

Abstract
The reduction-oxidation state of NAD+/NADH is critical for cellular health with NAD+ and its metabolites playing critical roles in aging and pathologies. Given the inherent autooxidation of reduced dinucleotides (i.e. NADH/NADPH), and the well-established differential stability, the accurate measurement of NAD+ and its metabolites is technically challenging. Moreover, sample processing, normalization and measurement strategies can profoundly alter results. Here we developed a rapid and sensitive liquid chromatography mass spectrometry-based method to quantify the NAD+ metabolome with careful consideration of these intrinsic chemical instabilities. Utilizing this method we assess NAD+ metabolite stabilities and determine the presence and concentrations of NAD+ metabolites in clinically relevant human samples including cerebrospinal fluid, erythrocytes, and primate skeletal muscle.
Keywords: NAD+ /NADH metabolism, NAD+ metabolome, LC-MS/MS, Metabolomics
1. Introduction
Nicotinamide adenine dinucleotide (NAD+) metabolism is a critical mediator of metabolic pathways that are implicated in the aging process and age-related pathologies. NAD+ declines with age in numerous tissues in rodent laboratory models and in humans [1]. Increasing the cellular NAD+ concentration via the administration of NAD+ precursors such as nicotinamide mononucleotide (NMN) or nicotinamide riboside (NR) effectively ameliorates dysfunction in numerous rodent models of age-related diseases [1,2]. NR administration increases blood NAD+ levels in humans [3] and had few if any side effects in a small randomized double-blind placebo clinical trial of middle-aged adults (56–79 years old) [4] or in a randomized placebo-controlled, double-blind and parallel-group designed clinical trial with forty healthy sedentary men with a daily dose of 2g/day [5]. Accurate assessment of NAD+ metabolites in multiple tissues has become important as more randomized controlled trials and observational studies are designed and conducted to facilitate our understanding of its complex biology in health and disease.
There are three pathways of NAD+ synthesis in biological systems including the de novo, Preiss-Handler, and salvage pathways (Scheme 1) [6,7]. The de novo synthesis of NAD+ begins with the conversion of dietary tryptophan to quinolinic acid (QA) by Indoleamine 2,3-dioxygenase (IDO). QA is converted to nicotinic acid mononucleotide (NAMN) by quinolinate phosphoribosyl transferase (QPRT). NAMN can also be synthesized via nicotinic acid riboside (NAR) by nicotinamide riboside kinases 1 and 2 (NRK1/2) or indirectly via nicotinic acid (NA) by nicotinate phosphoribosyltransferase (NAPRT). NAMN is then adenylylated to NAAD via nicotinamide mononucleotide adenyl-transferases 1–3 (NMNATs) before NAD+ synthesis via NADsynthetase (NADS). The predominant NAD+ synthesis pathway is the salvage pathway, where NAD+ catalysis by numerous enzymes (i.e. PARPs, SIRTs, CD38) yields nicotinamide (NAM), which is recycled to NMN by nicotinamide ribosyltransferase (NAMPT) and adenylylated to NAD+ by NMNATs. Importantly, NAMPT is considered the rate-limiting step of the salvage pathway and is implicated in the age-dependent decline of NAD+ [8]. Thus, bypassing NAMPT for NAD+ synthesis with precursors NMN or NR is an important therapeutic strategy to restore cellular NAD+.
Scheme 1. NAD+ Biosynthesis Pathways.
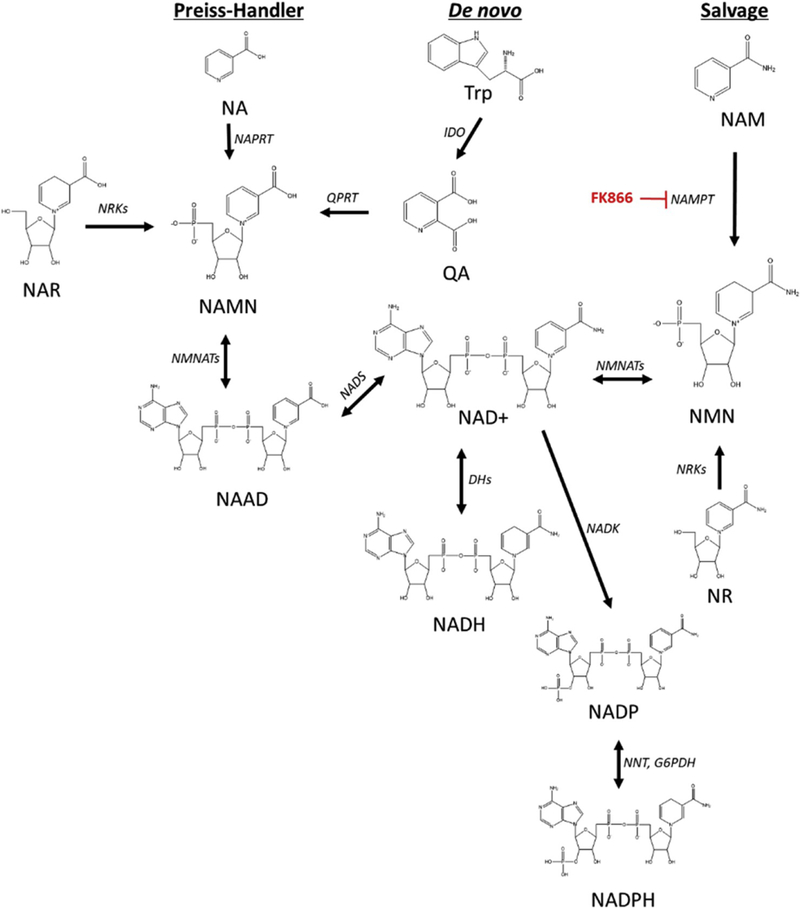
Three main pathways of NAD+ metabolite biosynthesis are the Preiss-Handler, de novo, and salvage pathways [6,7]. Chemical structures and synthesis enzymes (italics) for each metabolite are depicted. Nicotinic acid (NA), Quinolinic acid (QA), NA mononucleotide (NAMN), NA adenine dinucleotide (NAAD), NA riboside (NAR), nicotinamide adenine dinucleotide (NAD+), NAD+, reduced (NADH), NADphosphate, oxidized (NADP), NADphosphate, reduced (NADPH), nicotinamide mononucleotide (NMN), nicotinamide (NAM). Quinolinate phosphoribosyl transferase (QPRT), nicotinate phosphoribosyltransferase (NAPRT), nicotinamide riboside kinases (NRKs), Dehydrogenases (DHs), nicotinamide mononucleotide adenyl-transferases 1–3 (NMNATs), NADsynthetase (NADS), Indoleamine 2,3-dioxygenase (IDO), NADkinase (NADK), Nicotinamid riboside (NR), nicotinamide phosphoribosyl transferase (NAMPT), NAD(P) transhydrogenase (NNT), FK866 is a NAMPT inhibitor.
