

Abstract
Resistance to Gemcitabine (GEM) is a crucial problem in treatment of pancreatic cancer. Many studies indicate the direct impact of glycolytic enzyme on chemoresistance. However, it still has not been known whether Enolase 1 (ENO1), a multifunctional glycolytic enzyme, is a potential target to overcome GEM resistance in pancreatic ductal adenocarcinoma (PDAC). In this study, we showed that ENO1 high expression was associated with poor prognosis of PDAC patients. Moreover, we investigated the impacts of ENO1 silencing on hypoxia induced GEM chemoresistance in CFPAC-1 and MiaPaCa-2 cells. The results showed that, targeting ENO1 using ENO1-shRNA could sensitize hypoxia induced chemoresistance in pancreatic cancer cells by modulation of redox homeostasis, the mechanisms appear to be associated with influences on proliferation, apoptosis, and cell cycle regulated by increased intracellular reactive oxygen species (ROS). We demonstrated that targeting ENO1 could be a potential strategy for overcoming hypoxia induced GEM chemoresistance in PDAC cells.
Keywords: Phosphopyruvate hydratase, pancreatic neoplasms, gemcitabine, chemoresistance, redox homeostasis
Introduction
Pancreatic ductal adenocarcinoma (PDAC) has increased rapidly in recent years and became one of the most fatal forms of human cancer with under overall 5% five-year survival rate for all stages [1,2]. This poor prognosis mainly relates to the hard early diagnosis, and refractory to all treatment methods including chemotherapy [3]. Gemcitabine (GEM) is a nucleoside analog of deoxycytidine that has been used as a chemotherapeutic agent since its approval in 1996. It is now used as the standard first line treatment choice for PDAC chemotherapy. However, as a hallmark of PDAC, GEM resistance fails to improve survival rate in treated patients. Deciphering GEM resistance mechanisms and suggesting innovative strategies aimed to overcome GEM resistance may increase the efficacy of chemotherapy [4].
Most tumor cells are subjected to hypoxia microenvironment, which contributes to tumor chemoresistance [5-7]. Tumor cells exposured to hypoxia demonstrate increased levels of glycolysis, which are associated with increased levels of glycolytic enzyme activity. It has been described that some glucose enzymes have correlation with hypoxia induced drug resistance in cancer [8-10], such as hexokinase II [11], pyruvate kinase M1 (PKM1) and pyruvate kinase M2 (PKM2) [12-14].
Enolase 1 (ENO1), also known as pyruvate dehydrogenase 1, is a multifunctional glycolytic enzyme with upregulated expression at the mRNA and/or protein level in PDAC cell lines and tissues, which involved in the tumorigenesis, invasion and metastasis of PDAC [15-17]. Furthermore, proteomics analysis revealed that ENO1 is upregulated in many different drug-resistant cancer cells, such as docetaxel-resistant advanced nasopharyngeal carcinoma cells [18], cisplatin-resistant head and neck squamous cell carcinoma cells [19] and doxorubicin-resistant myelogenous leukemia cells [20]. ENO1 is also involved in the process of cell adhesion mediated drug resistance of Non-Hodgkin’s lymphoma [21] and tamoxifen resistance of breast cancer [22]. Taken together, these results suggest that ENO1 should be a potential target to overcome drug resistance in cancer.
Therefore, in the present study, we investigated whether inhibition of ENO1 expression through short-hairpin RNAs (shRNAs) would promote Gem chemosensitivity in PDAC cells in hypoxia conditions, our results demonstrated that ENO1 silencing could effectively improve the suppression of cell growth and induce cell apoptosis by Gem, we also observed that ENO1 silencing had impact on cellular redox homeostasis by improving intracellular ROS concentration. These results suggested that targeting ENO1 could be a potential strategy for overcoming Gem chemoresistance in PDAC.
Material and methods
Immunohistochemistry (IHC) staining
Tissue microarray (Shanghai Outdo Biotech Co. Ltd, HPan-Ade120Sur-01) was used for IHC staining, containing 57 pairs of human PDAC and adjacent non-cancer tissues, 6 human PDAC tissues. All cases were staged according to the seventh edition of the pancreatic cancer TNM staging system of the American Association of Cancer [23], including 5 stage I, 45 stage II, 4 stage IV and 9 without stages. This study was approved by the Ethics Committee of Shanghai Ninth People’s Hospital, Shanghai Jiaotong University School of Medicine.
IHC staining of the tissue microarray was performed according to the previous reported protocols [24]. IHC staining was graded by one blinded observer with two semiquantitative measurements: staining intensity (0-4) and percentage of stained cells (0= no staining, 1= less than 25%, 2=25%-50%, 3=50%-75%, and 4=75%-100%). A combined IHC score was calculated as the product of staining intensity and percentage of stained cells. Low expression was defined as a score 0-4 while high expression at score 6-16.
Cell lines and cell culture
The human pancreatic cancer cell lines CFPAC-1 and MiaPaCa-2 cells were obtained from American Type Culture Collection (ATCC). MiaPaCa-2 cells were grown in Dulbecco’s Modified Eagle Medium (DMEM; Hyclone, Massachusetts, USA), and CFPAC-1 cells were grown in RPMI-1640 medium (Hyclone, Massachusetts, USA), both supplemented with 10% fetal bovine serum (FBS; Gibcol, New York, USA), and 0.01% penicillin-streptomycin. Both cells were maintained in a humidified tissue culture incubator at 37°C with 21% O2 and 5% CO2. Hypoxia conditions were simulated in a SCI-TIVE N-N hypoxia workstation (Baker Ruskinn) at 1% O2 and 5% CO2.
