

Abstract
Recent studies have implicated autophagy in several inflammatory diseases involving aberrant endothelial cell (EC) responses, such as acute lung injury (ALI). However, the mechanistic basis for a role of autophagy in EC inflammation and permeability remain poorly understood. In this study, we impaired autophagy by silencing the essential Beclin1 autophagy gene in human pulmonary artery EC. This resulted in reduced expression of proinflammatory genes in response to thrombin, a procoagulant and proinflammatory mediator whose concentration is elevated in many diseases including sepsis and ALI. These (Beclin1-depleted) cells also displayed a marked decrease in NF-κB activity secondary to impaired DNA binding of RelA/p65 in the nucleus, but exhibited normal IκBα degradation in the cytosol. Further analysis showed that Beclin1 knockdown was associated with impaired RelA/p65 translocation to the nucleus. Additionally, Beclin1 knockdown attenuated thrombin-induced phosphorylation of RelA/p65 at Ser536, a critical event necessary for the transcriptional activity of RelA/p65. Beclin1 silencing also protected against thrombin-induced EC barrier disruption by preventing the loss of VE-cadherin at adherens junctions. Moreover, Beclin1 knockdown reduced thrombin-induced phosphorylation/inactivation of actin depolymerizing protein Cofilin1 and thereby actin stress fiber formation required for EC permeability as well as RelA/p65 nuclear translocation. Together, these data identify Beclin1 as a novel mechanistic link between autophagy and EC dysfunction (inflammation and permeability).
Keywords: Endothelial cells, NF-κB, adhesion molecules, permeability, autophagy
INTRODUCTION
Endothelial cell (EC)4 inflammation and permeability are prominent features of endothelial dysfunction associated with many inflammatory disease states, including pulmonary diseases such as acute lung injury and acute respiratory distress syndrome (ALI/ARDS) (1-4). Activation of NF-κB, a master regulator of inflammation, and loss of adherens junctions (AJs) are the major mechanisms of EC inflammation and permeability, respectively (5, 6). In unstimulated cells, NF-κB is retained as an inactive complex in the cytoplasm through its interaction with its inhibitory protein IκBα (5, 7). The binding of IκBα masks the nuclear localization signal of the RelA/p65 subunit of NF-κB, thereby preventing its translocation to the nucleus. In stimulated cells, NF-κB is released from IκBα secondary to its phosphorylation at Ser32 and Ser36 by IκBβ kinase (IKKβ). Phosphorylated IκBα undergoes rapid ubiquitination and proteasome-mediated degradation, and the released NF-κB [predominantly RelA/p65 homodimer in EC (8, 9)] migrates to the nucleus where it binds to the promoter of its target genes. In addition, phosphorylation of RelA/p65 at Ser536 enhances the transcriptional capacity of NF-κB bound to the promoter (5, 7). Activated NF-κB causes EC to acquire proadhesive and proinflammatory phenotypes by promoting the expression of adhesion molecules (ICAM-1, VCAM-1, E-selectin), cytokines (TNFα, IL-1β, IL-6), and chemokines (IL-8, MCP-1) (10-15).
Vascular endothelial (VE)-cadherin plays a central role in establishing endothelial cell-to-cell adhesion via its ability to form Ca2+-dependent homophilic cis and trans dimers at AJs. AJs thus formed serve to maintain EC barrier integrity, and allow minimal filtration of fluids and selective passage of molecules such as electrolytes, ions and proteins (16, 17). Loss of VE-cadherin from the cell surface is a critical determinant of AJ disassembly and EC permeability caused by proinflammatory mediators (1, 17-20). This is further aided by contractile forces generated by actin-myosin interaction (actin stress fiber formation) (1, 18, 19). Importantly, actin stress fibers also play a critical role in facilitating thrombin-induced RelA/p65 translocation to the nucleus and thereby EC inflammation (12, 21-23). The coordinate and concerted actions of these events (induction of proinflammatory genes via NF-κB activation and disruption of endothelial AJs via VE-cadherin disassembly, aided by actin-myosin interactions) facilitate adhesion and transendothelial migration of inflammatory cells, particularly neutrophils (PMN) (4, 11, 24, 25), and increase endothelial permeability (1, 18, 19) associated with ALI/ARDS (2, 22, 25-28).
Autophagy is an evolutionarily conserved cellular process characterized by the formation of a double-membrane vesicle, called the autophagosome, which ensures clearance of damaged intracellular components (organelles and proteins) by delivering them to lysosomes for degradation (29, 30). Recent studies have revealed novel roles of autophagy in embryogenesis, development, cell death, immunity and inflammation and provided evidence of associations between autophagic dysfunction and disease (29-31). It is becoming increasingly clear that autophagy has both protective and injurious effects on many diseases. For example, autophagy induction is critical for survival during the perinatal period of relative starvation and serves a protective function in ischemic hearts (32-34). Similarly, loss of autophagy in the central nervous system causes neurodegeneration (35, 36). Aberrant regulation of autophagy is associated with aging and human diseases, including cancer, neurodegeneration, IBD, sepsis, and pulmonary diseases (31, 35, 37-42). We have recently shown that inhibition of autophagy via 3-methyladenine (3-MA), administered either prophylactically or therapeutically, reduced lung vascular leakage and tissue edema (43); however, it remains unclear how autophagy is mechanistically linked to EC inflammation and permeability, two major pathogenic features of ALI.
Autophagy is accomplished in several sequential stages (initiation, nucleation, elongation, and maturation) and requires the participation of a large number (> 30) of autophagy-related proteins (Atg genes) (29, 30). Beclin1, an ortholog of the Atg6/vacuolar protein sorting (Vps)-30 protein in yeast, represents one such protein that has a central role in autophagosome formation and maturation (44). As part of the class III phosphatidylinositol 3-kinase (PI3K) complex, Beclin1 serves an important function in mediating the localization of other autophagy proteins to pre-autophagosomal structures (45). In addition to its role in autophagy, Beclin1 is also implicated in regulating apoptosis via its ability to interact with anti-apoptotic Bcl-2 family members through its BH3 domain (44). This interaction impairs the ability of Beclin1 to mediate the assembly of pre-autophagosomal structures, thereby inhibiting autophagy (46). Thus, the conditions that disrupt or favor Beclin1-Bcl2 complex play a key role in determining whether cells undergo autophagy or apoptosis (44, 47). Recently, it has been shown that Beclin1 is induced in a RelA/p65-dependent manner to promote autophagy in various cell types including EC (48-50). However, it is unclear if Beclin1 is also linked to NF-κB activation to cause EC inflammation. In this study, we demonstrate a novel role of Beclin1 in mediating NF-κB activation and EC inflammation. Our experiments also identify a previously unrecognized role of Beclin1 in causing loss of EC barrier integrity by its ability to promote VE-cadherin disassembly. Together, these data identify Beclin1 as a possible mechanistic link between autophagy and EC dysfunction.