Multiple liquid chromatography tandem mass spectrometry (LC-MS/MS) methods have been developed recently for the assessment of NAD+ metabolites in whole blood [9], cells [10] and mouse tissues [11]. However, these methods have long run times [9,10], complicated sample preparation [9–11], and in some cases lack internal standards [11]. Herein, we report the development of a rapid LC-MS/MS method for the determination of NAD+ and its metabolites with a simple, optimized sample handling procedure and its application to multiple biological matrices including cultured cells, human erythrocytes (red blood cells (RBCs)), cerebrospinal fluid (CSF) and primate skeletal muscle.
2. Materials and methods
2.1. Materials
Ammonium acetate, sodium chloride (NaCl), β-nicotinamide adenine dinucleotide hydrate (NAD+), β-nicotinamide adenine dinucleo tide, reduced disodium salt hydrate (NADH), nicotinic acid adenine dinucleotide sodium salt (NAAD), β-nicotinamide adenine dinucleotide phosphate reduced tetrasodium (NADPH), Adenosine 5′ diphosphoribose (ADPR), nicotinic acid mononucleotide (NAMN), nicotinamide mononucleotide (NMN), nicotinamide (NAM), β-nicotinamide adenine dinucleotide phosphate (NADP), β-nicotinamide adenine dinucleotide phosphate reduced tetrasodium (NADPH), FK-866 were obtained from Sigma-Aldrich (St. Louis, MO, USA or Munich, Germany), de-ionized water was obtained from a Milli-Q system (Millipore, Molsheim, France). Nicotinamide Riboside was obtained from Chromadex (Irvine, CA). All other chemicals used were of analytical grade. Metabolite Yeast Extract (U-13C, 98%)(Cat No. ISO1) was purchased from Cambridge isotope Laboratories, Inc.Red blood cells from Baltimore longitudinal study of aging (BLSA) participants were collected in the morning after an overnight fast, where 5 ml of venous blood was collected into a vacutainer tube containing EDTA, centrifuged at 2700×g for 30 min within 2 h. The plasma was removed and collected and the buffy coat was discarded. The RBCs were washed three times with PBS, mixed gently and centrifuged at 2700×g for 4 min. The RBCs were collected and subsequently snap frozen on dry ice and stored at –80 °C. Skeletal muscle samples were collected from rhesus monkeys (Macaca mulatta) housed continuously at the NIH Animal Center, Poolesville, MD. Biopsies of the vastus lateralis were obtained from anesthetized monkeys from ongoing NIA studies and flash frozen. All animal procedures for this study were reviewed and approved by the Animal Care and Use Committee (ACUC) at the Biomedical Research Center (NIA/NIH). 10 mL of human cerebrospinal fluid (pooled gender) was purchased from BioIVT (Westbury, NY).
2.2. Internal standard
Dry extract of > 2 × 109 Pichia pastoris cells (~15mg), from Cambridge Isotope Laboratories (Metabolite Yeast Extract (U-13C, 98%)) was dissolved in 2mL of deoxygenated buffered ethanol (75% ethanol/25% 1 mM HEPES, pH 7.1). The resulting solution was dispensed in 50 μl aliquots and snap-frozen and stored at –80 °C.
The internal standard was prepared on the day of the experiment, where a previously aliquoted 50 μl of internal standard was diluted to 300 μl with a deoxygenated buffered ethanol solution, or to 150 μl with methanol for the CSF.
2.3. Cell culture and sample preparation
HEK-293T cells were cultured (37 °C, 5% CO2) in Dulbecco’s modified Eagle’s medium (DMEM) supplemented 10% (v/v) with fetal bovine serum (FBS) and antibiotics (100 units ml–1 penicillin, 0.1 mg ml–1 streptomycin) (GIBCO). Cells were washed in ice cold PBS and were then trypsinized dissociated and resuspended in cold PBS. An aliquot was removed for cell counting and the remainder of the cells were pelleted by centrifugation at 4°C. PBS was aspirated, and cells were snap-frozen in liquid nitrogen before extraction for LC-MS/MS.
1 × 106 HEK-293T cell pellets were solubilized in 140 μl of preheated (80°C) buffered ethanol solution (3:1) (ethanol:HEPES [1mM, pH 7.1]) or deoxygenated buffered ethanol or 80% methanol and 10 μl of internal standard mixed on a thermomixer at 1000 rpm for 3 min at 80 °C. Samples were then centrifuged at 4 °C for 10 min at 13,200×g to remove the protein pellet. The supernatant was collected and placed in an autosampler vial for analysis.