Transfection of shRNAs
ShRNAs (TRCN0000029324) targeting the human ENO1 mRNA obtained from the human library MISSION® TRCHs 1.0 were purchased from Sigma-Aldrich (SHCLNG NM_001428) (St. Louis, MO, USA) with the targeted sequence 5’-CCGGCGTACCGCTTCCTTAGAACTTCTCGAGAAGTTCTAAGGAAGCGGTACGTTTTT-3’. MiaPaCa-2 and CFPAC-1 cells grown to 80-90% confluency were transiently transfected with ENO1-shRNA for 24 hours. The control shRNA (SHC002, Sigma) generates a scrambled shRNA sequence that does not target any known human gene. The efficiency of transfection was evaluated by quantitative realtime PCR and western blot analysis in transiently transfected cells after 24 hours.
Real-time PCR analysis
Total RNA was extracted from MiaPaCa-2 and CFPAC-1 cells using Trizol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Reverse transcription was performed using PrimeScriptTM RT reagent Kit (Takara, Dalian, China) for 15 min at 37°C, followed by 5 s at 85°C, and 60 min at 4°C. Real-time polymerase chain reaction (PCR) was performed using SYBR premix Ex as the tag (Takara, Dalian, China) according to the manufacturer’s instructions on an ABI PRISM 7500 (Applied Biosystems, Foster City, CA, USA). After PCR, 5 μl of PCR products were run on a 1% agarose gel and evaluated by relative pixel densitometry. The abundance of transcript was expressed relative to the control of glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
The sequences of primers used were: ENO1: Forward: 5’-TTGGCAGAAGTTCACAGCCA-3’, Reverse: 5’-CTGAAGAGACTCGGTCACGG-3’; GAPDH: Forward: 5’-TGACTTCAACAGCGACACCCA-3’, Reverse: 5’-CACCCTGTTGCTGTAGCCAAA-3’.
Western blot
Cells were subjected for protein isolation with lysis buffer (Thermo) supplied with protease inhibitor. 20 μg total protein was separated by SDS-PAGE experiment, followed by transferring to nitrocellulose filter (NC) membranes (Millipore) using a semi-dry transfer system (Bio-Rad, Hercules, California, USA). The membranes were blocked with 5% non-fat milk for 1 h, then incubated with primary antibodies overnight at 4°C. The membranes were incubated with horseradish peroxidase-linked secondary antibodies (Santa Cruz Biotechnology) for 1 h at room temperature. Blots were detected with ECL reagents (Millipore) and exposed to chemiluminescent imaging system for 5 min. Quantitative analysis was carried out with image analysis software Quantity One (Bio-Rad). Primary antibodies against ENO1 (ab155955) were purchased from Abcam (Cambridge, MA, USA).
Cell proliferation assays
Cell proliferation was monitored by analyzing relative survival cell number with CCK-8 Cell Proliferation/Viability Assay Kit (Sigma) in according to the guidelines. To evaluate the effectiveness of ENO1 silencing with ENO1-shRNA as an adjuvant therapy, we tested whether short term (24 hours) treatment with ENO1-shRNA would sensitize MiaPaCa-2 and CFPAC-1 cells to subsequent treatment with different concentration of GEM (Eli Lilly and Co, Indianapolis, IN). After 48 hours of incubation with GEM, IC50 was calculated. Experiments were replicated for three times.
Drugs were added as indicated 5 groups for cell proliferation curves: no treatment (control group), or ENO1 control-shRNA incubation (ENO1 control-shRNA group), or ENO1-shRNA incubation (ENO1-shRNA group) for 24 hours with normal condition followed by 72 hours with hypoxia conditions. No treatment for 24 hours with normal condition followed by 72 hours treatment with GEM (10-2 mol/L) with hypoxia conditions (GEM group), and ENO1-shRNA incubation for 24 hours with normal condition followed by 72 hours treatment with GEM (10-2 mol/L) with hypoxia conditions (ENO1-shRNA+GEM group).
Cellular colony formation assays
Transduced MiaPaCa-2 and CFPAC-1 cells were suspended in complete medium supplied with 0.45% low melting point agarose (Invitrogen). The cells were placed in 6-well cell culture plates containing 2 ml complete medium and agarose (0.75%) on the bottom layer. The plates were incubated at 37°C for treatments as described previously with hypoxia conditions. Two weeks later, colony numbers were obtained by staining the cell colonies with 0.005% crystal violet.
Cell apoptosis analysis
CFPAC-1 and MiaPaCa-2 cells were plated at 2×105/ml cells/well in a six-well plate, and then treated as described previously with hypoxia conditions for 48 h. Cells were collected and washed with PBS, stained using an Annexin-V-FITC Kit (Invitrogen), and analyzed by flow cytometry (BD Biosciences FACSCalibur). All experiments were replicated for three times.
Cell cycle analysis
MiaPaCa-2 and CFPAC-1 cells were trypsinized following treatments, and were spun down and washed once with PBS. Cell pellets were resuspended in 10 mg/ml PI (Sigma) for 30 min in darkness, and triturated with a 1 ml syringe attached to a 25G needle to lyse the cells and release intact nuclei. Nuclei were analyzed on flow cytometry (BD Biosciences FACSCalibur). Cell cycle and statistical analyses were performed using FlowJo version 10.