MATERIALS AND METHODS
Reagents:
Human α-thrombin was purchased from Enzyme Research Laboratories (South Bend, IN). Diethylaminoethyl (DEAE)-dextran was obtained from Sigma-Aldrich Chemical Company (St. Louis, MO). A rabbit polyclonal anti-Beclin1 antibody (3738S) was purchased from Cell Signaling Technology (Beverly, MA). Polyclonal antibodies to VCAM-1 (SC-8304), RelA/p65 (SC-8008), IκBα (SC-371), and β-actin (SC-47778) were from Santa Cruz Biotechnology (Santa Cruz, CA). An anti-phospho-(Ser536)-RelA/p65 (3033S) was from Cell Signaling Technology (Beverly, MA). Antibodies to VE-cadherin were obtained from Abcam (AB33168, Cambridge, MA) and BD Biosciences (BD555661, San Jose, CA). A rabbit polyclonal anti-Cofilin1 (clone D3F9, 5175S) antibody and a rabbit polyclonal anti-phospho-(Ser3)-Cofilin1 antibody (3311S) were obtained from Cell Signaling Technology (Beverly, MA). An anti-GAPDH antibody (SC-32233) and an anti-Lamin B antibody (SC-6216) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Plasmid maxi kit was from QIAGEN Inc. (Valencia, CA). All other materials were purchased from Thermo Fisher Scientific (Waltham, MA).
Endothelial Cell Culture:
Human pulmonary artery endothelial cells (HPAEC) were obtained from Lonza (Walkersville, MD) and cultured in gelatin-coated flasks as described (13). Briefly, cells were grown to confluency in endothelial basal medium 2 (EBM2) containing bullet kit additives (BioWhittaker, Walkersville, MD) and 10% FBS at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. For treatment, HPAEC were incubated in same serum-free EBM2 medium for 1-2 h prior to thrombin challenge. HPAEC between passages 3 and 7 were used.
RNAi Knockdown of Beclin1:
SMARTpool short-interfering RNA duplexes specific for human Beclin1 (si-Beclin1) and a nonspecific siRNA control (si-Con) were obtained from Dharmacon (Lafayette, CO). HPAEC were transfected with si-Beclin1 or si-Con using DharmaFect1 siRNA Transfection Reagent (Dharmacon) essentially as described (51). Briefly, 50-100 nM siRNA was mixed with DharmaFect1 and incubated for 24h with cells that are 50–60% confluent. At 48 h, after transfection, cells were used for experiments measuring the effect of Beclin1 knockdown on EC inflammation and barrier disruption.
Reporter Gene Transfection and Luciferase Assay:
The reporter plasmid pNF-κBLUC (Stratagene, La Jolla, CA) containing 5 copies of consensus NF-κB sequences linked to a minimal E1B promoter-Firefly luciferase gene was used to determine the transcriptional activity of NF-κB. The pTKRLUC plasmid (Promega Corp., Madison, WI) containing Renilla luciferase gene driven by the constitutively active thymidine kinase promoter was used to normalize the transfection efficiencies. To determine the effect of Beclin1 knockdown on NF-κB transcriptional activity, cells were first transfected with Beclin1 siRNA (si-Beclin1) using DharmaFect1 siRNA Transfection Reagent as described above. Twenty-four hours later, cells were again transfected with pNF-κBLUC and pTKRLUC using DEAE-dextran as described (23, 51). Briefly, DEAE-dextran (50 μg/ml) in serum-free EBM2 was mixed with 5 μg pNF-icB-LUC and 0.125 μg pTKRLUC. The resulting mixture was applied onto cells that were already transfected with si-Beclin1 or si-Con. After 1 h, cells were exposed to 10% dimethyl sulfoxide (DMSO) in serum-free EBM2 for 4 min, and then washed 2x with PBS and allowed to grow in EBM2-10% FBS. Twenty-four hours later, cells were treated with thrombin and then lysed in passive reporter lysis buffer (Promega Biotech, Madison, WI). The cell extracts were assayed for Firefly and Renilla luciferase activities using Dual Luciferase Reporter Assay System (Promega Biotech, Madison, WI) and the data were expressed as a ratio of Firefly to Renilla luciferase activity.
Cell Lysis, Immunoprecipitation and Immunoblotting:
After appropriate treatments, cells were lysed in radioimmune precipitation (RIPA) buffer (50 mMTris-HCl, pH 7.4, 150 mM NaCl, 5 mM NaF, 0.25 mM EDTA, pH 8.0, 1% deoxycholic acid, 1% Triton-X, 1 mM sodium orthovanadate supplemented with protease inhibitor cocktail [Sigma]) or phosphorylation lysis buffer (50 mM HEPES, 150 mM NaCl, 200 μM sodium orthovanadate, 10 mM sodium pyrophosphate, 100 mM sodium fluoride, 1 mM EDTA, 1.5 mM magnesium chloride, 10% glycerol, 0.5 to 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride [PMSF], and protease inhibitor cocktail as described (Fazal et al. 2009). Cell lysates were resolved on SDS-PAGE and transferred onto nitrocellulose membranes and the membranes were incubated with 5% (w/v) nonfat dry milk in TBST (10 mM Tris (pH 8.0), 150 mM NaCl, and 0.05% Tween 20) for 1 h at room temperature to block the residual binding sites on them. The membranes were then incubated with appropriate Abs and developed using an ECL method as previously described (12). For immunoprecipitation, cell lysates were prepared in 500 μl of NP-40 lysis buffer (1% NP-40, 50 mM Tris HCl pH 8.0, protease inhibitor cocktail) and then subjected to preclearing with 50 μl of protein G microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) for 1 hour at 4°C. The precleared lysate was subjected to immunoprecipitation by incubating with 0.6-1 μg of appropriate antibody and 50 μl of the protein G microbeads at 4°C overnight with gentle shaking as described (52). The immunoprecipitates were added to μ magnetic columns (Miltenyi Biotec, Bergisch Gladbach, Germany) and washed four times with the same volume of ice cold NP-40 buffer with 150 mM NaCl. Boiled SDS sample buffer was added to the columns for 5 min to elute the immunoprecipitated proteins, and the extracted proteins were analyzed by immunoblotting as described above. Representative blots presented in the results section come from the same membrane which may have more samples in various groups.
Immunofluorescence:
Confluent HPAEC monolayers grown on gelatin-coated coverslips were subjected to immunofluorescence staining as described (43). To localize F-actin filaments, the cells were incubated with Alexa Fluor 488-phalloidin for 20 min at room temperature. VE-cadherin antibody (BD Biosciences, San Jose, CA) was used to visualize AJs. DNA staining with Hoechst Dye was used to visualize nuclei. An anti-LC3 antibody was used to visualize LC3 puncta as a measure of autophagosome formation. The coverslips were mounted on the slide using Vectashield mounting media (Vector Laboratories, Lincolnshire, IL) and the images were acquired using an Axio Imager M2m confocal microscope (Zeiss). To quantify LC3 and phalloidin staining intensity, lines were drawn around individual cells and the average fluorescence was measured using the ‘Measure’ tool in ImageJ. The fluorescence value for each cell was averaged and graphed. Two methods were used to quantify barrier integrity from VE-cadherin staining. The number of cells with disrupted adherens junctions (AJs) was calculated by counting the number of cells in a field with discontinuous cell surface VE-cadherin staining and normalized to the total number of cells in the field. The percentage of cells with a disrupted VE-cadherin staining border was averaged and graphed. Gap formation was quantified using the ‘Measure’ tool in ImageJ. Gaps, or areas without cell coverage, were outlined and measured, and the area of gaps in each field was normalized to the size of the field. The percentage of gap area was averaged and graphed.