2.3.1. Cell treatment with NAMPT inhibitor FK866 or nicotinamide riboside (NR)
HEK293T cells cultured as detailed above were treated with either nicotinamide riboside (1mM, 24 h) or NAMPT inhibitor FK866 (100 nM, 24 h). 1×106 cells were used for analysis of each sample (n = 5 independent cell culture experiments).
2.4. Sample preparation of RBC’s, biological fluids and skeletal muscle
2.4.1. RBC’s, plasma and serum
20 μl of plasma-EDTA, serum or 15 μl RBCs were solubilized in 120 or 125 μl, respectively, of preheated (80°C) deoxygenated buffered ethanol solution (3:1) (ethanol:HEPES [1 mM, pH 7.1]) including 10 μl of internal standard and mixed on a thermomixer for 3 min at 80 °C. Samples were then centrifuged at 4°C for 10 min at 13,200×g to remove the protein pellet. The supernatant was collected and placed in an autosampler vial for analysis. Three quality controls with spiked standards was used for determination of the relative concentrations of NAD+ and its metabolites.
2.4.2. CSF
20 μl of CSF was solubilized in 60 μl of methanol, including the addition of 5 μl of internal standard. Samples were then centrifuged at 4 °C for 10 min at 13,200×g to remove the protein pellet. The supernatant was collected and placed in an autosampler vial for analysis. Three quality controls with spiked standards was used for determination of the relative concentrations of NAD+ and its metabolites.
2.4.3. Skeletal muscle
7 mg of non-human primate skeletal musclewas suspended in 140 μl of buffered ethanol (3:1) (ethanol/HEPES buffer [1 mM, pH 7.1]) (1:20 ratio) and 10 μl internal standard. The resulting solution was added to Precellys® soft tissue homogenization tubes and snap frozen in liquid nitrogen. Samples were homogenized at 6500 RPM for 2 × 20 s in a liquid nitrogen cooled Precellys 24. The samples were mixed on a thermomixer for 3 min at 80 °C. Samples were then centrifuged at 17000×g for 15 min at 4 °C and the supernatant transferred for LC/MS injection. A quality control with spiked standards was used for determination of the relative concentrations of NAD+ and its metabolites.
2.5. LC-MS/MS method for NADmetabolites
Separation of the NAD+ metabolites was accomplished using a C-18 Security guard cartridge (2.1 × 4 mm) and an Accucore HILIC column (2.1 × 150 mm, 2.6 μm, Thermo) at 32 °C. Mobile phase A consisted of ammonium acetate [7.5 mM, pH 7.86] and mobile phase B was acetonitrile. The following linear gradient was run for 14.0 min at a flow rate of 0.4 ml/min: 0–1 min 90% B, 1.5 min 72.5% B, 2.5 min 67.5% B, 8.0 min 20% B, 10 min 20% B, 10.1 min 90% B. Calibration curves were prepared in buffered ethanol by a 0.5 serial dilution of standards from 20 μg/ml to 0.156 μg/ml for NAD+; 10 μg/ml to 0.078 μg/ml for NADP; 8 μg/ml to 0.0625 μ/ml for NADPH; 5 μg/ml to 0.039 μg/ml for AcCoA; 4 μg/ml to 0.03125 μg/ml for NADH; 2.5 μg/ml to 0.019 μg/ml for NR; 2 μg/ml to 0.015625 μg/ml for NAAD, NMN, NAMN, ADPR; 1 μg/ml to 0.0078 μg/ml NAMN, NA. The injection volume per sample was 10 μL. Samples were kept at 4 °C in the autosampler tray prior to injection.
Relative values for the metabolites were determined using area ratios of the targeted metabolites and the corresponding internal standard using the following heavy standards: C13-NAD, C13-NADH, C13-ADPR, C13-AcCoA (Suppl Table 1b). Calibration curves were carried out in standard solutions. Matrix effects were accounted for in each targeted matrix by adjusting calculated levels based on three quality controls (low, middle, high). All studies were carried out with a daily calibration curve and quality controls as well as a pooled sample from the entire study for each matrix.
Data were acquired using a Nexera XR HPLC (Shimadzu) coupled with a QTRAP 5500 (SCIEX) and were analyzed with Analyst 1.6 (SCIEX). The positive ion mode data was obtained using multiple reaction monitoring (MRM). The instrumental source setting for curtain gas, ion spray voltage, temperature, ion source gas 1, ion source gas 2 were 30 psi, 5500 V, 500 C, 65 psi, and 55 psi, respectively. The collision activated dissociation was set to medium and the entrance potential was 10 V. The standards (Supplemental Table 1A) and internal standards (Supplemental Table 1B) were characterized using the MRM ion transitions, declustering potentials (DP), collision energies (CE), and collision cell exit potentials (CXP) listed in Supplemental Table 1.
