [DOI] [PubMed] [Google Scholar]
- 16.Kobold S, Völk S, Clauditz T, Küpper NJ, Minner S, Tufman A, Düwell P, Lindner M, Koch I, Heidegger S, Rothenfuer S, Schnurr M, Huber RM, Wilczak W, Endres S. Interleukin-22 is frequently expressed in small- and large-cell lung cancer and promotes growth in chemotherapy-resistant cancer cells. J Thorac Oncol. 2013;8:1032–1042. doi: 10.1097/JTO.0b013e31829923c8. [DOI] [PubMed] [Google Scholar]
- 17.Bi Y, Cao J, Jin S, Lv L, Qi L, Liu F, Geng J, Yu Y. Interleukin-22 promotes lung cancer cell proliferation and migration via the IL-22R1/STAT3 and IL-22R1/AKT signaling pathways. Mol Cell Biochem. 2016;415:1–11. doi: 10.1007/s11010-016-2663-8. [DOI] [PubMed] [Google Scholar]
- 18.Kim K, Kim G, Kim JY, Yun HJ, Lim SC, Choi HS. Interleukin-22 promotes epithelial cell transformation and breast tumorigenesis via MAP3K8 activation. Carcinogenesis. 2014;35:1352–61. doi: 10.1093/carcin/bgu044. [DOI] [PubMed] [Google Scholar]
- 19.Fukui H, Zhang X, Sun C, Hara K, Kikuchi S, Yamasaki T, Kondo T, Tomita T, Oshima T, Watari J, Imura J, Fujimori T, Sasako M, Miwa H. IL-22 produced by cancer-associated fibroblasts promotes gastric cancer cell invasion via STAT3 and ERK signaling. Br J Cancer. 2014;111:763–771. doi: 10.1038/bjc.2014.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu F, Pan X, Zhou L, Zhou J, Chen B, Shi J, Gao W, Lu L. Genetic polymorphisms and plasma levels of interleukin-22 contribute to the development of nonsmall cell lung cancer. DNA Cell Biol. 2014;33:705–714. doi: 10.1089/dna.2014.2432. [DOI] [PubMed] [Google Scholar]
- 21.Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, Philp NJ, Pestell RG, Lisanti MP. Mitochondrial metabolism in cancer metastasis: visualizing tumor cell mitochondria and the “reverse Warburg effect” in positive lymph node tissue. Cell Cycle. 2012;11:1445–1454. doi: 10.4161/cc.19841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Liu J. Tumor stroma as targets for cancer therapy. Pharmacol Ther. 2013;137:200–215. doi: 10.1016/j.pharmthera.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]