Nuclear Extract Preparation and Measurement of RelA/p65 DNA Binding Activity:
After appropriate treatments, cells were washed once with ice-cold Tris-buffered saline and scraped with cell lifter in 1 ml PBS. Cells were centrifuged at 2000 rpm for 2 min at 4°C. The pellet was resuspended in 400 μl of buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM [DTT], and 0.5 mM PMSF). After 20 min on ice, NP-40 was added to a final concentration of 0.6%, and the samples were centrifuged 1 min at 10,000 rpm to collect the supernatants containing the cytoplasmic proteins. The pelleted nuclei were resuspended and homogenized in 100 μl of buffer B (20 mM HEPES [pH 7.9], 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, and 1 mM PMSF). After 30 min at 4°C, lysates were centrifuged at 10,000 rpm for 5 min and supernatants containing the nuclear proteins were collected. The protein concentration of the nuclear extract was measured using a Bio-Rad protein determination kit (Bio-Rad Laboratories). Equal amount of nuclear proteins were used to determine RelA/p65 nuclear translocation by immunoblotting or its DNA binding activity using an ELISA-based DNA binding assay kit (Cayman Chemical, Ann Arbor, MI) as described (53).
Assessment of Endothelial Permeability by Transendothelial Electrical Resistance (TER):
The endothelial barrier integrity was assessed by measuring transendothelial electrical resistance (TER) across confluent monolayers using the highly sensitive Electrical Cell-Substrate Impedance Sensing (ECIS) system (Applied Biophysics, Troy, NY) as described (43, 53). Briefly, confluent HPAEC grown on gelatin-coated gold microelectrodes in EBM2 containing 10% FBS. After 24 h, culture medium was replaced with EBM2 containing 1% FBS and 2 h later thrombin was added and the TER was measured over a period of 4 h. TER was measured over time and normalized to baseline resistance.
Statistical Analysis:
Multiple groups were analyzed by one-way ANOVA, followed by Tukey post-test. When two groups were analyzed, a Student’s t test was performed. All statistical analyses were performed using GraphPad Prism 6 (GraphPad Software, San Diego) and data presented as mean ± SE. A p value < 0.05 was considered statistically significant.
RESULTS
Beclin1 Silencing Inhibits Autophagic Activity in EC.
We investigated whether knockdown of Beclin1 influences autophagic activity in EC. Cells were transfected with short interfering RNA (si-RNA) targeting Beclin1 (si-Beclin1) or control si-RNA (si-Con). Following transfection, cells were challenged with thrombin and analyzed for autophagy induction by monitoring LC3 puncta. Cells transfected with si-Beclin1 showed an impressive knockdown (depletion) of Beclin1 irrespective of whether they were stimulated with thrombin or left untreated (Fig. 1A). Figure 1B and 1C show that thrombin enhanced autophagy in cells transfected with si-Con as evidenced by an increased number of LC3 puncta in these cells. In contrast, both basal and thrombin-induced autophagic activity was lost in Beclin1-depleted cells (Fig. 1B-C). These data show that thrombin increases autophagic activity in EC and that Beclin1 is essential for both constitutive and thrombin-stimulated autophagy in these cells.
Figure 1. Beclin1 is essential for autophagosome formation in EC.
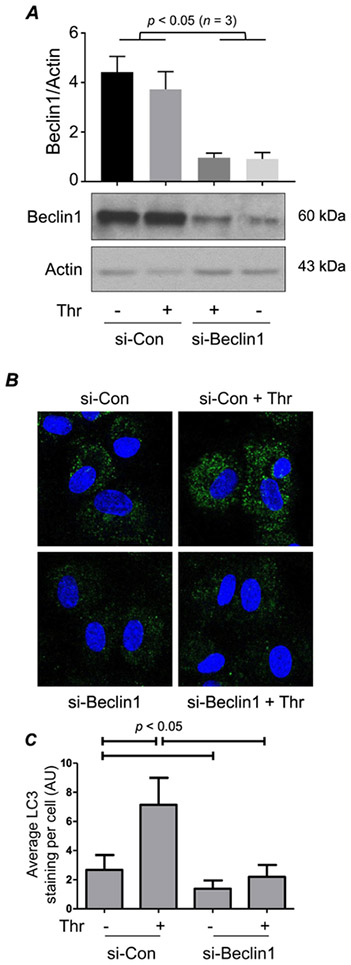
(A) HPAEC were transfected with Control or Beclin1 siRNA (si-Con or si-Beclin1) for 48 h prior to 1 h thrombin (Thr; 5 U/ml) treatment. Cells were lysed and Western blot analysis was performed to determine Beclin1 and Actin levels. The bar graph represents the effect of siRNA on Beclin1 level normalized to Actin. Data are mean + S.E. (n = 3 for each condition) and were analyzed by Student’s t-test. (B) HPAEC were transfected with si-Con or si-Beclin1. Forty-eight hours later, cells were treated with thrombin for 1 h, and then fixed and stained with anti-LC3 antibody (green) to mark autophagosomes and DAPI (blue) to mark nuclei. Each image is representative of three experiments. (C) Mean fluorescence intensity of LC3 puncta/cell was determined by analyzing 32-36 cells. Values are reported in arbitrary units (AU).
Beclin1 Silencing Inhibits NF-κB Activity and EC Inflammation.
To ascertain the role of Beclin1 in the mechanism of thrombin-induced EC inflammation, we first evaluated the effect of Beclin1 knockdown on NF-κB-dependent reporter gene activity. Results showed that thrombin induced a marked increase in NF-κB activity and that this response was significantly attenuated in Beclin1-depleted cells (Fig 2A). We also tested if Beclin1 is a stimulus-specific or general mediator of NF-κB. We found that NF-κB activity induced by lipopolysaccharides (LPS) was also sensitive to Beclin1 knockdown (Fig. S1 in Supplemental Data). These data identify Beclin1 as a critical regulator of NF-κB activation in EC.
Figure 2. Silencing of Beclin1 attenuates thrombin-induced NF-κB activity and inflammatory gene expression in EC.
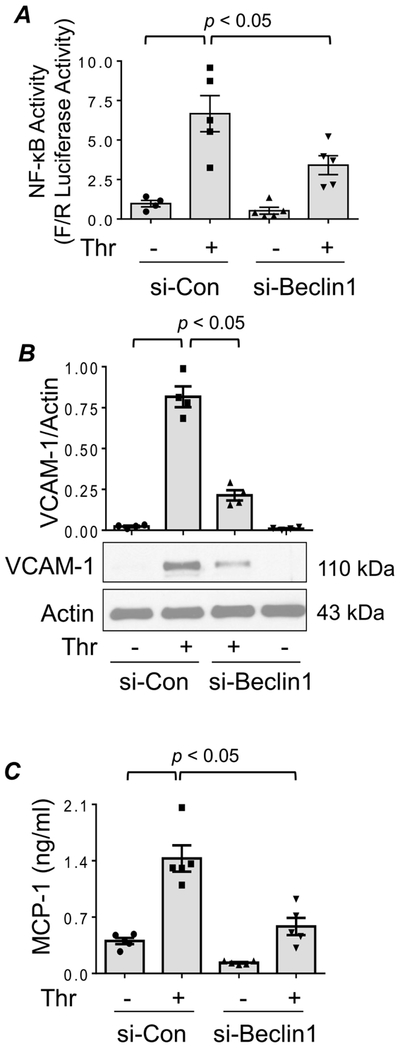
(A) HPAEC were transfected with si-Con or si-Beclin1 using DharmaFect1. After 24 h, cells were again transfected with NF-κBLUC and Renilla-LUC constructs using DEAE-dextran as described in Materials and Methods. Cells were treated with thrombin for 6 h and the cell extracts were assayed for Firefly and Renilla luciferase activities. Renilla luciferase was used as an internal control for transfection efficiency. Bars indicate mean ± SEM (n = 4-6) for each condition, and were analyzed by ANOVA. (B) HPAEC were transfected with si-Con or si-Beclin1 for 48 h prior to treatment with thrombin for 6 h. Cells were lysed and Western blot analysis was performed to determine VCAM-1 levels. The levels of Actin were used to monitor protein loading. Bars indicate mean ± SEM (n = 4) for each condition, and were analyzed by ANOVA. (C) Cell supernatants were collected after 48 h transfection and 6 h thrombin treatment, and ELISA was performed to determine MCP-1 release. Bars indicate mean ± SEM (n = 4–6) for each condition, and data was analyzed by ANOVA.