3. Results and discussion
3.1. LC-MS/MS method for NAD+ metabolites
The increasing number of publications on the development of chromatographic methods for the assessment of several NAD+ metabolites from the salvage and de novo pathways reflect the growing interest in understanding the role of NAD+ metabolic pathways in aging and age-related chronic diseases [9–12]. In the published methods, many NAD+ metabolites have been resolved and their concentrations determined, however, to date, none of the methods have reported on the stability of NAD(H) and NADP(H). It has been well established that NAD(P)H is unstable in acidic environments, while NAD(P) is less stable under alkaline conditions [13], and that NAD(P)H is prone to autooxidation. As time, pH and temperature are critical mediators of NAD+ metabolome stability, a rapid assessment of metabolites is critical for accurate measurement of the NAD+ metabolome. As a result, a novel LC-MS/MS method was developed using an Accucore HILIC column with a linear gradient with a total run time of 14 min (Fig. 1), less than half the run time of other currently available methods [9–11]. In addition, the sample preparation does not require drying steps (stream drying [10,11] or speed vac [9]), increasing the stability of the NAD+ metabolites [14,15]. The Internal Standard (IS) used in this study was yeast cultured in C13-glucose, similar to previous studies [9]. However, in this case, the IS of isotopically labeled NAD+ metabolites was commercially available from Cambridge isotope Laboratories. In our study, C13–labeled NAD+, NADH, ADPR and AcCoA were observed and their multiple reaction monitoring (MRM) transitions are reported in Table 1B. Since not all isotopic labels were observed, the C13–metabolite with the strongest correlation coefficient for a given metabolite was used as the internal standard.
Fig. 1.
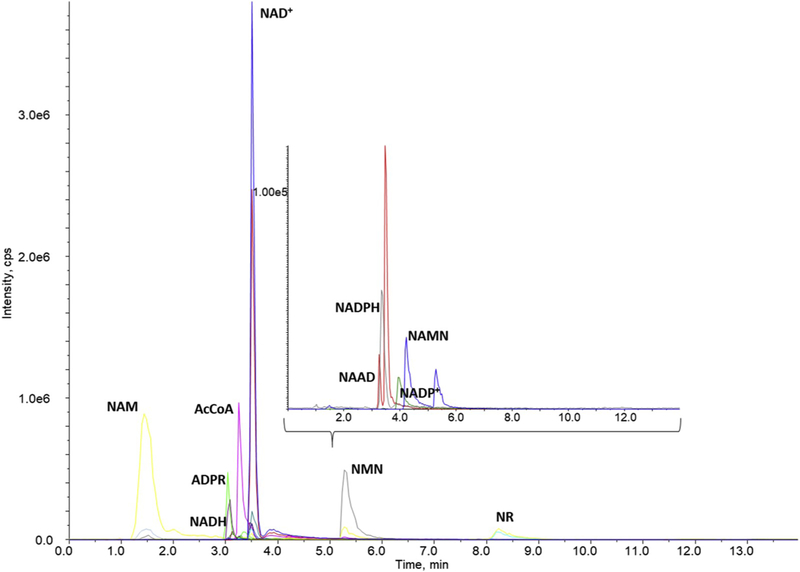
Separation of the NAD+ metabolites in standard solution using an Accucore HILIC column (2.1 × 150 mm, 2.6 μm, Thermo) at 32 °C with a chromatographic run time of 14 min. Mobile phase A consisted of 7.5 mM ammonium acetate with 1.54 mM ammonium hydroxide and mobile phase B was acetonitrile. The chromatogram is presented with intensity in cps (y-axis) and time (min) on the x-axis. The inset represents an expanded view of the peaks observed below 1.0e5 (y-axis) in a similar timescale.
Table 1.
Calculated concentrations of metabolites in the NAD+ metabolome in multiple matrices: HEK-293 cells, human red blood cells, non-human primate skeletal muscle and human cerebrospinal fluid.
| Cells (μM) | RBC (μM) | Muscle (nmol/g) | CSF (μM) | |
|---|---|---|---|---|
| NAD | 104.65 ± 13.35 | 25.40 ± 3.88 | 226.8 ± 4.3 | 0.072 ± 0.008 |
| NADH | 9.30 ± 2.07 | 3.18 ± 0.22 | 45.2 ± 3.1 | BQ |
| ADPR | 3.17 ± 0.14 | 3.11 ± 0.36 | 29.2 ± 11.8 | BQ |
| NADP | 27.01 ± 7.42 | 5.57 ± 0.59 | 3.76 ± 0.71 | ND |
| NADPH | 23.25 ± 3.73 | 5.50 ± 0.67 | BQ | ND |
| NAAD | BQ | BQ | 2.39 ± 1.00 | ND |
| NMN | 1.13 ± 0.32 | 0.81 ± 0.10 | 0.42 ± 0.05 | 0.081 ± 0.001 |
| NAMN | 0.61 ± 0.15 | 0.52 ± 0.12 | BQ | ND |
| NR | BQ | 0.37 ± 0.06 | BQ | BQ |
| NAM | 10.03 ± 1.32 | 14.39 ± 0.72 | 68.1 ± 8.9 | ND |
| AcCoA | 23.32 ± 4.03 | 1.31 ± 0.06 | 1.25 ± 0.47 | BQ |
The baseline chromatographic resolution of the NAD+ metabolites was necessary for several of the metabolites because certain metabolites were observed in multiple MRM transitions. For example, NAD+ is observed in the NAAD transition (Suppl. Figure 1), and NAM is observed in the transition of NA. In both cases, a baseline resolution is necessary for the accurate measurement of endogenous metabolites. While in our study NAAD and NAD+ were resolved, baseline resolution was not achieved with nicotinic acid (NA) and nicotinamide (NAM). Further, picolinic acid, an isomer of nicotinic acid with the carboxylic acid in the 2-position as opposed to the 3-position of the pyridine ring, also co-eluted with NA, therefore NA was not reported in our study.
