

SUMMARY
Progression through states of pluripotency is required for cells in early mammalian embryos to transition away from heightened self-renewal and toward competency for lineage specification. Here, we use a CRISPR mutagenesis screen in mouse embryonic stem cells (ESCs) to identify unexpected roles for nuclear export and intracellular Ca2+ homeostasis during the exit out of the naive state of pluripotency. Mutation of a plasma membrane Ca2+ pump encoded by Atp2b1 increased intracellular Ca2+ such that it overcame effects of intracellular Ca2+ reduction, which is required for naive exit. Persistent self-renewal of ESCs was supported both in Atp2b1−/− Tcf7l1−/− double-knockout ESCs passaged in defined media alone (no LIF or inhibitors) and in wild-type cells passaged in media containing only calcitonin and a GSK3 inhibitor. These new findings suggest a central role for intracellular Ca2+ in safe-guarding naive pluripotency.
Graphical Abstract
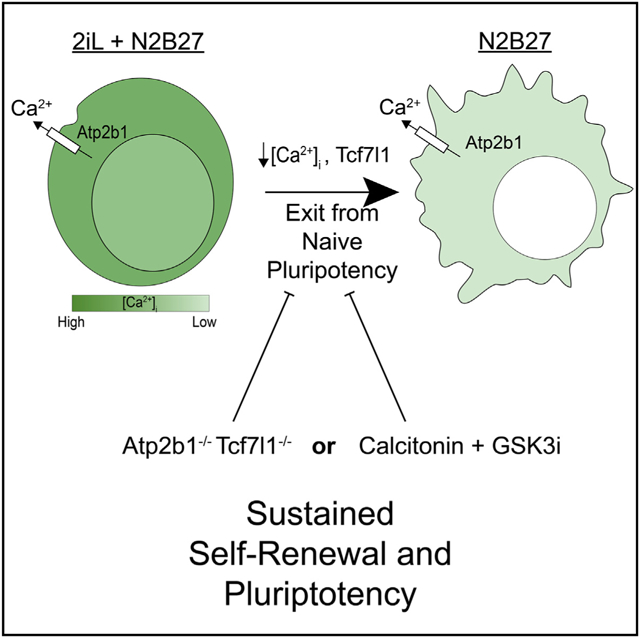
In Brief
MacDougall et al. identify Ranbp3 and Atp2b1 as regulators of naive pluripotency exit. Intracellular Ca2+ levels decrease during exit, and increasing Ca2+ levels in combination with GSK3i or Tcf7l1−/− sustains self-renewal and pluripotency in the absence of inhibitors or LIF.
INTRODUCTION
In early mammalian embryos, the cells destined to contribute to the adult must expand in number while remaining pluripotent. Embryos proceed through substantially different environments as they transition from a free embryo to one that has successfully implanted in the uterus. As a part of the embryo, the pluripotent cells must also progress through different cellular environments throughout this process. An appreciation for these dynamics has recently coincided with the characterization of distinct states of pluripotency (Morgani et al., 2017; Shahbazi et al., 2017) and the concept that progression through pluripotent states enables cells to maintain pluripotency in the diverse cellular environments they encounter during early embryogenesis.
The concept of different pluripotent states was struck forward with the identification of a ground state of pluripotency by Ying and Smith. Mouse embryonic stem cells (ESCs) propagated and retained pluripotency in chemically defined N2B27 media supplemented with only two inhibitors (2i) to Mek (MEKi) and Gsk3 (GSK3i) kinases (Ying et al., 2008). Addition of LIF cytokine activated Jak/Stat3 signaling, and it stimulated self-renewal in what have become standard 2iL conditions for ESCs (Morgani et al., 2017; Smith et al., 1988; Williams et al., 1988; Wray et al., 2011). Analyses of gene expression and epigenetic characteristics indicate that ESCs in these in vitro conditions correspond to cells in the inner cell mass of the blastocyst, in the so-called naive state of pluripotency (Morgani et al., 2017).