Intracellular ROS measurement
Intracellular ROS was measured using the ROS detection kit (Beyotime Company, ShangHai, China) according to the manufacturer’s instructions. Briefly, after treatment, cells were loaded with 10 μmol/L 2,7-Dichlorodi-hydrofluorescein diacetate (DCFH-DA) to incubate for 60 min at 37°C, cells were trypsinized and washed three times with PBS and re-suspended in 0.5 ml PBS. The intracellular fluorescence of DCFH was detected (λ excitation =500±15 nm, λ emission =530±20 nm) using a Synergy HT microplate reader (Biotec Instruments).
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity
Cell NADPH oxidase activity was carried out by a quantitative colourimetry detection kit (GENMED, Shanghai), and measured using an enzyme micro-plated reader (Biotec Instruments) at 550 nm according to the manufacturer’s instructions. The specific NADPH oxidase activity was calculated by the total activity minus the non-specific activity.
NADP/NADPH ratio
The intracellular NADP/NADPH ratio was detected by NADP/NADPH quantification colorimetric kit (Biovision Incorporated, USA), assays were performed following the kit protocol. NADP/NADPH ratio was calculated after monitoring the absorbance at 450 nm with an enzyme micro-plated reader (Biotec Instruments).
Reduced glutathione (GSH) levels
After treatments, the supernatant of cells were collected and measured by colorimetric method following the manufacturer’s instructions of GSH assay kit (Nanjing Jiancheng bioengineering institute, Nanjing, China). Sample fluorescence was monitored with an enzyme micro-plated reader (Biotec Instruments) at 405 nm, and GSH concentration was calculated.
Statistical analysis
All the values were expressed as mean ± S.D. from at least three independent experiments. The difference between two groups was analyzed by the parametric unpaired Student’s t-test. To analyze the difference among three or more groups, one-way ANOVA analysis was implemented. The statistical analysis was performed by GraphPad Prism 6 and p values less than 0.05 are considered significant.
Results
ENO1 high expression was associated with poor prognosis of PDAC patients
Our IHC staining results showed that the ENO1 expression level was related to the prognosis of PDAC patients. ENO1 expression was up-regulated in human PCAC tissues, ENO1 high expression rate was 77.2% (44/57) in human PDAC tissues but 31.6% (18/57) in adjacent non-cancer tissues (Figure 1A, 1B). Moreover, high expression of ENO1 predicted shorter overall survival (OS). The median survival time of high ENO1 expression PDAC patients was 8 months (95% Confidence Interval (CI) of ratio: 0.1557 to 0.4567), in contrast, the median survival time of low ENO1 expression PDAC patients was 30 months (95% CI of ratio: 2.19 to 6.422) (P<0.001, Figure 1C).
Figure 1.
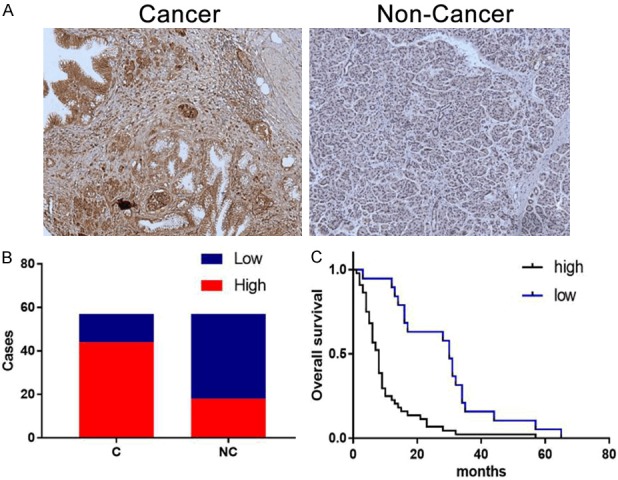
High ENO1 expression predicted poor prognosis in pancreatic cancer. A. Representative images showed the expression of eno1 protein in human pdac and adjacent non-cancer tissues using ihc staining. Brown staining indicated eno1 expression. B. Bar plots represented high eno1 expression in human pdac tissues (n=57). C. High expression of eno1 predicted shorter overall survival (os) of pdac patients, ***P<0.001.
ENO1 inhibition by ENO1-shRNA in pancreatic cancer cell lines
Compared with the blank group after transfection of 48 hours, both ENO1-shRNA transfected MiaPaCa-2 and CFPAC-1 cells showed significantly reduced ENO1 mRNA expression (P<0.01, Figure not shown), and also significantly reduced relative ENO1 protein expression (P<0.001, Figure not shown). No inhibition of ENO1 mRNA or protein expression were detected in control shRNA group.
ENO1 silencing impaired on cell proliferation to enhance GEM chemosensitivity of pancreatic cancer cell lines
To investigate whether ENO1 contributed to cell proliferation and hypoxia induced drug resistance in human pancreatic cancer cells, we evaluated the effectiveness of ENO1 silencing as an adjuvant therapy in MiaPaCa-2 and CFPAC-1 cells. We investigated whether short term (24 hour) treatment with ENO1-shRNA would sensitize hypoxia induced drug resistance cells to subsequent treatment with GEM, after 48 hour incubation with GEM, we found that ENO1 silencing with ENO1-shRNA significantly enhanced the chemosensitivity of both MiaPaCa-2 and CFPAC-1 cells with significant decreases in IC50 (Figure 2A, 2B). Next, we analyzed the cell proliferation curve, the results indicated that ENO1 inhibition by ENO1-shRNA in MiaPaCa-2 and CFPAC-1 cells resulted in significant inhibition in cell growth when compared to control groups (P<0.001, Figure 2C, 2D). We noticed that ENO1-shRNA and GEM combination treatment inhibited pancreatic cancer cell growth significantly stronger than ENO1-shRNA or GEM single treatment. Consistent with our cell growth data, ENO1 silencing greatly attenuated the colony numbers of MiaPaCa-2 and CFPAC-1 cells (P<0.01, P<0.001, Figure 2E-G).