3.2. Changes in the NAD+ metabolome with FK866 and NR treatment
The developed method was applied to HEK-293T cells, where it was demonstrated that 5×105 cells were sufficient to enable the identification of all targeted NAD+ metabolites, with the exception of NR and NAAD, which are known to be present at very low cellular concentrations [9]. We also determined whether the modulation of the NAD+ metabolome would result in the expected changes in NAD+ and/or its metabolites. Modulation of any of the pathways outlined in Scheme 1, should result in changes in NAD+ and/or NAD+ metabolites. For example, the addition of an NAMPT inhibitor should decrease the recycling of NAM and subsequently NAD+ and downstream NAD+ metabolites, as NAMPT is a rate-limiting step in the salvage pathway [8], which accounts for approximately 80% of NAD+ synthesis [16]. We used FK-866, a known specific NAMPT inhibitor with nanomolar affinity against NAMPT. As expected, the incubation of HEK-293T cells with 100 nM of FK866 for 24 h, resulted in a significant decrease in nearly all NAD+ metabolites (Fig. 2). These alterations demonstrate the importance of the salvage pathway for a multitude of NAD+ metabolites which play diverse roles in cell signaling and metabolism. Conversely, the addition of 1 mM NR to the cells for 24 h resulted in significant increases in several metabolites, including a two-fold increase in NAD+(Fig. 2), a ~30% increase in NADP and a ten-fold increase in NR.
Fig. 2.
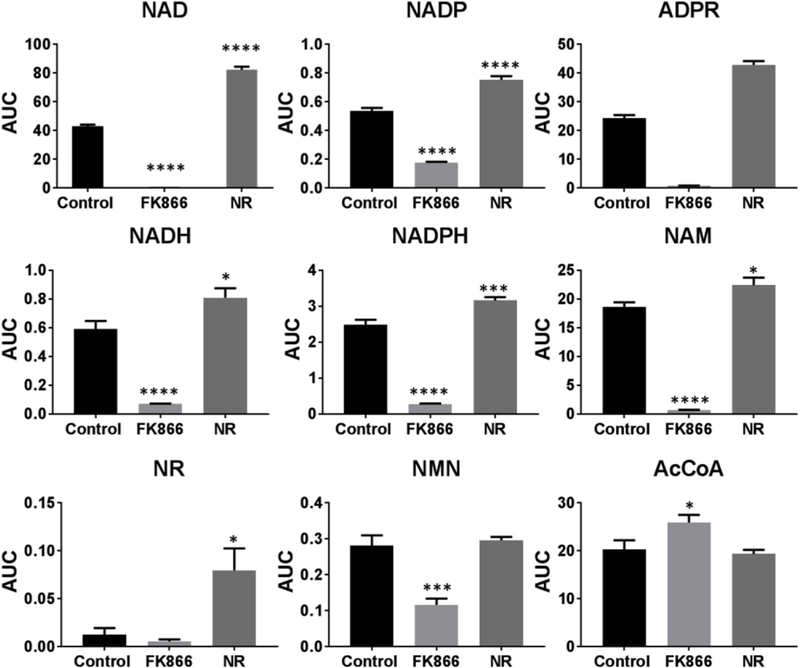
Changes in the area under the curve (AUC) of NAD+ and its metabolites in HEK293T cells alone (control)with 100 nM of FK866 (FK-866) orl mM NR (NR) for 24 h.
3.3. NAD+ metabolite stability
The autosampler stability (4 °C) of endogenous NAD+ metabolites was assessed over 24 h using multiple extraction buffers including buffered ethanol, deoxygenated buffered ethanol and 80% methanol in HEK-293T cells. Initial analysis demonstrated that signal intensities were overall similar between the three buffers, with deoxygenated buffered ethanol having a slight increase in signal intensity for 6 of the 11 metabolites, and methanol having increased signal intensity for NAD+, with the remaining 4 metabolites (NADH, NADP, NR and AcCOA) being relatively equivalent across all buffers. The majority of the metabolites targeted were stable over time, however, NADH was only stable for 18 h. Comparing the three different extraction buffers for stability, we found that while deoxygenated buffered ethanol offered some improvement on the NADH stability over time compared to buffered ethanol in cells, the stability was similar to that observed with methanol. Improvements in the stability of NADH were attempted with changes in pH and ionic strength (potassium chloride or sodium chloride) (data not shown), however, stability was limited to 18 h postextraction in all cases. As a result, all remaining studies were carried out in deoxygenated buffered ethanol, with the exception of CSF which was carried out in methanol as improved signal intensity was observed in CSF, and samples were analyzed within 16 h of sample preparation.The propensity for NADH autooxidation emphasizes the requirement of a rapid method for accurate measurements.
3.4. Application to multiple matrices
Due to the potential clinical importance of NAD+ replenishment, the developed method was applied to clinically relevant matrices, including plasma, human red blood cells (RBCs), cerebrospinal fluid (CSF) and non-human primate skeletal muscle tissue. In plasma, NAD+, NAM, ADPR and AcCoA were above the detection limit of our method, while NADH, NMN, NADP and NADPH were at or below detection, similar to a previously published report [3]. In contrast with plasma, several studies have reported quantitative levels of NAD+ metabolites in whole blood [9], however in RBCs only the NAD+ concentration has been previously reported [17].The intra-day precision of the method was determined for each matrix (Suppl Table 2). The overall precision averaged to 8.0% for the HEK-293T cells and RBCs and 5% for CSF matrix, with the majority of quality controls falling within 10% of expected values.