Exit from the naive state is obligatory for competent responses to lineage specification signals during gastrulation (Morgani et al., 2017; Smith, 2017). Removal of 2iL and addition of basic fibroblast growth factor (Fgf) and activin A rapidly converts naive ESCs to the primed state of pluripotency (Buecker et al., 2014; Hayashi et al., 2011), which is used to categorize epiblast cells from post-implantation embryos, mouse epiblast-derived stem cells (EpiSCs) derived from implanted embryos (Brons et al., 2007; Tesar et al., 2007), and human ESCs in traditional culture conditions (Thomson et al., 1998). Intermediate pluripotent states, called poised and formative, have recently been characterized for cells exiting naive pluripotency and are defined by gene expression, microRNA activities, histone modifications, and DNA methylation characteristics (Du et al., 2018; Kinoshita and Smith, 2018; Morgani et al., 2017; Smith, 2017).
Factors mediating the exit from naive pluripotency have been identified, and some of their mechanisms have been elucidated. Tcf7l1 is a transcription factor that targets naive genes (Esrrb and Nanog) for repression in ESCs, and its activity is effectively inhibited with Wnt/β-catenin signaling or GSK3i (Martello et al., 2012; Pereira et al., 2006; Shy et al., 2013; Wray et al., 2011; Yi et al., 2011). In mouse embryos, Tcf7l1 is required for the timely response of epiblast cells to lineage specification signals during gastrulation (Hoffman et al., 2013) and is considered the critical regulator of the effect of GSK3i in 2iL conditions (Wray et al., 2011; Yi et al., 2011). In addition to the Gsk3/Tcf7l1 axis and LIF/Jak/Stat3 signaling (Martello et al., 2013), the relocation of Oct4 binding by Otx2 (Buecker et al., 2014), the mTorc1 pathway control of Gsk3 and Tfe3 transcription factor localization (Betschinger et al., 2013; Villegas et al., 2019), the activities of DNA methylcytosine oxidases Tet1 and Tet2 (Fidalgo et al., 2016; Finley et al., 2018), and unique microRNA dynamics (Du et al., 2018) have all been genetically demonstrated to play important roles in the exit from naive pluripotency.
Interestingly, there is little evidence that genetically interrupting the mechanisms necessary for the exit from naive pluripotency could be sufficient to enable self-renewal without exogenously added inhibitors or cytokines. For example, Tcf7l1−/− mutants delay the exit from the naive state, but they cannot sustain self-renewal in N2B27 without 2i conditions (Wray et al., 2011; Yi et al., 2011; Ying et al., 2008). In this study, we use a genome-wide CRISPR mutagenesis screen to probe the genetic underpinnings of naive pluripotency, and we examine whether cessation of self-renewal is inexorable upon withdrawal of 2iL. The screen identified several genes important for exiting the naive state, including those involved in nuclear export (Ranbp3) and cellular Ca2+ homeostasis (Atp2b1). Surprisingly, reduction of intracellular Ca2+ was required for exiting naive pluripotency, and inactivating Atp2b1 mutations inhibited naive exit by increasing intra-cellular Ca2+ levels in ESCs. Moreover, Atp2b1−/− Tcf7l1−/− double-mutant ESCs were able to self-renew in N2B27 media alone without 2iL, indicating a central role for intracellular Ca2+ in pluripotency. Finally, maintaining high intracellular Ca2+ with calcitonin combined with GSK3i was sufficient to maintain self-renewal in wild-type ESCs, demonstrating a robust effect of the combination. Given the importance of Ca2+ signaling for the trophectoderm and endometrium during implantation (Armant, 2005; Tosti et al., 2016; Wang et al., 2013), these new findings make it tempting to speculate that intracellular Ca2+ is used in pluripotent cells to coordinate transitions between states of pluripotency within the embryo.