Given that NF-κB is an essential regulator of EC inflammation (5), we examined if the knockdown of Beclin1 exerts a similar protective effect on proinflammatory gene expression in EC. Thrombin stimulation of EC transfected with si-Con resulted in increased levels of proinflammatory mediators (VCAM-1 and MCP-1) whereas in EC transfected with si-Beclin1 levels of these mediators were significantly reduced (Fig. 2B-C). A similar inhibitory effect of Beclin1 knockdown was also observed on LPS-induced increase in IL-6 and MCP-1 levels (Fig. S2 in Supplemental Data). These findings are consistent with the effect of Beclin1 knockdown on NF-κB activity. These data implicate a role of the Beclin1/autophagy axis in mediating EC inflammation via activation of NF-κB.
Beclin1 Silencing Inhibits NF-κB Activity by Preventing DNA Binding of RelA/p65 in the Nucleus without Affecting IκBα Degradation in the Cytoplasm.
We determined if Beclin1 controls NF-κB activity by regulating its DNA binding activity in the nucleus. It should be noted that thrombin-induced NF-κB complexes are predominantly composed of RelA/p65 homodimer (8, 21) and that maximal RelA/p65 DNA binding occurs at 1 h after thrombin stimulation (Bijli et al AJP Lung). We, therefore, used this time point to assess the role of Beclin1 in RelA/p65 binding to DNA in response to thrombin. Analysis of nuclear extracts from si-Con transfected cells showed increased DNA binding of RelA/p65 upon thrombin challenge; however, this response was significantly attenuated in si-Beclin1 transfected cells (Fig. 3A). Because degradation of IκBα in the cytoplasm is essential for RelA/p65 DNA binding in the nucleus and maximally occurs at 1 h after thrombin challenge (Bijli et al AJP Lung), we next determined the effect of Beclin1 silencing on IκBα degradation under similar conditions of thrombin treatment. Intriguingly, Beclin1 knockdown failed to inhibit thrombin-induced IκBα degradation (Fig. 3B), unlike its effect on RelA/p65 nuclear DNA binding (Fig. 3A). These results show that the effect of Beclin1 knockdown of RelA/p65 nuclear DNA binding occurs downstream of IκBα degradation and may be related to impaired nuclear translocation of RelA/p65.
Figure 3. (A-B) Knockdown of Beclin1 inhibits RelA/p65 nuclear DNA binding without affecting IκBα degradation.

HPAEC were transfected with si-Con or si-Beclin1 48 h prior to treatment with thrombin for 1 h. (A) An ELISA-based assay was used to assess RelA/p65 DNA binding activity as described in the Materials and Methods. Bars indicate mean ± SEM (n = 3) for each condition, and were analyzed by ANOVA. (B) Total cell lysates were prepared and Western blots were performed to determine IκBα and RelA/p65 levels. Bars indicate mean ± SEM (n = 3) for each condition, and were analyzed by ANOVA. (C-F) Knockdown of Beclin1 prevents RelA/p65 nuclear translocation via inhibition of cofilin1 phosphorylation and actin stress fiber formation. HPAEC were transfected with si-Con or si-Beclin1 48 h prior to treatment with thrombin for 1 h. (C) Nuclear extracts were analyzed by immunoblotting for RelA/p65 and Lamin B levels. Bars indicate mean ± SEM (n = 3) for each condition, and were analyzed by ANOVA. (D) Total cell lysates were analyzed by Western blot to determine the phosphorylation status of cofilin1 at Ser3. Total levels of cofilin1 were used to monitor protein loading. Bars indicate mean ± SEM (n = 3) for each condition, and were analyzed by ANOVA. (E) Cells were fixed and stained with Phalloidin-488 (green) to mark actin filaments and DAPI (blue) to mark nuclei. Each image is representative of three experiments. (F) Mean fluorescence intensity of actin stress fibers (phalloidin staining)/cell was determined by analyzing 35-39 cells. Values are reported in arbitrary units (AU), and were analyzed by ANOVA.
Beclin1 Silencing Inhibits DNA Binding of RelA/p65 by Preventing its Translocation to the Nucleus.
The above data led us to investigate the possibility that the protective effect of Beclin1 knockdown on RelA/p65 DNA binding is secondary to an impairment in the nuclear translocation of RelA/p65. Cells transfected with si-Con or si-Beclin1 were treated with thrombin for 1 h, which we have previously shown causes maximal RelA/p65 nuclear translocation (Bijli et al. AJP Lung). Immunoblotting of nuclear extracts from these cells showed a significant decrease in thrombin-induced RelA/p65 nuclear translocation in Beclin1-depeleted cells (Fig. 3C). These data reveal an important function of Beclin1 in controlling NF-κB activation by its ability to regulate RelA/p65 trafficking to the nucleus.
Beclin1 Silencing Impairs RelA/p65 Nuclear Translocation by Inhibiting Cofilin-1 Phosphorylation and Actin Stress Fiber Formation.
We have previously reported that phosphorylation/inactivation of Cofilin1 and the associated changes in actin cytoskeleton are necessary for thrombin-induced nuclear translocation of RelA/p65 without affecting IκBα degradation (12, 21). To test the possibility that Beclin1 controls RelA/p65 nuclear localization by mediating Cofilin1 phosphorylation at Ser3 and actin stress fiber formation, we evaluated the effect of Beclin1 knockdown on these responses. Results showed that Beclin1 knockdown was effective in inhibiting thrombin-induced Ser3 phosphorylation/inactivation of Cofilin1 and stress fiber formation (Fig. 3 D-F). These results support the notion that Beclin1 mediates thrombin-induced RelA/p65 nuclear translocation, at least in part, by causing Cofilin1-dependent changes in the actin cytoskeleton.
Beclin1 Silencing Inhibits RelA/p65 Transcriptional Activity by Preventing its Phosphorylation.
We and others have shown that phosphorylation of Ser536 within the transactivation domain 1 of RelA/p65 is a critical event in conferring transcriptional competency to the bound RelA/p65 in EC (5, 51, 54). We determined if Beclin1 also contributes to NF-κB activity by mediating Ser536 phosphorylation of RelA/p65. Because thrombin-induced Ser536 phosphorylation of RelA/p65 also occurs maximally at 1 h after stimulation (Bijli et al. AJP Lung), we used the same treatment conditions to determine the effect of Beclin1 knockdown on this response. Thrombin challenge of cells transfected with si-Con resulted in RelA/p65 at Ser536, as expected; however, knockdown of Beclin1 inhibited this response (Fig. 4). Together, these data show that Beclin1 controls EC inflammation by its ability to facilitate the nuclear translocation and thereby DNA binding of RelA/p65 after its release from IκBα in the cytoplasm, and to promote the phosphorylation at Ser536 and thereby transcriptional capacity of the bound RelA/p65.
Figure 4. Beclin1 knockdown inhibits thrombin-induced RelA/p65 phosphorylation at Ser536.

HPAEC were transfected with si-Con or si-Beclin1 48 h prior to treatment for 1 h with thrombin. Cells were lysed and analyzed by Western blot to determine the phosphorylation status of RelA/p65 at Ser536. Total levels of RelA/p65 were used to monitor protein loading. Bars indicate mean ± SEM (n = 3) for each condition, and were analyzed by ANOVA.