In order to determine whether sample processing resulted in changes in the stability of the NAD+ metabolites, benchtop stability was assessed for each matrix, by placing the sample at room temperature for the indicated times before extraction. In the cellular matrix (Fig. 3A), NADH and NADPH levels decreased rapidly within 30 min, as expected, however, surprisingly NAD+ and ADPR were only stable at room temperature for 10 min, with greater than 80% decrease within 30 min. Conversely, NAM and NMN levels increased after 1 h, consistent with the degradation observed with NAD+ and other higher molecular weight metabolites. Similar results were observed with RBCs (Fig. 3B) and skeletal muscle (Fig. 3C), with the exception that NMN levels decreased in these matrices. Intriguingly, NADP+ was stable for 60 min in both RBCs and HEK-293T cells. Distinct from the other matrices, in the CSF, only two metabolites were in the linear range (NAD+ and NMN). NAD+ had a less pronounced decrease over an hour relative to the other matrices, while NMN had similar decreases (> 80%) (Fig. 3D). The increased stability of NAD+ in CSF may be due to the already low levels of the metabolite in CSF, the rapid drop of metabolite stability after a single freeze-thaw cycle or the relative lack of NAD+ consuming enzymes present in other matrices (e.g. PARPs and sirtuins). It should be noted, that the RBC and CSF samples were a pooled gender sample, and therefore the results need to be validated in a larger cohort of well-characterized people in future studies. These observations suggest that it is imperative to optimize sample collection and extraction procedures to ensure the preservation of endogenous levels of NAD+ and related metabolites.
Fig. 3.
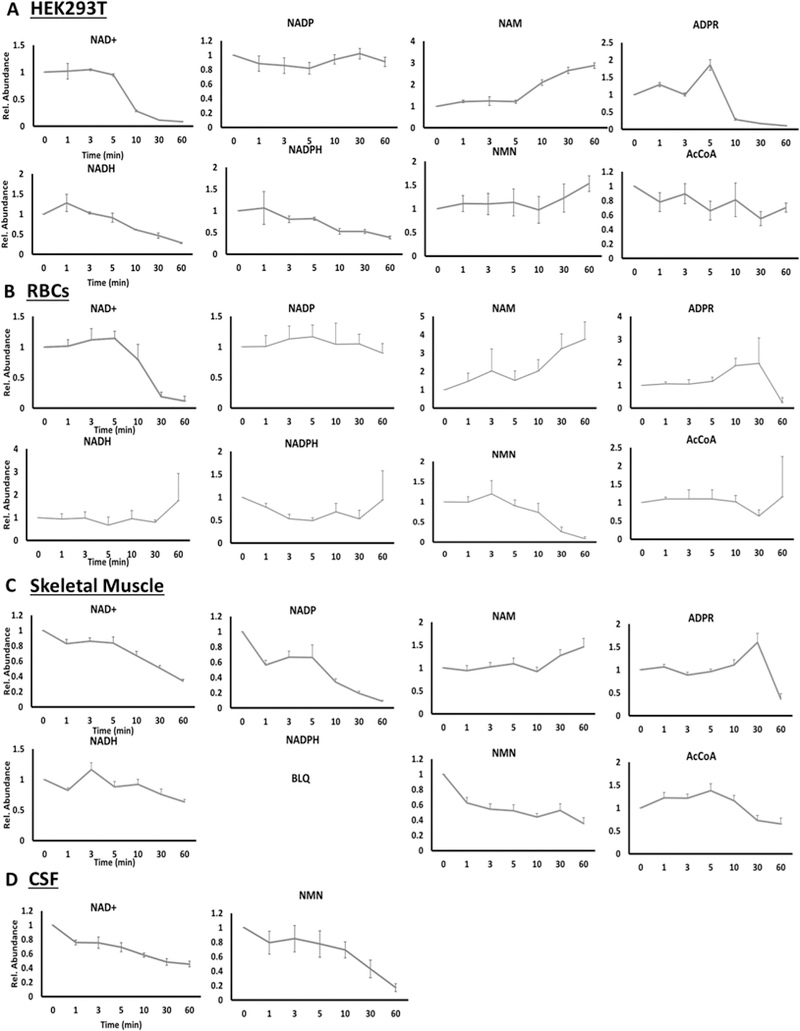
Benchtop Stability of NADmetabolome in HEK-293 Cells (n = 8) (A), human RBCs (n = 5) (B), primate skeletal muscle (C) and human CSF (n = 3) (D). Concentrations of reported metabolites were measured after the indicated time (min) that the matrix was left at room temperature, prior to extraction for analysis.
The relative concentrations of all the targeted NAD+ metabolites in cells, RBCs, primate skeletal muscle and CSF is reported in Table 1. Cellular NAD+ concentrations were obtained following calculations based on Trammel and Brenner [9] and were similar to those reported by in HeLa cells using LC MS/MS and in HEK-293T cells (105 μM NAD+ vs ~100μM, respectively) using a genetically encoded subcellular NAD+ biosensor [9,18]. In RBCs, NAAD was the only metabolite that was below the linear range, which is consistent with a recent study indicating that NAAD is only detectable in human blood after treatment with the NAD+ precursor, nicotinamide riboside [3]. NAD+ was 25 μM, and NADH was 3 μM similar to previously reported values [17]. This resulted in a ~11:1 ratio between NAD+/NADH for cells and ~8:1 for RBCs, which is in agreement with thermodynamically calculated values [19]. The NADP/NADPH ratio was ~1:1 for both HEK-293T cells and RBCs. These values are different from those expected thermodynamically (NADP/NADPH = 0.01 [18]) but are consistent with a recent publication reporting similar ratios for NADP/NADPH in human plasma [12]. However, of the metabolites tested, NADPH was the only metabolite that had precision at or below 17% for both the cells and RBCs, indicating that the reported concentrations may be underestimating endogenous NADPH levels, as NADPH is prone to autooxidation [15].