Figure 2.
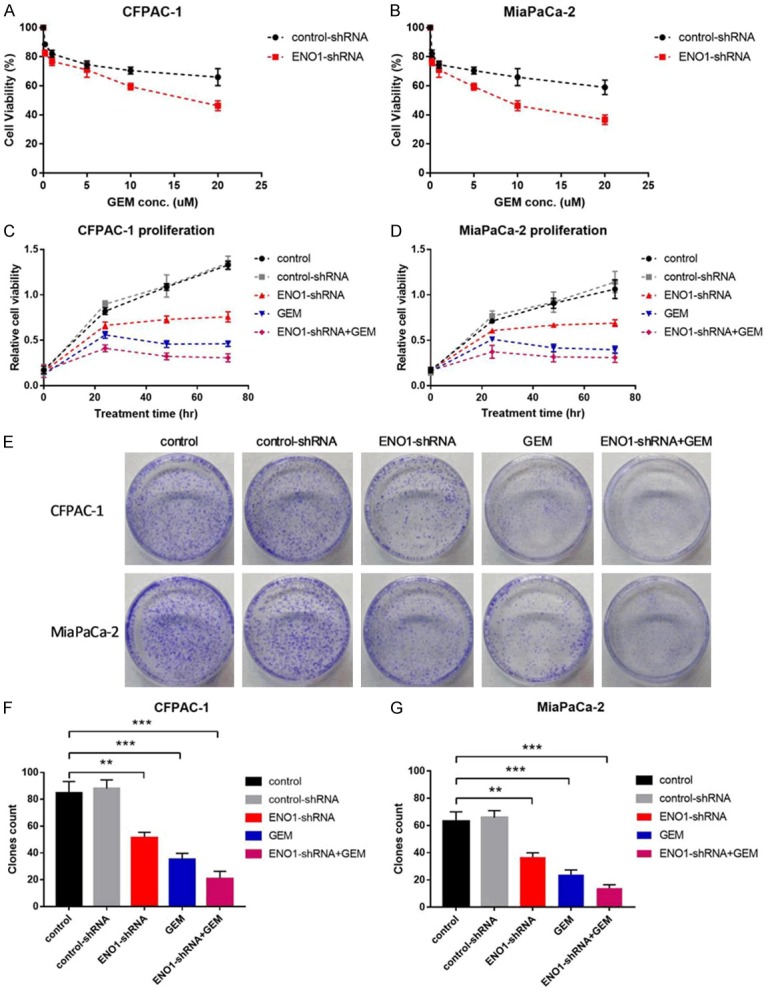
ENO1 silencing impaired on cell proliferation to enhance GEM chemosensitivity in pancreatic cancer cells. A, B. CFPAC-1 and MiaPaCa-2 cells were transiently transfected with ENO1-shRNA for 24 h, subsequently treated with different concentration of GEM for 48 h, IC50 was calculated. C, D. CFPAC-1 and MiaPaCa-2 cells viability with different treatment for ENO1 control-shRNA, ENO1-shRNA, GEM or ENO1-shRNA+GEM under hypoxia conditions were measured by CCK-8 assay. E-G. Cellular colony formation assays of CFPAC-1 and MiaPaCa-2 cells with different treatment under hypoxic conditions. Results are from representative experiments in triplicate and shown as the mean ± S.D. **P<0.01, ***P<0.001.
ENO1 silencing affected on cell apoptosis and cell cycle to enhance GEM chemosensitivity of pancreatic cancer cell lines
To further investigate the growth inhibition, we compared the cell apoptosis and cell cycle profiles of ENO1 silencing pancreatic cancer cells with controls by flow cytometry. Flow cytometric analysis showed that ENO1 silencing increased apoptosis rate of MiaPaCa-2 and CFPAC-1 cells, and enhanced the effect of GEM (P<0.01, P<0.001, Figure 3A-C). ENO1 silencing dramatically increased the cell population in the G0/G1 phase, whereas it reduced the cell population in the S phases, which resulted in a transient G1/S phase block (P<0.01, P<0.001, Figure 3D-F).
Figure 3.

ENO1 silencing affected on cell apoptosis to enhance GEM chemosensitivity in pancreatic cancer cells. A-C. CFPAC-1 and MiaPaCa-2 cells were treated with ENO1 control-shRNA, ENO1-shRNA, GEM or ENO1-shRNA+GEM under hypoxia conditions for 48 h, Cell apoptosis was determined by flow cytometry analysis. Results are from representative experiments in triplicate and shown as the mean ± S.D. **P<0.01, ***P<0.001. ENO1 silencing affected on cell cycle to enhance GEM chemosensitivity in pancreatic cancer cells. D-F. CFPAC-1 and MiaPaCa-2 cells were treated with ENO1 control-shRNA, ENO1-shRNA, GEM or ENO1-shRNA+GEM under hypoxia conditions for 48 h. Cell cycle was analyzed by flow cytometry. Results are from representative experiments in triplicate and shown as the mean ± S.D. **P<0.01, ***P<0.001.
