

Abstract
Pancreatic adenocarcinoma (PDAC) is mostly refractory to immunotherapies. In this issue of Immunity, Li et al. (2018) generate a library of clonal PDAC tumors to examine the tumor-intrinsic features shaping the anti-tumor immune response and find that tumor cell-derived CXCL1 directly blunts T cell infiltration and reduces responsiveness to immunotherapy.
Pancreatic adenocarcinoma (PDAC) represents one of the most aggressive cancer types with no improvements to outcomes over several decades, resulting in a dismal prognosis. While immunotherapies, notably immune checkpoint blockade, have improved survival in numerous cancer types, PDAC is mostly refractory to these interventions. A particular research focus in PDAC has been the fibrotic stroma, a physical barrier to immune infiltration. Little attention has been given to the impact of tumor-cell-intrinsic alterations on anti-tumor immunity, particularly T cell exclusion from the tumor microenvironment (TME). In this issue of Immunity, Li et al. (2018) leveraged intra-tumor heterogeneity to generate a library of clonal PDAC tumors that enabled the examination of tumor-cell-intrinsic alterations associated with responsiveness to immunotherapy (Li et al., 2018). As previously observed for other cancer types, sensitivity to immunotherapy correlated with infiltration of tumor-reactive CD8+ T cells into the TME. However, in contrast to reports in melanoma or ovarian cancer, pancreatic tumor cells were found to actively recruit immune suppressive cell types to blunt anti-tumor immune responses in a dominant fashion.
Immune checkpoint blockade (ICB) therapies have changed the treatment landscape for many cancers. When effective, patients observe long-term survival, mediated by the induction of a tumor-specific memory T cell response. However, only 20%–40%of patients exhibit significant responses to ICB, even when considering cancer types such as melanoma that are associated with increased responsiveness to ICB. Clinical studies have provided strong evidence that a pre-existing anti-tumor immune response, characterized by CD8+ T cell infiltration, is required for therapeutic benefit from ICB (Gandhi et al., 2018; Tumeh et al., 2014); these tumors are referred to as T cell inflamed. Understanding the mechanisms that underlie poor T cell infiltration into the tumor are therefore key to develop rational treatment strategies for cancers that do not respond to ICB.
In cancer types such as melanoma or lung cancer, identification of such mechanisms has been achieved by comparing human patient samples of responders and non-responders following checkpoint blockade therapy (Koyama et al., 2016; Spranger et al., 2015). However, the dismal response rates to immunotherapy observed in PDAC make this approach unfeasible. Therefore, Li et al. (2018) set out to investigate whether intra-tumor heterogeneity in PDAC could be used to identify tumor cell-intrinsic alterations associated with response to immunotherapy.
The authors used murine PDAC cell line isolated from a mouse model of pancreatic cancer (PDX-Cre:KrasLSLG12D: p53LSLR172H; abbreviated KPC), which they sub-cloned and screened in vivo for a T-cell-inflamed (7/17) and non-T-cell-inflamed phenotype (8/17). These diverse clones, derived from the same parental cell line, suggest a high degree of intra-tumor heterogeneity. T cell infiltration was also examined in human PDAC samples, confirming that a T-cell-inflamed and non-T-cell-inflamed phenotype can also be observed in patients. T cell infiltration was found to be predominantly driven by CD8+ T cells, consistent with previous reports of the T-cell-inflamed TME from other cancer types (Spranger et al., 2015). The authors subsequently excluded neo-antigens, microbiota, or tissue microenvironment as potential drivers of T cell exclusion, suggesting that tumor-cell-intrinsic pathways were mediating T cell exclusion. Indeed, alterations in signaling pathways have been reported to be associated with the non-T-cell-inflamed TME in melanoma, lung cancer, and ovarian cancer (Peng et al., 2015; Spranger et al., 2015; Spranger et al., 2017). It is worth pointing out that tumor growth was not directly affected by the degree of T cell infiltration, recapitulating results in other cancer models. However, metastatic potential was higher in mice bearing non-T-cell-inflamed tumors compared to T-cell-inflamed counterparts. This observation highlights that immune evasion mediated by tumor-cell-intrinsic alterations might affect the immune response in established tumor lesions, as well as on small, newly formed metastatic lesions.
To understand whether the T cell infiltrate is tumor-reactive, the authors examined the T cell infiltrate in T-cell-inflamed and non-T-cell-inflamed PDAC tumors. Markers for T cell dysfunction, including the increased expression of PD-1, Lag-3, CTLA-4, and Tim-3 (Wherry, 2011) were detected on T cells isolated from the T-cell-inflamed but no activation markers were detected on the few T cells isolated from non-T-cell-inflamed tumors. Consistently, when exposed to combination immunotherapy (gemcitabine, nabpaclitaxel, anti-CD40 mAb, anti-CTLA-4 mAb, and anti-PD-1 mAb), T-cell-inflamed tumors regressed, whereas non-T-cell-inflamed tumors did not. Notably, complete tumor regression was also observed when the therapy was reduced to anti-CD40 mAb, anti-CTLA-4 mAb, and anti-PD-1 mAb, highlighting the potential of ICB as a stand-alone therapeutic approach. The authors also found a strong association between the lack of T cell infiltration, lack of DC1 dendritic cells accumulation, and increase in suppressive myeloid cell populations.