The impaired nuclear translocation and phosphorylation of RelA/p65 following Beclin1 knockdown prompted us to determine if Beclin1 associates with RelA/p65 to regulate these events. To test this possibility, we immunoprecipitated Beclin1 or RelA/p65 from control and thrombin-challenged cells and then analyzed the Beclin1 and Rel/p65 immunoprecipitates for the presence of RelA/p65 and Beclin1 respectively by immunoblotting. No association of Beclin1 with RelA/p65 or its upstream regulator IKK;β was noted (Fig. 5). These data exclude a direct association between Beclin1 with NF-κB complex and points to the existence of an intermediate protein that may serve to link Beclin1 to NF-κB pathway.
Figure 5. Beclin1 does not directly interact with NF-κB pathway components.

HPAEC were treated with thrombin for indicated time points and lysed. RelA (A) or Beclin1 (B) were immunoprecipitated from cell lysates by incubation with RelA, Beclin1, or non-specific IgG control antibodies. Immunoprecipitated fractions were analyzed by Western blot to determine Beclin1, RelA or IKKβ levels. Each blot is representative of three experiments. FI, flow through; Inp, input (total cell lysate).
Beclin1 Silencing Restores EC Barrier Integrity.
The ability of Beclin1 to regulate actin filament formation (Fig. 3 E-F) suggests a role for it in the mechanism of EC barrier disruption. To address this possibility, we determined the effect of Beclin1 knockdown on EC barrier integrity by monitoring the dynamic changes in transendothelial electrical resistance (TER). As reported in previous studies (43, 53, 55, 56), thrombin induced a rapid decrease in TER (Fig. 6A) in cells transfected with si-Con which was followed by a gradual recovery with maximal recovery occurring by 3-4 h. Knockdown of Beclin1 had a small protective effect on the acute decrease in TER (within 0.5 h after thrombin challenge), but showed a more efficient and enhanced TER recovery (Fig. 6A). Interestingly, the effect of Beclin1 silencing on thrombin-induced TER changes is similar to that of autophagy inhibitor 3-methyladenine (3-MA) (43). These data further support a role for the Beclin1/autophagy axis in EC barrier disruption. Because the initial decrease and later recovery of TER is characterized by the disassembly and subsequently reassembly of AJs, we next determined the effect of Beclin1 knockdown on the initial loss and later recovery of VE-cadherin at AJs after thrombin challenge. Knockdown of Beclin1 only partially protected against the acute disruption of AJs and the resultant interendothelial gap formation, but was more effective in accelerating the reannealing of AJs and thereby closing the interendothelial gaps (Fig. 6B-G). These results identify a novel role of Beclin1 in regulating EC barrier dysfunction by virtue of impairing the restoration of VE-cadherin at AJs.
Figure 6. Beclin1 knockdown enhances endothelial barrier recovery in response to thrombin.
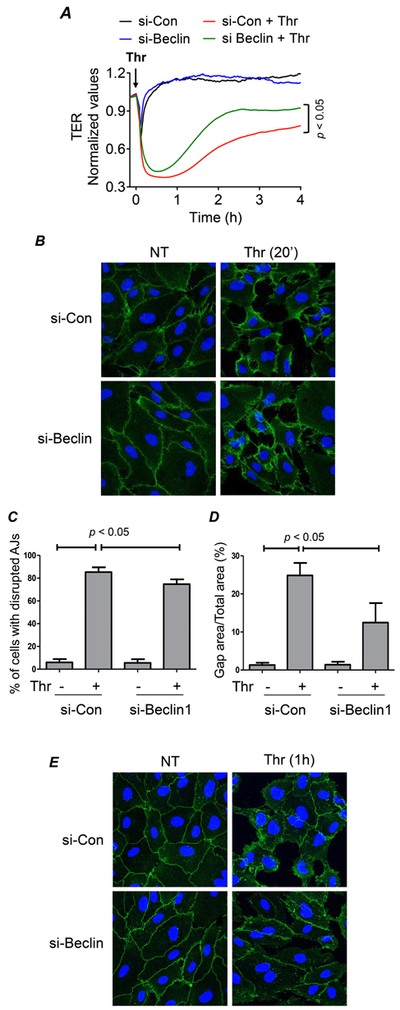

(A) HPAEC were transfected with si-Con or si-Beclin1 for 48 h and transferred to gold electrode plates and allowed to grow to confluency. Cells were treated with thrombin (indicated by arrow) and transendothelial resistance (TER) was measured over 4 h by Electric Cell-substrate Impedance Sensing (ECIS). Resistance was normalized to the values at 0 h, n = 3 for each condition. Data was analyzed by Student’s t-test. (B&E) HPAEC were grown on cover slips and transfected with si-Con or si-Beclin1 48 h prior to treatment with thrombin. After treatment, fixed and non-permeabilized cells were stained with anti-VE-cadherin antibody (green) and DAPI (blue). Each image is representative of three experiments. (C&F) The percentage of cells with disrupted adherens junctions (AJs) was reported by quantifying the number of cells with a discontinuous border of VE-cadherin staining normalized to the total number of cells in a field. Data are mean + S.E. (n = 3-4 fields with 45-65 cells in each field/condition) and were analyzed by ANOVA. (D&G) Gap formation between cells was measured by calculating the area without cell coverage in a given field normalized to the total area of the field. Data are mean + S.E. (n = 3-4 fields with 45-65 cells in each field/condition) and were analyzed by ANOVA.
DISCUSSION
Beclin1 is a key regulator of autophagy by its ability to initiate autophagosome formation via recruitment of other autophagy proteins to pre-autophagosomal structures (44, 45, 57). We have recently shown that autophagy is an important component of lung vascular injury and that targeting it via 3-methyladenine (3-MA) protects against ALI (43). However, the mechanistic basis for a role of autophagy, particularly the involvement of Beclin1 in EC inflammation and permeability, two important pathogenic features of ALI, remain poorly understood. Recently, it has been shown to be regulated by NF-κB (50); however, the role of Beclin1 in regulating NF-κB activation and thereby EC inflammation remains unclear. By using si-RNA-mediated silencing of Beclin1, we now provide evidence that autophagy-deficient EC exhibit dampened inflammatory responses when exposed to thrombin. These (Beclin1-depleted) cells also display a marked decrease in NF-κB activity secondary to impaired nuclear translocation and phosphorylation of RelA/p65. Moreover, Beclin1-depeleted EC show improved barrier function secondary to enhanced VE-cadherin reassembly at AJs and reduced actin stress fiber formation following thrombin challenge. Together, these data reveal a novel function of the Beclin1 in the mechanism of EC inflammation and permeability.
The relationship between autophagy and NF-κB is complex and appears to be context-dependent. Autophagy is implicated in many cellular responses regulated by NF-κB including cell survival, differentiation, senescence, inflammation, and immunity (58). As such, many upstream regulators are shared by autophagy and NF-κB; however, depending upon the context they may be regulated differentially by the same upstream signals (50, 58, 59). In a majority of cases, RelA/p65 promotes autophagy through induction of Beclin1 and autophagy in turn may be engaged to terminate or promote NF-κB signaling depending upon the cellular context and the stimulus (48, 50, 58-60). While the autophagy and NF-κB interplay has been studied in some detail in cancer cells (49, 58, 60), this relationship is poorly described in the context of the endothelium. A recent report by Zeng et al. (50) show that stimulated ischemia/reperfusion (sI/R) in EC activates RelA/p65 to induce Beclin1 and thereby autophagy. Beclin1-autophagy axis thus engaged serves to exacerbate sI/R-induced EC injury and death (50, 61). Thus, our findings that Beclin1 is engaged by thrombin to mediate RelA/p65 activation and EC inflammation are not only novel but also suggest the existence of a feed-forward mechanism whereby engagement of Beclin1 not only activates RelA/p65, but the latter also promotes Beclin1 expression, thereby amplifying the Beclin1-RelA/p65-autophagy axis. While such a possibility needs to be rigorously tested and requires a separate comprehensive study, it is supported by our previous studies showing that rapamycin, an inhibitor of Mechanistic Target of Rapamycin (MTOR) and a potent inducer of autophagy, augmented thrombin-induced EC inflammation by causing a rapid and persistent activation of RelA/p65 (62).