ENO1 silencing impacted cellular redox homeostasis of pancreatic cancer cell lines
Moreover, we observed cellular redox homeostasis of CFPAC-1 and MiaPaCa-2 treated with ENO1-shRNA and gemcitabine (Figure 4). The results indicated that intracellular ROS concentration was up-regulated with ENO1-shRNA or ENO1-shRNA+GEM treatment compared with control group in both cells (P<0.01, P<0.001, Figure 4A, 4B). The activity of NADPH oxidase, which convert NADPH to NADP and create O2 -, was also up-regulated in ENO1-shRNA group and ENO1-shRNA+GEM group (P<0.05, P<0.01, P<0.001, Figure 4C, 4D). As a result, ENO1-shRNA or ENO1-shRNA+GEM treatment dramatically increased the ratio of intracellular NADP/NADPH in CFPAC-1 and MiaPaCa-2 cells (P<0.01, Figure 4E, 4F). Finally, we analyzed GSH concentration and found that ENO1 silencing had no significant impact on GSH, but ENO1-shRNA+GEM treatment demonstrated decreased GSH concentration (P<0.05, Figure 4G, 4H). Taken together, these evidence demonstrated that ENO1 silencing impacts cellular redox homeostasis by improved intracellular ROS concentration.
Figure 4.
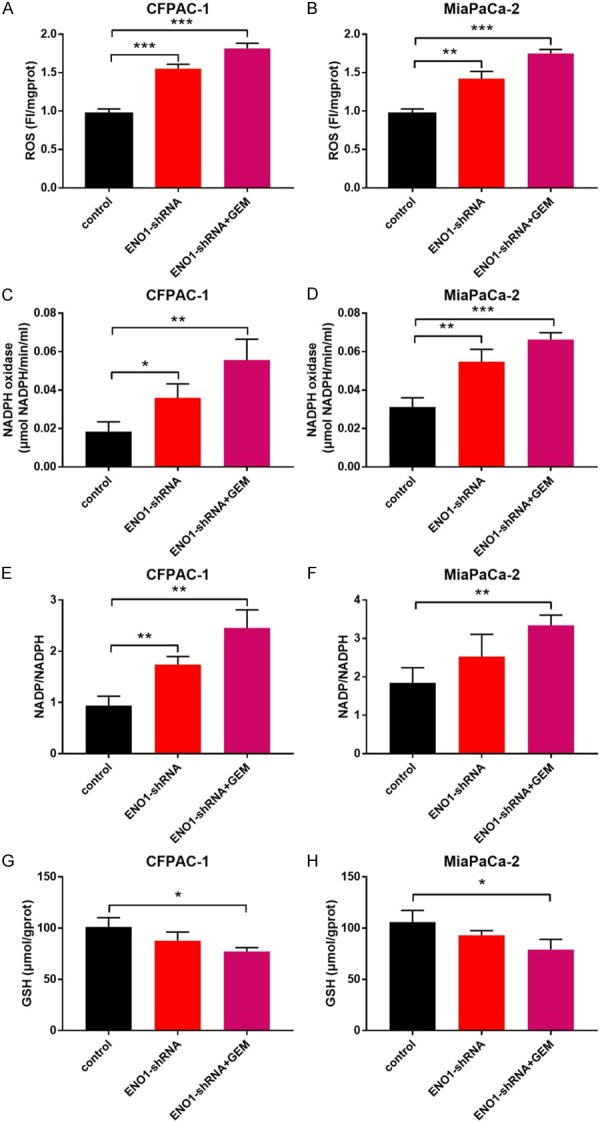
ENO1 silencing impacted cellular redox homeostasis in pancreatic cancer cell lines. CFPAC-1 and MiaPaCa-2 cells treated with ENO1-shRNA or ENO1-shRNA+GEM under hypoxia conditions for 48 h. A, B. Intracellular ROS was measured using a ROS detection kit. C, D. NADPH oxidase activity was measured using a quantitative colourimetry detection kit. E, F. Intracellular NADP/NADPH ratio was detected by NADP/NADPH quantification colorimetric kit. G, H. The supernatant of pancreatic cancer cells were collected and measured by colorimetric by GSH assay kit. Results are from representative experiments in triplicate and shown as the mean ± S.D. *P<0.05, **P<0.01, ***P<0.001.
Discussion
Glycolysis is enhanced in most tumor cells which is named Warburg effect, tumor cells mostly subjected to hypoxia microenvironment, can reprogram metabolic procedure associated with increased levels of glycolytic enzyme to enhance glycolysis pathway [25,26]. ENO1, also called alpha-enolase, is a key glycolytic enzyme catalyzing the conversion of 2-phospho-D-glycerate to phosphoenolpyruvate. It is known that ENO1 expression was significantly up-regulated at the mRNA and/or protein level in human PDAC tissues, PDAC cell lines and animal model tissues [15-17,24]. Our results showed that the expression of ENO1 in PDAC tissues was increased compared with adjacent non-cancer tissues. ENO1 high expression rate was 77.2% in human PDAC tissues but 31.6% in adjacent non-cancer tissues. Our results showed that ENO1 expression was associated with the prognosis of PDAC patients, Patients with higher ENO1 expression had significant shorter survival time than those with lower ENO1 expression, high expression of ENO1 predicted shorter OS. We also found the ENO1 level in peripheral blood of PDAC patients was closely associated with the prognosis of PDAC in our previous study [24]. Our results were in line with some additional studies that the ENO1 high level contributed to poor prognosis of pancreatic cancers [27,28].