RESULTS
Identification of Genes Required for Exit from Naive Pluripotency with CRISPR-Cas9
To identify genes important for transitions between pluripotency states, multiple genome-wide CRISPR mutagenesis screens have been conducted. Screens have used cells harboring fluorescent reporters and isolated mutant cells that aberrantly retain or lose reporter activity as a means to identify genes important for transitions (Baeg et al., 2005; Betschinger et al., 2013; Li et al., 2018; Villegas et al., 2019; Yang et al., 2012). A screen recently completed by Yusa and colleagues was effective at identifying a large number of genes that play a role in the exit from naive pluripotency (Li et al., 2018). For the present study, we aimed for a more stringent test and the identification of genes important for a sustained, long-term maintenance of the naive state in proliferating cells. For selection of exit-resistant mutants, we took advantage of the poor colony-forming ability of primed cells when plated in 2iL (Figure 1A). Removal of 2iL and addition of Fgf2 (12 ng/mL) has previously been demonstrated to stimulate exit from naive pluripotency (Buecker et al., 2014; Tsukiyama and Ohinata, 2014). Selection using this conversion of media was validated by loss of greater than 90% of naive colony-forming units within 4 days of Fgf2 treatment (Figure 1A). As expected, Tcf7l1−/− ESCs retain the ability to form colonies in naive conditions even after 5 days in Fgf2-containing media (Figure 1A, bottom). Based on this pilot experiment, we concluded that this replating procedure of switching between 2iL and Fgf2-containing media would provide sufficient selection of mutants defective at exiting the naive state.
Figure 1. CRISPR Screen for Genes Required for Exit from Naive Pluripotency.

(A) (Top) Schematic illustrating the replating assay used to enrich for mutants with defects in naive exit. Naive state ESCs in 2iL are transitioned toward epiblast-like cells (EpiLCs) by withdrawing 2iL and adding Fgf2. After naive exit, EpiLCs are competent for lineage specification. (Bottom) Exit from the naive state is assessed by colony-forming ability in 2iL condition. Colony formation drops for wild-type cells after switching media to −2iL + Fgf2 media. Tcf7l1−/− ESCs serve as a well-characterized, positive control for delayed naive exit. Data represent the mean colony count ± SD of n = 2–3.
(B) (Top) Schematic of the CRISPR screen flow illustrating that Cas9-expressing ESCs are transduced with a lentiviral sgRNA library and subjected to two rounds of −2iL/+Fgf2-to-2iL replating for positive selection of mutant phenotypes. (Bottom) The distributions of normalized sgRNA counts in the plasmid library and both biological replicates of the screen before and after selection are plotted. n = 2 represents independent lentivirus preparations. Related to Table S1 and Figure S1.
(C) Sequencing data for the internal control gene, Tcf7l1, are shown as reads counts for each of the 5 individual sgRNAs in the sgRNA library, for both biological replicates (n = 2), before and after selection. Data are presented as boxplots with overlaid swarm plots of the individual data points.
(D) MAGeCK was used to create a robust ranking algorithm (RRA) score for each gene targeted in the screen, taking into account the effect of all sgRNAs targeting that specific gene. The ranked RRA score distribution of gene level data with FDR cutoffs demarcated and shaded and the top 10 genes plotted as points. n = 2. Related to Table S2.
(E) MAGeCK-RRA was used to score Gene Ontology, biological process terms from all sgRNA data. The table shows well-characterized and poorly understood Gene Ontology, biological process terms that meet a cutoff of FDR < 10%. Related to Table S3.
To mutate genes, a stable Cas9-expressing ESC line was transduced with a library consisting of 87,897 single guide RNAs (sgRNAs) targeting the 19,150 protein coding genes in the mouse genome (Koike-Yusa et al., 2014). Cells were functionally confirmed to express high Cas9 activity following transfection of sgRNA expression plasmids (Figure S1A). Expression of Cas9 did not alter naive ESC morphology (Figure S1B). The entire sgRNA library was introduced into Cas9-expressing cells by lentivirus transductions in two biological replicates. Cells were subjected to puromycin selection to eliminate nontransduced ESCs prior to mutant selection by replating (Figure 1B; Table S1).