We determined the mechanism by which Beclin1 regulates RelA/p65 activation in the endothelium. We first focused on the involvement of Beclin1 in cytosolic activation of RelA/p65 (i.e. the release of RelA/p65 from its inhibitor IκBα) by monitoring the status of IκBα degradation in Beclin1-depleted cells. We noted that thrombin-induced IκBα degradation was insensitive to Beclin1 knockdown. Interestingly, however, Beclin1 silencing was effective in preventing thrombin-induced nuclear activation of RelA/p65 as assessed by DNA binding activity of RelA/p65 in the nucleus. The decrease in nuclear DNA binding of RelA/p65 despite its release from IκBα in the cytosol pointed to a possible defect in the nuclear localization of the released RelA/p65. Indeed, thrombin-induced nuclear translocation of RelA/p65 was impaired in Beclin1-depleted EC. These data unveil a new role of Beclin1 in regulating thrombin-induced nuclear translocation of RelA/p65 in EC.
We previously showed the existence of an actin cytoskeleton-dependent mechanism of RelA/p65 nuclear translocation (21). These studies established that thrombin-induced RelA/p65 nuclear translocation and EC inflammation requires dynamic changes in actin cytoskeleton (i.e. actin stress fiber formation) regulated by RhoA/ROCK/LIMK1 and subsequent cofilin-1 phosphorylation/inactivation (12, 21, 23). To test the possibility that Beclin1 engages this mechanism to facilitate RelA/p65 nuclear translocation, we determined the status of cofilin1-dependent changes in actin cytoskeleton in Beclin1-depleted EC. Consistent with this possibility, Beclin1-depleted cells showed a marked reduction in thrombin-induced cofilin1 phosphorylation/inactivation and actin stress fiber formation. These data support the notion that Beclin1 controls EC inflammation in part by its ability to regulate RelA/p65 nuclear translocation via a mechanism that relies on cofilin1-dependent changes in actin cytoskeleton.
The involvement of both RhoA/ROCK/LIMK1 pathway and Beclin1 in Cofilin1 phosphorylation suggests the existence of a cross-talk between these pathways. Indeed a recent study has shown that Beclin1 serves to activate RhoA/ROCK/LIMK pathway to promote programmed death ligand-2 (PD-L2)-dependent osteosarcoma cell invasion and metastasis (63). In another study, Gurkar et al. (64) have shown that Beclin1 is engaged downstream of ROCK1 to mediate autophagy caused by nutrient deprivation. These reports support our observation that Beclin1 is linked to Cofilin1; however, additional studies are required to establish whether Beclin1 acts as an upstream regulator or downstream effector of RhoA/ROCKLIMK pathway to mediate thrombin-induced Cofilin1 phosphorylation in endothelial cells.
We also addressed the role of Beclin1 in a phosphorylation-dependent mechanism of RelA/p65 activation. Studies have shown that signal-induced phosphorylation of RelA/p65 on serine residues including Ser536 is an additional regulatory pathway that enhances the transcriptional capacity of RelA/p65 (5, 65). We previously showed that thrombin induces Ser536 phosphorylation to increase the transcriptional potential of RelA/p65 in EC (5, 54, 66). These findings led us to examine if Beclin1 is also involved in mediating Ser536 phosphorylation of RelA/p65. We found that thrombin-induced phosphorylation of RelA/p65 at Ser536 was significantly decreased in EC depleted of Beclin1, implicating a role of Beclin1 in controlling the transcriptional activity of RelA/p65. Collectively, our findings reveal that Beclin1 regulates thrombin-induced RelA/p65 activation by a dual mechanism whereby it serves to facilitate the nuclear translocation and consequently DNA binding of the released RelA/p65 and also phosphorylation of RelA/p65 to increase its transcriptional capacity. Consistent with its role in RelA/p65 activation, Beclin1 silencing was associated with a significant decrease in proinflammatory gene expression in EC. Thus, these data identify Beclin1 as a critical mediator of EC inflammation by its ability to control RelA/p65 nuclear translocation and phosphorylation.
The protective effect of Beclin1 silencing on thrombin-induced actin stress fiber formation pointed to a role for Beclin1 in the mechanism of EC permeability. Indeed, Beclin1-depleted cells showed enhanced barrier recovery after thrombin challenge. Consistent with this, Beclin1 silencing was associated with enhanced reassembly of VE-cadherin at AJs after thrombin challenge. These data are in accord with our earlier report showing a similar barrier enhancing effect of autophagy inhibitor 3-methyladenine (3-MA) in thrombin-treated EC (54, 67), and unveil a new role of Beclin1 in the mechanism of EC barrier disruption.
In summary, this study show that Beclin1 controls NF-κB activation and thereby EC inflammation by a dual mechanism that serves to facilitate the translocation of RelA/p65 for its DNA binding in the nucleus after its release from IκBα in the cytoplasm, and to increase the transcriptional capacity of the bound RelA/p65 through it phosphorylation at Ser536. Additionally, our data reveal a hitherto unknown role of Beclin1 in the mechanism of EC permeability by its ability to regulate AJs and actin cytoskeleton influence the reassembly of AJs and barrier recovery.
Supplementary Material
Highlights.
Thrombin-induced autophagy in endothelial cells is mediated by Beclin1
Beclin1 is a critical determinant of endothelial cell inflammation via its ability to activate NF-κB
Beclin1 regulates endothelial barrier dysfunction by preventing the reassembly of adherens junctions
Footnotes
This work was supported by NHLBI grants HL116632, HL130870, and HL138538, and NIGMS grant GM130463. This work was also supported in part by National Institute of Environmental Health Sciences Center (EHSC) Grant ES-01247.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Abbreviations used in this paper: EC, endothelial cell; VCAM-1, vascular cell adhesion molecule-1; ICAM-1, intercellular adhesion molecule-1; PMN, polymorphonuclear leukocytes; NF-κB, nuclear factor-κB; IKKβ, IkappaB kinase; IL-6, interleukin 6; IL-1B, interleukin 1 beta; TNF-α, tumor necrosis factor alpha; LC3, microtubule-associated protein 1A/1B-light chain 3; HPAEC, human pulmonary artery EC; PAGE, polyacrylamide gel electrophoresis; LUC, luciferase; PBS, phosphate-buffered saline; TBS, Tris-buffered saline; PMSF, phenylmethylsulfonyl fluoride.