We next confirmed whether ENO1 is a potential target to overcome drug resistance of pancreatic cancer. Our results showed that ENO1 silencing using ENO1-shRNA can drastically enhance chemosensitivity of pancreatic cancer cells for GEM treatment in vitro. It was reported that ENO1 participated in chemoresistance in some other cancers, such as cell adhesion mediated drug resistance of Non-Hodgkin’s lymphoma [21], tamoxifen resistance of breast cancer [22] and cisplatin resistance of gastric cancer [29]. RNAi-mediated knockdown of ENO1 increased the sensitivity of tumor cells to antitubulin chemotherapeutics [30]. Our findings suggested that targeting ENO1 using ENO1-shRNA, effectively improve the suppression of cell growth and induce cell apoptosis by Gem. Some additional studies also proved targeting ENO1 inhibited cell growth and induced cell death [31-33].
The effects of ROS to cancer cells are complex and perhaps context-dependent. In general, ROS level is elevated in cancer cells that contributes to the tumor initiation, promotion and progression. ROS has tumor suppressing effects as well, excessive production of ROS could result in damages of mitochondria and cell membranes, as well as activation of various cell-death pathways and causing cell death [34,35]. In fact, a variety of chemotherapic agents including Gem induce ROS generation as an additional anticancer mechanism [36]. At the same time, cancer cells go to develop a highly regulated antioxidant defense system to tolerate oxidative damage of the higher ROS level. The cellular antioxidant defense system consists of various antioxidant enzymes, most of these enzymes are under the control of the transcription factor NF-E2-related factor-2 (Nrf2) [37]. In pancreatic cancer, Kras induce low intracellular ROS levels which may contribute to intrinsic resistance to Gem [38]. Therefore, manipulating ROS levels by redox modulation is a way to enhance chemosensitivity without causing significant toxicity to normal cells [39].
In the present study, our results showed that ENO1 silencing treatment appeared to effectively increase accumulation of ROS and decrease NADPH through increased NADPH oxidase activities which normally function to remove harmful ROS. It is possible that ENO1 silencing enhances chemosensitivity to Gem in part through increasing ROS generation and decreasing cellular antioxidant defense, resulting in a redox imbalance to facilitate apoptosis and cell death in pancreatic cancer cells, while the exact mechanism and contribution of ROS to ENO1 silencing-induced chemosensitivity remains unclear and need further research. It was pointed out that as a moonlight-like protein, ENO1 located at the cell wall may protect sessile cells from oxidative stress [40], and ENO1 knockdown rescued oxidative phosphorylation in pancreatic cells [41]. Our data supported ENO1 high expression of pancreatic cancer may lead to low ROS concentration, which could contribute to reduce chemosensitivity to Gem.
Conclusion
Our findings suggested that targeting ENO1 using ENO1-shRNA, can sensitize hypoxia induced resistance in pancreatic cancer cells by modulation of ROS homeostasis, the mechanisms might associate with influences on proliferation, apoptosis, and cell cycle regulated by increased intracellular ROS. Such an adjuvant treatment could be an effective therapeutic option to synergize with current GEM therapy. The data presented in this study may help to elucidate ENO1 mediated adjuvant treatment to enhance the cytotoxicity of GEM towards chemoresistant pancreatic cancer.
Acknowledgements
Fundamental research program funding of Ninth People’s Hospital affiliated to Shanghai Jiao Tong University School of Medicine (No. JYZZ044).
Informed consent was issued by all participants included in the study.
Disclosure of conflict of interest
None.
Abbreviations
- DMEM
Dulbecco’s Modified Eagle Medium
- DCFH-DA
2,7-Dichlorodi-hydrofluorescein diacetate
- ENO1
enolase 1
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GEM
gemcitabine
- GSH
glutathione
- IHC
immunohistochemistry
- NADPH
nicotinamide adenine dinucleotide phosphate
- Nrf2
NF-E2-related factor-2
- OS
overall survival
- PC
pancreatic cancers
- PDAC
pancreatic ductal adenocarcinoma
- PKM1
pyruvate kinase M1
- PKM2
pyruvate kinase M2
- ROS
reactive oxygen species
- shRNA
short-hairpin RNA
- SA-β-gal
senescence-associated beta-galactosidase
References
- 1.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 2.Kenner BJ, Chari ST, Cleeter DF, Go VL. Early detection of sporadic pancreatic cancer: strategic map for innovation--a white paper. Pancreas. 2015;44:693–712. doi: 10.1097/MPA.0000000000000369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brentnall TA. Progress in the earlier detection of pancreatic cancer. J. Clin. Oncol. 2016;34:1973–1974. doi: 10.1200/JCO.2016.66.5265. [DOI] [PubMed] [Google Scholar]