Next-generation sequencing (NGS) of sgRNA amplified from genomic DNA was used to assess frequency of each individual sgRNA in populations of cells. Prior to replating for selection of the mutant phenotype, the ESC mutant library displayed only a slightly reduced complexity of sgRNA sequences relative to the DNA used to generate lentivirus (Figure 1B; compare blue to green). Some reduction was expected due to mutation of essential genes in 2iL conditions, and the complexity of the ESC mutant library indicates a potential genome-wide mutagenesis. As expected, the complexity of the sgRNA sequences was greatly reduced by the replating selection scheme, consistent with those samples having been enriched with mutants possessing enhanced replating capability (Figure 1B; red). Whereas the replicates of the ESC mutant library were similar to each other, enriched mutant samples were less similar, which is indicative of successful selection (Figures S1C and S1D; Table S1). We also confirmed that there was no genomic location bias that occurred as a result of screening (Figure S1E; Table S1). Thus, the relative increased frequency of sgRNA targeting specific genes in enriched mutants was used to infer a potential effect of the targeted gene on replating capability. Tcf7l1 was used as an internal control for the selection based on effects of Tcf7l1−/− cells (Figure 1A). All five of the Tcf7l1-targeting sgRNAs in the library were significantly increased by the mutant selection process (Figure 1C), demonstrating the enrichment of mutants with defects in the exit from naive pluripotency.
To identify genes from changes in frequencies of sgRNAs after replating selection, we used the robust ranking algorithm (RRA) from MAGeCK (Li et al., 2014, 2015). The frequencies of all sgRNAs targeting a gene are considered together to reduce false positives from bystander effects and to give each gene an RRA enrichment score (Table S2). The distribution of RRA scores showed relatively few genes passing typically used cutoff for positives (Figure 1D; Table S2; 35 genes with false discovery rate [FDR] < 10%). Of these 35 genes, 28 were among the 563 genes identified by the CRISPR screen based on reporter gene activity (Table S2; Li et al., 2018). This analysis identified several categories with well-documented roles in pluripotency and naive pluripotency, including regulation of mitogen-activated protein kinase (Fgf/Mek/Erk) signaling and Stat3 activity (Figure 1E; Table S3). Given the roles of these categories with respect to LIF and MEKi from 2iL conditions, we interpreted their identification as an indication that the screen hit relevant gene targets. Notably, categories with poorly understood roles in naive pluripotency, such as endosome-lysosome vesicular trafficking and nuclear export, were also identified with this analysis.
To independently validate individual candidate genes identified by the screen, we used an arrayed approach, which directly measured mutagenesis of the targeted gene. In separate transient transfections, sgRNAs targeting the top 40 candidate genes and control sgRNAs were introduced into naive ESCs (Clarke et al., 2018), a starting level of mutagenesis (indel frequency) was determined by NGS, and cells were subjected to the naive exit replating procedure (Figure S2A). Because Cas9 was expressed transiently, changes to indel frequencies after replating are caused by selective advantage or disadvantage of cells harboring indel mutations among the mixed population of cells (Figures S2A and S2B; Table S4). 26 of the 40 candidates displayed an increased indel frequency after two rounds of replating (Figures S2B and S2C; Table S4). Eleven genes displayed a reduced indel frequency after two rounds of replating, including the olfactory receptor OLFR478 gene, which is not expressed in pluripotent cells. Notably, a set of genes not clearly linked to processes known to control exit from the naive state were validated; these include the E3 ubiquitin ligase Fbxw7, the plasma membrane Ca2+ transporter Atp2b1, and a nuclear export protein Ranbp3.
Clonal, Functional Validation of Selected Candidate Genes
Recent research has elucidated some genetic determinants of fitness selection on cell competition among pluripotent cells (Bowling et al., 2018). Given the screen utilized selective advantage to identify genes, it was possible that some of the gene hits enhance cell competition rather than pluripotency, per se. To eliminate effects resulting from a genetically mixed population of cells and directly assess pluripotency, we generated clonal mutant ESC lines from the previously generated Rex1-GFPd2 insertion cells (Wray et al., 2011). Nine candidate genes were chosen based on their validation (Figure S2; Table S4) and our assignment of the genes into either an mTorc1 cluster (Tsc1 and Rragc), a vesicle trafficking cluster (Rab7, Vps11, and Vps35), a cluster of unexplored genes (Fbxw7, Atp2b1, and Ranbp3), or the positive control (Tcf7l1). For each of these genes, individual mutant lines were generated by transient CRISPRCas9 activity, isolation of clones, DNA sequence verification of mutations, and expansion of cultures in 2iL media. Each of the mutant lines and the original Rex1-GFPd2 line (“wild-type”) displayed similarly high levels of GFP fluorescence in naive 2iL conditions (Figures 2 and S3; Table S5).