REFERENCES
- 1.Mehta D, and Malik AB. 2006. Signaling mechanisms regulating endothelial permeability. Physiol Rev 86: 279–367. [DOI] [PubMed] [Google Scholar]
- 2.Bhattacharya J, and Matthay MA. 2013. Regulation and repair of the alveolar-capillary barrier in acute lung injury. Annu Rev Physiol 75: 593–615. [DOI] [PubMed] [Google Scholar]
- 3.Garcia JG 2009. Concepts in microvascular endothelial barrier regulation in health and disease. Microvasc Res 77: 1–3. [DOI] [PubMed] [Google Scholar]
- 4.Liu SF, and Malik AB. 2006. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol 290: L622–L645. [DOI] [PubMed] [Google Scholar]
- 5.Rahman A, and Fazal F. 2011. Blocking NF-κB: an inflammatory issue. Proc Am Thorac Soc 8: 497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dejana E, and Vestweber D. 2013. The role of VE-cadherin in vascular morphogenesis and permeability control. Prog Mol Biol Transl Sci 116: 119–144. [DOI] [PubMed] [Google Scholar]
- 7.Ghosh S, and Hayden MS. 2008. New regulators of NF-kappaB in inflammation. Nat Rev Immunol 8: 837–848. [DOI] [PubMed] [Google Scholar]
- 8.Rahman A, Anwar KN, True AL, and Malik AB. 1999. Thrombin-induced p65 homodimer binding to downstream NF-kappa B site of the promoter mediates endothelial ICAM-1 expression and neutrophil adhesion. J Immunol 162: 5466–5476. [PubMed] [Google Scholar]
- 9.Ledebur HC, and Parks TP. 1995. Transcriptional regulation of the intercellular adhesion molecule-1 gene by inflammatory cytokines in human endothelial cells. Essential roles of a variant NF-kappa B site and p65 homodimers. J Biol Chem 270: 933–943. [DOI] [PubMed] [Google Scholar]
- 10.Ye X, Ding J, Zhou X, Chen G, and Liu SF. 2008. Divergent roles of endothelial NF-kappaB in multiple organ injury and bacterial clearance in mouse models of sepsis. J Exp Med 205: 1303–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rahman A, and Fazal F. 2009. Hug tightly and say goodbye: role of endothelial ICAM-1 in leukocyte transmigration. Antioxid Redox Signal 11: 823–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fazal F, Bijli KM, Minhajuddin M, Rein T, Finkelstein JN, and Rahman A. 2009. Essential role of cofilin-1 in regulating thrombin-induced RelA/p65 nuclear translocation and intercellular adhesion molecule 1 (ICAM-1) expression in endothelial cells. J Biol Chem 284: 21047–21056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Minhajuddin M, Bijli KM, Fazal F, Sassano A, Nakayama KI, Hay N, Platanias LC, and Rahman A. 2009. Protein kinase C-delta and phosphatidylinositol 3-kinase/Akt activate mammalian target of rapamycin to modulate NF-kappaB activation and intercellular adhesion molecule-1 (ICAM-1) expression in endothelial cells. J Biol Chem 284: 4052–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.You J, Peng W, Lin X, Huang QL, and Lin JY. 2010. PLC/CAMK IV-NF-kappaB involved in the receptor for advanced glycation end products mediated signaling pathway in human endothelial cells. Mol Cell Endocrinol 320: 111–117. [DOI] [PubMed] [Google Scholar]
- 15.Paria BC, Malik AB, Kwiatek AM, Rahman A, May MJ, Ghosh S, and Tiruppathi C. 2003. Tumor necrosis factor-alpha induces nuclear factor-kappaB-dependent TRPC1 expression in endothelial cells. J Biol Chem 278: 37195–37203. [DOI] [PubMed] [Google Scholar]
- 16.Mehta D, Ravindran K, and Kuebler WM. 2014. Novel regulators of endothelial barrier function. Am J Physiol Lung Cell Mol Physiol 307: L924–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giannotta M, Trani M, and Dejana E. 2013. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell 26: 441–454. [DOI] [PubMed] [Google Scholar]
- 18.Tiruppathi C, Ahmmed GU, Vogel SM, and Malik AB. 2006. Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation 13: 693–708. [DOI] [PubMed] [Google Scholar]
- 19.Dudek SM, and Garcia JG. 2001. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol 91: 1487–1500. [DOI] [PubMed] [Google Scholar]
- 20.Tian Y, Gawlak G, O'Donnell JJ, Birukova AA, and Birukov KG. 2016. Activation of Vascular Endothelial Growth Factor (VEGF) Receptor 2 Mediates Endothelial Permeability Caused by Cyclic Stretch. J Biol Chem 291: 10032–10045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fazal F, Minhajuddin M, Bijli KM, McGrath JL, and Rahman A. 2007. Evidence for actin cytoskeleton-dependent and -independent pathways for RelA/p65 nuclear translocation in endothelial cells. J Biol Chem 282: 3940–3950. [DOI] [PubMed] [Google Scholar]
- 22.Fazal F, Bijli KM, Murrill M, Leonard A, Minhajuddin M, Anwar KN, Finkelstein JN, Watterson DM, and Rahman A. 2013. Critical role of non-muscle myosin light chain kinase in thrombin-induced endothelial cell inflammation and lung PMN infiltration. PLoS One 8: e59965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leonard A, Marando C, Rahman A, and Fazal F. 2013. Thrombin selectively engages LIM kinase 1 and slingshot-1L phosphatase to regulate NF-κB activation and endothelial cell inflammation. Am J Physiol Lung Cell Mol Physiol 305: L651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Springer TA 1994. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 76: 301–314. [DOI] [PubMed] [Google Scholar]
- 25.Gao R, Ma Z, Hu Y, Chen J, Shetty S, and Fu J. 2015. Sirt1 restrains lung inflammasome activation in a murine model of sepsis. Am J Physiol Lung Cell Mol Physiol 308: L847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tiruppathi C, Soni D, Wang DM, Xue J, Singh V, Thippegowda PB, Cheppudira BP, Mishra RK, Debroy A, Gian Z, Bachmaier K, Zhao YY, Christman JW, Vogel SM, Ma A, and Malik AB. 2014. The transcription factor DREAM represses the deubiquitinase A20 and mediates inflammation. Nat Immunol 15: 239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lucas R, Verin AD, Black SM, and Catravas JD. 2009. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem Pharmacol 77:1763–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gong H, Rehman J, Tang H, Wary K, Mittal M, Chaturvedi P, Zhao YY, Chatturvedi P, Zhao Y, Komarova YA, Komorova YA, Vogel SM, and Malik AB. 2015. HIF2α signaling inhibits adherens junctional disruption in acute lung injury. J Clin Invest 125: 652–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mizushima N 2007. Autophagy: process and function. Genes Dev 21: 2861–2873. [DOI] [PubMed] [Google Scholar]
- 30.Xie Z, and Klionsky DJ. 2007. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 9: 1102–1109. [DOI] [PubMed] [Google Scholar]
- 31.Shintani T, and Klionsky DJ. 2004. Autophagy in health and disease: a double-edged sword. Science 306: 990–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, and Mizushima N. 2004. The role of autophagy during the early neonatal starvation period. Nature 432: 1032–1036. [DOI] [PubMed] [Google Scholar]
- 33.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, and Sadoshima J. 2007. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 100: 914–922. [DOI] [PubMed] [Google Scholar]
- 34.Efeyan A, Zoncu R, Chang S, Gumper I, Snitkin H, Wolfson RL, Kirak O, Sabatini DD, and Sabatini DM. 2013. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 493: 679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, and Tanaka K. 2006. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441: 880–884. [DOI] [PubMed] [Google Scholar]