To further understand whether T cell activation of tumor-infiltration T cells is required for response to immunotherapy, the authors turned to two tumor cell clones (7160c5 and 6422c5) that had an intermediate degree of T cell infiltration and similar numbers of tumor-infiltrating T cells. However, T cells from these tumors exhibited drastically different activation states: T cells isolated from 7160c5 displayed an activated phenotype, whereas those isolated from 6422c5 did not. As observed in the T-cell-inflamed tumors, T cell activation was accompanied by expression of surface molecules associated with T cell dysfunction in 7160c5. Consistent with the notion that only tumor-reactive T cells are predictive for response toward immunotherapy, 7160c5 responded to immunotherapy while 6422c5 did not. Notably, 6422c5 had high numbers of myeloid cells and low numbers of cross-presenting dendritic cells, similar to non-T-cell-inflamed KPC clones. This skewing of the antigen-presenting cell compartment with strong reduction in cross-presenting dendritic cells suggests that deficiencies T cell priming might contribute to the establishment of non-T-cell-inflamed tumors.
The presence of tumor-reactive T cells and their requirement for sensitivity toward immunotherapy suggested that tumor rejection could induce a protective anti-tumor memory response. Indeed, mice that exhibited complete responses following combination immunotherapy were capable of controlling the tumor if inoculated with the same tumor cell clone but also with a different clone. Interestingly, memory T cell responses elicited treatment of T-cell-inflamed tumors with ICB protected mice upon inoculation with other PDAC tumors, both T-cell-inflamed and non-T-cell-inflamed tumor cell clones. This observation suggests that tumor-associated self-antigens, shared between all tumor cell clones, are sufficient to mediate a productive anti-tumor immune response. This is of particular interest as the majority of PDAC patients have low numbers of non-synonymous mutations and thus likely express few neo-antigens. These results also suggest that the kinetics of memory T cell recruitment to tumors can supersede suppressor cell accumulation. This is in contrast to previous work that found that lack of DC1 DC recruitment was sufficient to circumvent memory T cell-mediated rejection of second tumors (Spranger et al., 2017). Further, these results support the above-stated notion that the non-T-cell-inflamed TME is partly mediated by failed T cell activation.
Skewing within the tumor-associated innate immune cell compartment can result in non-T-cell-inflamed tumors in melanoma and lung cancer (Koyama et al., 2016; Spranger et al., 2017). In melanoma, T cell exclusion was found to be dependent on failed infiltration of DC1 DCs, blunting T cell priming and effector T cell recruitment in a CXCL9/10-dependent fashion (Spranger et al., 2017). In lung cancer, increased neutrophil infiltration into lung cancer lesions was sufficient for the establishment of a non-T-cell-inflamed TME (Koyama et al., 2016). Li et al. (2018) found that T cell infiltration in PDAC tumors was strongly associated with the presence of intratumoral DC1 DC. Furthermore, genetic ablation of DC1 DC using BATF3−/− mice or blockade of CXCL9/10 signaling, using a blocking antibody against the receptor CXCR3, resulted in significant reduction in T cell infiltration. Surprisingly, this reduction in T cell infiltration had only modest effects on the response to immunotherapy, suggesting that relatively few tumor-infiltrating T cells can mediate an effective anti-tumor response, if these are tumor-antigen-specific T cells.
To explore whether the observed increase in myeloid cells is causative for the non-T-cell-inflamed TME, the authors implanted a mixture of T-cell-inflamed and a non-T-cell-inflamed tumor cell clones in a single site. The non-T-cell-inflamed tumor cell clone dominated the TME, characterized by poor T cell infiltration and high myeloid cell content. Transcriptional analysis of these tumors revealed increased expression of the chemokine CXCL1 in non-T cell-inflamed tumor cell clones. The authors go on to show that the promoter region of Cxcl1 was enriched in H3K4me3 histone marks in non-T-cell-inflamed tumor cell clones, rendering the locus accessible for transcription factors, in particular c-myc. C-myc is a transcription factor binding to E-boxes and was found to be of higher abundance in non-T-cell-inflamed tumors. The authors were able to show that both c-myc and epigenetic modulation of the locus can result in increased expression of CXCL1. Genetic deletion of CXCL1 from non-T-cell-inflamed clones resulted not only in establishment of a T-cell-inflamed phenotype but also sensitized these clone toward immunotherapeutic interventions. These observations support the notion that tumor-cell-derived chemokines are affected by tumor-cell-intrinsic alterations and are highly effective modulators on the tumor-immune microenvironment.