Primate skeletal muscle levels of the NAD+ metabolites were also determined. With our method, the calculated concentrations of NAD+ and NADH were determined to be 220 μM and 45 μM, respectively, assuming a density of 1.06 g/cm3 for skeletal muscle. While there are limited data available for NAD+ metabolites in skeletal muscle, levels of NAD+ and NADH have been reported in the range from ~1.5 to 1.9 mmol/kg dry wt muscle for NAD+ and ~0.08–0.20 mmol/kg dry wt muscle, using a bioluminescence-based method [20,21]. Conversion of these levels to molar amounts and assuming a dry weight to wet weight conversion of 4.2 [21], would suggest that the levels of NAD+ are between ~ 300 and 426 μM in skeletal muscle and NADH between 17 and 45 μM, similar to the values obtained with our method and a recent LC-MS/MS method [15]. The NAD+/NADH ratio in skeletal muscle was ~5, which was lower than what was found in RBCs and the cells. These findings are in line with recent reports detailing the relatively low NAD+/NADH ratio in skeletal muscle likely due to the abundance of mitochondria and the high concentration of NADH within the mitochondria in skeletal muscle [22]. In skeletal muscle, all remaining NAD+ metabolites were identified, with the exception of NAMN, NR and NADPH. The lack of NADPH is surprising, as a previous study had reported ~25 μM NADPH in skeletal muscle [20].The discrepancy with NADPH may be due to the necessity to weigh frozen tissue for normalization and the potential for autooxidation of this metabolite during the specimen processing [15], which would be consistent with the relatively poor precision observed for NADPH in both cells and RBCs.
In the CSF, the majority of the metabolites were below detection, with several below quantitation, including ADPR, AcCoA, NADH and NR (Table 1). However, among the detectable metabolites, NMN and NADwere the most abundant (81 and 72 nM, respectively) (Table 1). These surprisingly low circulating levels of metabolites in CSF may result from contamination by a small number of cells that have undergone cytolysis. Also, contrary to what was found in every other tested matrix, where NAD+ was found to be the major circulating metabolite, and NMN found to be ~1 order of magnitude lower in RBCs and ~2 orders of magnitude lower in cells/skeletal muscle, NMN was equivalent to NAD+ in CSF. While the exact reason for NMN abundance in CSF requires further study, we speculate that it may be driven by three possibilities (i) that there could be a few contaminating cells present in CSF, resulting in both low levels of NAD+ and NMN, (ii) that endogenous NAD+ is being degraded to NMN or (iii) that extracellular NAMPT; also known as pre-B cell colony-enhancing factor or visfatin, synthesizes NMN in the extracellular matrix [23]. In fact, one study identified eNAMPT in human CSF and correlated levels to body mass [24]. To date, extracellular NAD+ synthesis enzymes have not been described, although cellular release of NAD+ under stressful conditions have been reported [25]. Moreover, a recent study provides evidence that NAD+ is transported into mitochondria, contrary to the longstanding notion that the mitochondrial inner and outer membranes are impermeable to NAD+. In light of these findings, whether NAD+ can cross intact cellular plasma membranes, thus expanding a role for extracellular NAD+, warrants further investigation [26].
4. Conclusions
The method developed provides a rapid, simple and precise measurement of the NAD+ metabolome from a single sample that is applicable to multiple biological matrices, including skeletal muscle, RBCs and CSF. This is the first report on the levels in CSF. Moreover, the well-established intrinsic chemical stabilities of redox couples (NAD(P)/NAD(P)H) were assessed in these different matrices. Importantly, the rapid run time and minimal processing steps in this newly developed method, minimizes the putative metabolite alterations which may occur during sample collection and processing. Implementing the presented considerations and advantages of the current method will help facilitate the understanding of complex metabolic alterations implicated in physiological processes and the etiology of a range of metabolic and age-related disorders and may also assist in identifying potential treatment strategies.
Supplementary Material
Acknowledgements
This research was supported by the Intramural Research Program of the National Institutes on Aging, National Institutes of Health. The authors are grateful to the Baltimore Longitudinal Study of Aging study participants and staff for their dedication to these studies. V.Bohr has a CRADA with Chromadex.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ab.2019.02.019.