- 36.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, and Mizushima N. 2006. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441: 885–889. [DOI] [PubMed] [Google Scholar]
- 37.Brest P, Corcelle EA, Cesaro A, Chargui A, Belaïd A, Klionsky DJ, Vouret-Craviari V, Hebuterne X, Hofman P, and Mograbi B. 2010. Autophagy and Crohn’s disease: at the crossroads of infection, inflammation, immunity, and cancer. Curr Mol Med 10: 486–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ryter SW, Nakahira K, Haspel JA, and Choi AM. 2012. Autophagy in pulmonary diseases. Annu Rev Physiol 74: 377–401. [DOI] [PubMed] [Google Scholar]
- 39.Sun Y, Li C, Shu Y, Ju X, Zou Z, Wang H, Rao S, Guo F, Liu H, Nan W, Zhao Y, Yan Y, Tang J, Zhao C, Yang P, Liu K, Wang S, Lu H, Li X, Tan L, Gao R, Song J, Gao X, Tian X, Qin Y, Xu KF, Li D, Jin N, and Jiang C. 2012. Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Sci Signal 5: ra16. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Liu G, Dull RO, Schwartz DE, and Hu G. 2014. Autophagy in pulmonary macrophages mediates lung inflammatory injury via NLRP3 inflammasome activation during mechanical ventilation. Am J Physiol Lung Cell Mol Physiol 307: L173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jin L, Batra S, and Jeyaseelan S. 2017. Deletion of Nlrp3 Augments Survival during Polymicrobial Sepsis by Decreasing Autophagy and Enhancing Phagocytosis. J Immunol 198: 1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, and Choi AM. 2011. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Slavin SA, Leonard A, Grose V, Fazal F, and Rahman A. 2018. Autophagy inhibitor 3-methyladenine protects against endothelial cell barrier dysfunction in acute lung injury. Am J Physiol Lung Cell Mol Physiol 314: L388–L396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kang R, Zeh HJ, Lotze MT, and Tang D. 2011. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 18: 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kihara A, Kabeya Y, Ohsumi Y, and Yoshimori T. 2001. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep 2: 330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, and Levine B. 1998. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol 72: 8586–8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marquez RT, and Xu L. 2012. Bcl-2:Beclin 1 complex: multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am J Cancer Res 2: 214–221. [PMC free article] [PubMed] [Google Scholar]
- 48.Copetti T, Bertoli C, Dalla E, Demarchi F, and Schneider C. 2009. p65/RelA modulates BECN1 transcription and autophagy. Mol Cell Biol 29: 2594–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shu CW, Chang HT, Wu CS, Chen CH, Wu S, Chang HW, Kuo SY, Fu E, Liu PF, and Hsieh YD. 2016. RelA-Mediated BECN1 Expression Is Required for Reactive Oxygen Species-Induced Autophagy in Oral Cancer Cells Exposed to Low-Power Laser Irradiation. PLoS One 11: e0160586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zeng M, Wei X, Wu Z, Li W, Zheng Y, Li B, Meng X, Fu X, and Fei Y. 2016. Simulated ischemia/reperfusion-induced p65-Beclin 1-dependent autophagic cell death in human umbilical vein endothelial cells. Sci Rep 6: 37448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bijli KM, Fazal F, and Rahman A. 2012. Regulation of RelA/p65 and Endothelial Cell Inflammation by Proline-Rich Tyrosine Kinase 2. Am J Respir Cell Mol Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bijli KM, Fazal F, Minhajuddin M, and Rahman A. 2008. Activation of Syk by protein kinase C-delta regulates thrombin-induced intercellular adhesion molecule-1 expression in endothelial cells via tyrosine phosphorylation of RelA/p65. J Biol Chem 283: 14674–14684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bijli KM, Fazal F, Slavin SA, Leonard A, Grose V, Alexander WB, Smrcka AV, and Rahman A. 2016. Phospholipase C-epsilon signaling mediates endothelial cell inflammation and barrier disruption in acute lung injury. Am J Physiol Lung Cell Mol Physiol 311: L517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anwar KN, Fazal F, Malik AB, and Rahman A. 2004. RhoA/Rho-associated kinase pathway selectively regulates thrombin-induced intercellular adhesion molecule-1 expression in endothelial cells via activation of I kappa B kinase beta and phosphorylation of RelA/p65. J Immunol 173: 6965–6972. [DOI] [PubMed] [Google Scholar]
- 55.Tiruppathi C, Freichel M, Vogel SM, Paria BC, Mehta D, Flockerzi V, and Malik AB. 2002. Impairment of store-operated Ca2+ entry in TRPC4(−/−) mice interferes with increase in lung microvascular permeability. Circ Res 91: 70–76. [DOI] [PubMed] [Google Scholar]
- 56.Mehta D, Rahman A, and Malik AB. 2001. Protein kinase C-alpha signals rho-guanine nucleotide dissociation inhibitor phosphorylation and rho activation and regulates the endothelial cell barrier function. J Biol Chem 276: 22614–22620. [DOI] [PubMed] [Google Scholar]
- 57.He C, and Levine B. 2010. The Beclin 1 interactome. Curr Opin Cell Biol 22: 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trocoli A, and Djavaheri-Mergny M. 2011. The complex interplay between autophagy and NF-κB signaling pathways in cancer cells. Am J Cancer Res 1: 629–649. [PMC free article] [PubMed] [Google Scholar]
- 59.Nivon M, Richet E, Codogno P, Arrigo AP, and Kretz-Remy C. 2009. Autophagy activation by NFkappaB is essential for cell survival after heat shock. Autophagy 5: 766–783. [DOI] [PubMed] [Google Scholar]
- 60.Chen L, Liu D, Zhang Y, Zhang H, and Cheng H. 2015. The autophagy molecule Beclin 1 maintains persistent activity of NF-κB and Stat3 in HTLV-1-transformed T lymphocytes. Biochem Biophys Res Commun 465: 739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zeng M, Wei X, Wu Z, Li W, Li B, Zhen Y, Chen J, Wang P, and Fei Y. 2013. NF-κB-mediated induction of autophagy in cardiac ischemia/reperfusion injury. Biochem Biophys Res Commun 436: 180–185. [DOI] [PubMed] [Google Scholar]
- 62.Minhajuddin M, Fazal F, Bijli KM, Amin MR, and Rahman A. 2005. Inhibition of mammalian target of rapamycin potentiates thrombin-induced intercellular adhesion molecule-1 expression by accelerating and stabilizing NF-kappa B activation in endothelial cells. J Immunol 174: 5823–5829. [DOI] [PubMed] [Google Scholar]
- 63.Ren T, Zheng B, Huang Y, Wang S, Bao X, Liu K, and Guo W. 2019. Osteosarcoma cell intrinsic PD-L2 signals promote invasion and metastasis via the RhoA-ROCK-LIMK2 and autophagy pathways. Cell Death Dis 10: 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gurkar AU, Chu K, Raj L, Bouley R, Lee SH, Kim YB, Dunn SE, Mandinova A, and Lee SW. 2013. Identification of ROCK1 kinase as a critical regulator of Beclin1-mediated autophagy during metabolic stress. Nat Commun 4: 2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hayden MS, and Ghosh S. 2004. Signaling to NF-kappaB. Genes Dev 18: 2195–2224. [DOI] [PubMed] [Google Scholar]
- 66.Bijli KM, Fazal F, and Rahman A. 2012. Regulation of Rela/p65 and endothelial cell inflammation by proline-rich tyrosine kinase 2. Am J Respir Cell Mol Biol 47: 660–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chao CH, Chen HR, Chuang YC, and Yeh TM. 2018. Macrophage Migration Inhibitory Factor-Induced Autophagy Contributes to Thrombin-Triggered Endothelial Hyperpermeability in Sepsis. Shock 50: 103–111. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.