- 4.Binenbaum Y, Na’ara S, Gil Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist Updat. 2015;23:55–68. doi: 10.1016/j.drup.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 5.Rohwer N, Cramer T. Hypoxia-mediated drug resistance: novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist Updat. 2011;14:191–201. doi: 10.1016/j.drup.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Yokoi K, Fidler IJ. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin Cancer Res. 2004;10:2299–2306. doi: 10.1158/1078-0432.ccr-03-0488. [DOI] [PubMed] [Google Scholar]
- 7.Akakura N, Kobayashi M, Horiuchi I, Suzuki A, Wang J, Chen J, Niizeki H, Kawamura Ki, Hosokawa M, Asaka M. Constitutive expression of hypoxia-inducible factor-1alpha renders pancreatic cancer cells resistant to apoptosis induced by hypoxia and nutrient deprivation. Cancer Res. 2001;61:6548–6554. [PubMed] [Google Scholar]
- 8.Kasuya K, Tsuchida A, Nagakawa Y, Suzuki M, Abe Y, Itoi T, Serizawa H, Nagao T, Shimazu M, Aoki T. Hypoxia-inducible factor-1α expression and gemcitabine chemotherapy for pancreatic cancer. Oncol Rep. 2011;26:1399–1406. doi: 10.3892/or.2011.1457. [DOI] [PubMed] [Google Scholar]
- 9.Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271:32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- 10.Grasso C, Jansen G, Giovannetti E. Drug resistance in pancreatic cancer: impact of altered energy metabolism. Crit Rev Oncol Hematol. 2017;114:139–152. doi: 10.1016/j.critrevonc.2017.03.026. [DOI] [PubMed] [Google Scholar]
- 11.Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, Keating MJ, Huang P. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005;65:613–621. [PubMed] [Google Scholar]
- 12.Calabretta S, Bielli P, Passacantilli I, Pilozzi E. Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in pancreatic cancer cells. Oncogene. 2016;35:2031–2039. doi: 10.1038/onc.2015.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taniguchi K, Sakai M, Sugito N, Kuranaga Y, Kumazaki M, Shinohara H, Ueda H, Futamura M, Yoshida K, Uchiyama K, Akao Y. PKM1 is involved in resistance to anti-cancer drugs. Biochem Biophys Res Commun. 2016;473:174–180. doi: 10.1016/j.bbrc.2016.03.074. [DOI] [PubMed] [Google Scholar]
- 14.Kim DJ, Park YS, Kang MG, You YM, Jung Y, Koo H, Kim JA, Kim MJ, Hong SM, Lee KB, Jang JJ, Park KC, Yeom YI. Pyruvate kinase isoenzyme M2 is a therapeutic target of gemcitabine-resistant pancreatic cancer cells. Exp Cell Res. 2015;336:119–129. doi: 10.1016/j.yexcr.2015.05.017. [DOI] [PubMed] [Google Scholar]
- 15.Cappello P, Tomaino B, Chiarle R, Ceruti P, Novarino A, Castagnoli C, Migliorini P, Perconti G, Giallongo A, Milella M, Monsurrò V, Barbi S, Scarpa A, Nisticò P, Giovarelli M, Novelli F. An integrated humoral and cellular response is elicited in pancreatic cancer by alpha-enolase, a novel pancreatic ductal adenocarcinoma-associated antigen. Int J Cancer. 2009;125:639–648. doi: 10.1002/ijc.24355. [DOI] [PubMed] [Google Scholar]
- 16.Capello M, Ferri-Borgogno S, Cappello P, Novelli F. α-Enolase: a promising therapeutic and diagnostic tumor target. FEBS J. 2011;278:1064–1074. doi: 10.1111/j.1742-4658.2011.08025.x. [DOI] [PubMed] [Google Scholar]
- 17.Wang L, Liu HL, Li Y, Yuan P. Proteomic analysis of pancreatic intraepithelial neoplasia and pancreatic carcinoma in rat models. World J Gastroenterol. 2011;17:1434–1441. doi: 10.3748/wjg.v17.i11.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peng X, Gong FM, Ren M, Ai P, Wu S, Tang J, Hu X. Proteomic analysis of docetaxel resistance in human nasopharyngeal carcinoma cells using the two-dimensional gel electrophoresis method. Anticancer Drugs. 2016;27:748–755. doi: 10.1097/CAD.0000000000000388. [DOI] [PubMed] [Google Scholar]
- 19.Nishimura K, Tsuchiya Y, Okamoto H, Ijichi K, Gosho M, Fukayama M, Yoshikawa K, Ueda H, Bradford CR, Carey TE, Ogawa T. Identification of chemoresistant factors by protein expression analysis with iTRAQ for head and neck carcinoma. Br J Cancer. 2014;111:799–806. doi: 10.1038/bjc.2014.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qinghong S, Shen G, Lina S, Yueming Z, Xiaoou L, Jianlin W, Chengyan H, Hongjun L, Haifeng Z. Comparative proteomics analysis of differential proteins in respond to doxorubicin resistance in myelogenous leukemia cell lines. Proteome Sci. 2015;13:1–18. doi: 10.1186/s12953-014-0057-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu X, Miao X, Wu Y, Li C, Guo Y, Liu Y, Chen Y, Lu X, Wang Y, He S. ENO1 promotes tumor proliferation and cell adhesion mediated drug resistance (CAM-DR) in non-Hodgkin’s lymphomas. Exp Cell Res. 2015;335:216–223. doi: 10.1016/j.yexcr.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 22.Tu SH, Chang CC, Chen CS, Tam KW, Wang YJ, Lee CH, Lin HW, Cheng TC, Huang CS, Chu JS, Shih NY, Chen LC, Leu SJ, Ho YS, Wu CH. Increased expression of enolase alpha in human breast cancer confers tamoxifen resistance in human breast cancer cells. Breast Cancer Res Treat. 2010;121:539–553. doi: 10.1007/s10549-009-0492-0. [DOI] [PubMed] [Google Scholar]
- 23.Edge SB, Byrd DR, Compton CC. AJCC cancer staging manual. 7th edition. NY, New York: Springer; 2010. [Google Scholar]