References
- [1].Fang EF, Kassahun H, Croteau DL, Scheibye-Knudsen M, Marosi K, Lu H, Shamanna RA, Kalyanasundaram S, Bollineni RC, Wilson MA, et al. , NAD+ replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair, Cell Metabol. 24 (2016) 566–581, 10.1016/j.cmet.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yoshino J, Baur JA, Imai S-I, NAD+ intermediates: the biology and therapeutic potential ofNMNand NR, Cell Metabol. 27 (3) (2018) 513–528, 10.1016/j.cmet.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Trammell SAJ, Schmidt MS, Weidemann BJ, Redpath P, Jaksch F, Dellinger RW, Li Z, Dale Abel E, Migaud ME, Brenner C, Nicotinamide riboside is uniquely and orally bioavailable in mice and humans, Nat. Commun. 7 (2016) 12948, 10.1038/ncomms12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Martens CR, Denman BA, Mazzo MR, Armstrong ML, Reisdorph N, McQueen MB, Chonchol M, Seals DR, Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD+ in healthy middle-aged and older adults, Nat. Commun. 9 (2018) 1286, 10.1038/s41467-018-03421-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dollerup OL, Christensen B, Svart M, Schmidt MS, Sulek K, Ringgaard S, Stødkilde-Jørgensen H, Møller N, Brenner C, Treebak JT, Jessen N, A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: safety, insulin-sensitivity, and lipid-mobilizing effects, Am. J. Clin. Nutr. 108 (2) (2018) 343–353, 10.1093/ajcn/nqy132. [DOI] [PubMed] [Google Scholar]
- [6].Verden E, NAD+ in aging, metabolism, and neurodegeneration, Science 350 (6265) (2015) 1208–1213, 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- [7].Bogan KL, Brenner C, Nicotinicacid, nicotinamide, and nicotinamide riboside:a-molecularevaluation of NAD+ precursorvitamins in human nutrition, Annu. Rev. Nutr. 28 (2008) 115–130, 10.1146/annurev.nutr.28.061807.155443. [DOI] [PubMed] [Google Scholar]
- [8].Revollo JR, Grimm AA, Imai S-I, The NADBiosynthesis Pathway Mediated by nicotinamide Phosphoribosyltransferase Regulates Sir2 activity in mammalian cells, J. Biol. Chem. 279 (2004) 50754–50763, 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- [9].Trammell SA, Brenner C, Targeted, LCMS-based metabolomics for quantitative measurement of NAD(+) metabolites, Comput. Struct. Biotechnol. J. 4 (2013), 10.5936/csbj.201301012e201301012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bustamante S, Jayasena T, Richani D, Gilchrist RB, Wu LE, Sinclair DA, Singh Sachdev P, Braidy N, Quantifying the cellular NAD+ metabolome using a tandem liquidchromatography mass spectrometry approach, Metabolomics 14 (15) (2018), 10.1007/s11306-017-1310-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yaku K, Okabe K, Nakagawa T, Simultaneous measurement of NADmetabolome in aged mice tissue using liquid chromatography tandem-mass spectrometry, Biomed. Chromatogr. 32 (6) (2018), 10.1002/bmc.4205e4205. [DOI] [PubMed] [Google Scholar]
- [12].Clement J, Wong M, Poljak A, Sachdev P, Braidy N, The Plasma NAD+ Metabolome Is Dysregulated in ‘normal’ Ageing, Rejuvenation Research, 2018, 10.1089/rej.2018.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rover Junior L, Fernandes CJ, de Oliveira Neto G, Kubota LT, Katekawa E, Serrano SH, Study of NADH stability using ultraviolet-visible spectrophotometric analysis and factorial design, Anal. Biochem. 260 (1) (2018) 50–55. [DOI] [PubMed] [Google Scholar]
- [14].Davila A, Liu L, Chellappa K, Redpath P, Nakamaru-Ogiso E, Paolella LM, Zhang Z, Migaud ME, Rabinowitz JD, Baur JA, Nicotinamide adenine dinucleotide is transported into mammalian mitochondria, Elife 7 (2018), 10.7554/eLife.33246 pii: e33246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lu W, Wang L, Chen L, Hui S, Rabinowitz JD, Extraction and quantitation of NAD(P)(H), Antioxidants Redox Signal. (3) (2018) 167–179, 10.1089/ars.2017.7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chiarugi A, Dolle C, Felici R, Ziegler M, The NADmetabolome–a key determinant of cancer cell biology, Nat. Rev. Canc. 12 (2012) 741–752, 10.1038/nrc3340. [DOI] [PubMed] [Google Scholar]
- [17].Zerez CR, Lachant NA, Lee SJ, Tanaka KR, Decreased erythrocyte nicotinamide adenine dinucleotide redox potential and abnormal pyridine nucleotide content in sickle cell disease, Blood 71 (1988) 512–515. [PubMed] [Google Scholar]
- [18].Cambronne XA, Stewart ML, Kim D, Jones-Brunette AM, Morgan RK, Farrens DL, Cohen MS, Goodman RH, Biosensor reveals multiple sources for mitochondrial NAD+, Science 352 (6292) (2016) 1474–1477, 10.1126/science.aad5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nicholls DG, Ferguson SJ, Bioenergetics4, Academic Press, Elsevier, Amsterdam, 2013, pp. 27–51. [Google Scholar]
- [20].Sahlin K, NADH and NADPH in human skeletal muscle at rest and during ischaemia, Clin. Physiol. Funct. Imaging 3 (5) (1983) 477–485. [DOI] [PubMed] [Google Scholar]
- [21].White AT, Schenk S, NAD+ /NADH and skeletal muscle mitochondrial adaptations to exercise, Am. J. Physiol. Endocrinol. Metab. 303 (3) (2012) E308–E321, 10.1152/ajpendo.00054.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Goody MF, Henry CA, A need for NAD+ in muscle development, homeostasis, and aging, Skeletal Muscle 8 (1) (2018) 9, 10.1186/s13395-018-0154-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Grolla AA, Travelli C, Genazzani AA, Sethi JK, Extracellular nicotinamide phosphoribosyltransferase, a new cancer metabokine, Br. J. Pharmacol. 173 (14) (2016) 2182–2194, 10.1111/bph.13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hallschmid M, Randeva H, Tan BK, Kern W, Lehnert H, Relationship between cerebrospinal fluid visfatin (PBEF/Nampt) levels and adiposity in humans, Diabetes, Mar. 58 (3) (2009) 637–640, 10.2337/db08-1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Haaq F, Adriouch S, Brass A, Jung C, Moller S, Scheuplein F, Bannas P, Seman M, Koch-Nolte F, Extracellular NADand ATP: partners in immune cell modulation, Purinergic Signal. 3 (2007) 71–81, 10.1007/s11302-006-9038-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Davila A, Liu L, Chellappa K, Redpath P, Nakamaru-Ogiso E, Paolella LM, Zhang Z, Migaud ME, Rabinowitz JD, Baur JA, Nicotinamide adenine dinucleotide is transported into mammalian mitochondria, eLife 7 (2018), 10.7554/eLife.33246e33246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.