C-myc signaling has been associated with reduced T cell infiltration, mediated through direct inhibition of phagocytosis by macrophages (Casey et al., 2016). Thus, it should be addressed whether c-myc-dependent CXCL1 expression is a more generalizable method of immune suppression. Epigenetic alterations have also previously been linked to lack of T cell infiltration. Epigenetic silencing of CXCL9 and 10 was reported to mediate a non-T-cell-inflamed TME in ovarian cancer (Peng et al., 2015). This observation suggests the yin-yang like qualities of chemokines: they attract T cells but also mediate T cell exclusion through recruitment of T-cell-suppressing myeloid cells (see Figure 1). Understanding the molecular mechanism mediating the establishment of epigenetic marks specifically on the regulatory region of chemokines could open up new therapeutic avenues for directly targeting chemokine production by tumor cells.
Figure 1. The Chemokine Yin-Yang Mediating T-Cell-Inflamed and Non-T-Cell-Inflamed Tumors.
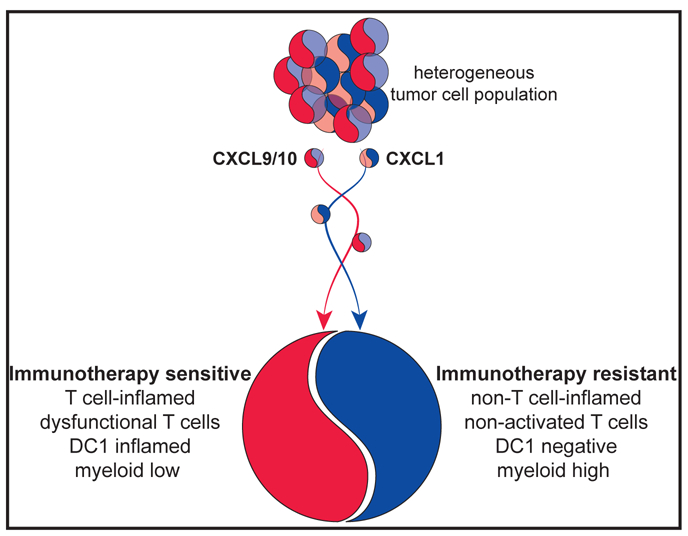
A heterogeneous tumor cell population comprises both tumor cell clones capable of producing myeloid-attracting chemokines (predominantly blue) and T-cell-attracting chemokines (predominantly red). Based on the expression level of both chemokine groups, the cancer cells will mediate a T-cell-inflamed (red) and non-T-cell-inflamed tumors (blue).
Li et al. (2018) have provided strong evidence that CXCL1-mediated recruitment of myeloid cells is directly associated with a non-T-cell-inflamed TME in PDAC. This phenotype is further associated with reduced infiltration of cross-presenting DC, additionally suggesting that chemokine-mediated recruitment of this cell type could also be affected by CXCL1 production or immune-suppressive myeloid cells. Obtaining a comprehensive picture of tumor-cell-intrinsic epigenetic regulation of chemokine production and the direct and indirect effects on the tumor immune environment in a cancer type-specific fashion might be highly informative for our understanding of anti-tumor immunity and evasion.
REFERENCES
- Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gutgemann I, Eilers M, and Felsher DW (2016). MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, Clingan P, Hochmair MJ, Powell SF, et al. ; KEYNOTE-189 Investigators (2018). Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 378, 2078–2092. [DOI] [PubMed] [Google Scholar]
- Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, Buczkowski KA, Liu Y, Awad MM, Denning WL, et al. (2016). STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res. 76, 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, Richman LP, Lin JH, Sun YH, Rech AJ, et al. (2018). Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 49, this issue, 178–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama S, Putzel GG, Rakhilin N, et al. (2017). The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 549, 282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W, et al. (2015). Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Bao R, and Gajewski TF (2015). Melanoma-intrinsic b-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235. [DOI] [PubMed] [Google Scholar]
- Spranger S, Dai D, Horton B, and Gajewski TF (2017). Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer cell 31, 711–723 e714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui P, Wiesner DL, Xu J, Zhang Y, Lee J, Van Dyken S, Lashua A, Yu C, Klein BS, Locksley RM, et al. (2018). Pulmonary neuroendocrine cells amplify allergic asthma responses. Science 360, eaan8546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shin-taku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Fernandes H, and Artis D (2018). Neuronal-immune system cross-talk in homeostasis. Science 359, 1465–1466. [DOI] [PubMed] [Google Scholar]
- Veiga-Fernandes H, and Pachnis V (2017). Neuroimmune regulation during intestinal development and homeostasis. Nat. Immunol. 18, 116–122. [DOI] [PubMed] [Google Scholar]
- Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour RE, Nyman J, Dionne D, Hofree M,Cuoco MS, Rodman C, Farouq D, et al. (2017). The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 549, 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ (2011). T cell exhaustion. Nat. Immunol. 12, 492–499. [DOI] [PubMed] [Google Scholar]
- Xiang YY, Wang S, Liu M, Hirota JA, Li J, Ju W, Fan Y, Kelly MM, Ye B, Orser B, et al. (2007). A GABAergic system in airway epithelium is essential for mucus overproduction in asthma. Nat. Med. 13, 862–867. [DOI] [PubMed] [Google Scholar]