- 24.Yin H, Wang L, Liu HL. ENO1 overexpression in pancreatic cancer patients and its clinical and diagnostic significance. Gastroenterol Res Pract. 2018;2018:3842198. doi: 10.1155/2018/3842198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sedoris KC, Thomas SD, Miller DM. Hypoxia induces differential translation of enolase/MBP-1. BMC Cancer. 2010;10:157. doi: 10.1186/1471-2407-10-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao J, Zhao R, Xue Y, Niu Z, Cui K, Yu F, Zhang B, Li S. Role of enolase-1 in response to hypoxia in breast cancer: exploring the mechanisms of action. Oncol Rep. 2013;29:1322–1332. doi: 10.3892/or.2013.2269. [DOI] [PubMed] [Google Scholar]
- 27.Hsiao KC, Shih NY, Fang HL, Huang TS, Kuo CC, Chu PY, Hung YM, Chou SW, Yang YY, Chang GC, Liu KJ. Surface α-enolase promotes extracellular matrix degradation and tumor metastasis and represents a new therapeutic target. PLoS One. 2013;8:e69354. doi: 10.1371/journal.pone.0069354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Principe M, Ceruti P, Shih NY, Chattaragada MS. Targeting of surface alpha-enolase inhibits the invasiveness of pancreatic cancer cells. Oncotarget. 2015;6:11098–11113. doi: 10.18632/oncotarget.3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qian X, Xu W, Xu J, Shi Q, Li J, Weng Y, Jiang Z, Feng L, Wang X, Zhou J, Jin H. Enolase 1 stimulates glycolysis to promote chemoresistance in gastric cancer. Oncotarget. 2017;8:47691–47708. doi: 10.18632/oncotarget.17868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Georges E, Bonneau AM, Prinos P. RNAi-mediated knockdown of α-enolase increases the sensitivity of tumor cells to antitubulin chemotherapeutics. Int J Biochem Mol Biol. 2011;2:303–308. [PMC free article] [PubMed] [Google Scholar]
- 31.Cappello P, Rolla S, Chiarle R, Principe M, Cavallo F, Perconti G, Feo S, Giovarelli M, Novelli F. Vaccination with ENO1 DNA prolongs survival of genetically engineered mice with pancreatic cancer. Gastroenterology. 2013;144:1098–1106. doi: 10.1053/j.gastro.2013.01.020. [DOI] [PubMed] [Google Scholar]
- 32.Ejeskär K, Krona C, Carén H, Zaibak F, Li L, Martinsson T, Ioannou PA. Introduction of in vitro transcribed ENO1 mRNA into neuroblastoma cells induces cell death. BMC Cancer. 2005;5:161. doi: 10.1186/1471-2407-5-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song Y, Luo Q, Long H, Hu Z, Que T, Zhang X, Li Z, Wang G, Yi L, Liu Z, Fang W, Qi S. Correction: alpha-enolase as a potential cancer prognostic marker promotes cell growth, migration, and invasion in glioma. Mol Cancer. 2015;13:235–237. doi: 10.1186/1476-4598-13-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okon IS, Zou MH. Mitochondrial ROS and cancer drug resistance: implications for therapy. Pharmacol Res. 2015;100:170–174. doi: 10.1016/j.phrs.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang L, Li J, Zong L, Chen X, Chen K, Jiang Z, Nan L, Li X, Li W, Shan T, Ma Q, Ma Z. Reactive oxygen species and targeted therapy for pancreatic cancer. Oxid Med Cell Longev. 2016;2016:1616781. doi: 10.1155/2016/1616781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ju HQ, Gocho T, Aguilar M, Wu M, Zhuang ZN, Fu J, Yanaga K, Huang P, Chiao PJ. Mechanisms of overcoming intrinsic resistance to gemcitabine in pancreatic ductal adenocarcinoma through the redox modulation. Mol Cancer Ther. 2015;14:788–798. doi: 10.1158/1535-7163.MCT-14-0420. [DOI] [PubMed] [Google Scholar]
- 37.Chio IIC, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, Wilson J, Sangar V, Hao Y, Öhlund D, Wright K, Filippini D, Lee EJ, Da Silva B, Schoepfer C, Wilkinson JE, Buscaglia JM, DeNicola GM, Tiriac H, Hammell M, Crawford HC, Schmidt EE, Thompson CB, Pappin DJ, Sonenberg N, Tuveson DA. NRF2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer. Cell. 2016;166:963–976. doi: 10.1016/j.cell.2016.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kong B, Qia CJ, Erkan M, Kleeff J, Michalski CW. Overview on how oncogenic Kras promotes pancreatic carcinogenesis by inducing low intracellular ROS levels. Front Physiol. 2013;4:246–257. doi: 10.3389/fphys.2013.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen R, Lai LA, Sullivan Y, Wong M, Wang L, Riddell J, Jung L, Pillarisetty VG, Brentnall TA, Pan S. Disrupting glutamine metabolic pathways to sensitize gemcitabine-resistant pancreatic cancer. Sci Rep. 2017;7:7950. doi: 10.1038/s41598-017-08436-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Serrano-Fujarte I, López-Romero E, Cuéllar-Cruz M. Moonlight-like proteins of the cell wall protect sessile cells of Candida from oxidative stress. Microb Pathog. 2016;90:22–33. doi: 10.1016/j.micpath.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 41.Capello M, Ferri-Borgogno S, Riganti C, Chattaragada MS, Principe M, Roux C, Zhou W, Petricoin EF, Cappello P, Novelli F. Targeting the Warburg effect in cancer cells through ENO1 knockdown rescues oxidative phosphorylation and induces growth arrest. Oncotarget. 2016;7:5598–5612. doi: 10.18632/oncotarget.6798. [DOI] [PMC free article] [PubMed] [Google Scholar]