Figure 2. Delayed Loss of Rex1-GFPd2 Naive State Reporter for Mutant ESC Lines.

Flow cytometry measurement of GFP fluorescence is shown for all cells after 48 h in either 2iL, −2iL/+Fgf2, −GSK3i, −GSK3i/−MEKi, or −LIF media (treatment). The mean Rex1-GFPd2 fluorescence ± SD is shown and relates to the representative flow histograms in Figure S2. Wild-type is overlaid on all mutant and control samples as the black, unfilled bars for ease of comparison. Mutants are organized by assignment to the clusters illustrated to the left. Related to Table S5. n = 2 biological replicates of >10,000 live-cell, singlet events were acquired for each treatment of each mutant. The error bars represent SD.
When 2iL is withdrawn and Fgf2 is added, Rex1-GFPd2 activity is lost in wild-type cells within 48 h, but it is retained in Tcf7l1−/− cells (Figures 2, S3A, and S3B; Table S5). We reasoned that mutations that prevent or delay exit from the naive state would maintain Rex1-GFPd2 similar to Tcf7l1−/− cells, whereas mutants that provide primarily a survival advantage would lose Rex1-GFPd2 fluorescence. All mutants maintained Rex1-GFPd2 above wild-type levels, and this was well correlated with replating efficiency in 2iL after 72 h in the absence of 2iL (Figures 2 and S3D). Many of the mutants maintained Rex1-GFPd2 quite weakly relative to the Tcf7l1−/− control (Figure 2), despite strong selection in the genetic validation experiment (Figure S2).
To determine whether the candidate genes could be stratified based on pathways impacted, we tested mutants for differential sensitivity to withdrawal of combinations of 2iL components (Figures S3C and S3D). Interestingly, none of the mutants displayed a very distinct pathway-specific sensitivity; resistance to individual component withdrawal was proportional to resistance to Fgf2 for most mutants (Figures 2 and S3D). The exception to this finding was the vesicular trafficking cluster, which appeared to be more resistant to removal of GSK3i than other components (Figures 2 and S3D; Table S5). The two mutants with the greatest resistance to reducing Rex1-GFPd2 harbored mutations in Atp2b1 and Ranbp3. These two genes do not have previously described roles in pluripotent cells; however, replating capability and retention of Rex-GFPd2 after challenge with Fgf2 indicated that they likely had important activities required for the exit from naive pluripotency. We selected candidate genes Atp2b1 and Ranbp3 for further mechanistic examinations into their roles in pluripotency.
RanBP3 Is Required for Proper Cessation of Stat3 and Wnt Signaling during Naive Exit
Ran-binding protein 3 (RanBP3), encoded by Ranbp3, has been shown to accelerate both Crm1-dependent and independent nuclear export via various processes involving the Ran guanosine triphosphatase (GTPase) (Koyama et al., 2014; Lindsay et al., 2001; Yoon et al., 2008). Previous reports show that nuclear accumulation of Ran was diminished in Ranbp3−/− cells (Yoon et al., 2008). Therefore, Ran localization was tested as a potential mediator of effects in Ranbp3−/− cells. As cells exit naive pluripotency, Ran immunofluorescence shifted from being primarily cytoplasmic in naive cells to nuclear after withdrawal of 2iL in wild-type and Tcf7l1−/− cells (Figure S4A). The relative nuclear localization of Ran was diminished in RanBP3−/− cells, consistent with effects in other cell types (Figure S4A; Yoon et al., 2008). To test whether an overall reduction of Ran activity could mediate effects of Ranbp3−/− mutations, loss of Rex1-GFPd2 activity was examined in cells expressing dominant-negative Ran mutants that inhibit nuclear import (G19V and Q69L) and export (T24N; Figure S4B; Izaurralde et al., 1997; Lounsbury et al., 1996; Ren et al., 1995). The lack of effect of Ran mutants (Figure S4B) and absence of Ran mutations from naive pluripotency screens (Hackett et al., 2018; Li et al., 2018) combined to indicate that exit from naive pluripotency was unlikely to be controlled by a switch in Ran activity but perhaps by a subset of targets constitutively regulated by RanBP3.
Newly generated Ranbp3−/− cells (Figures S4C–S4F) were used to test the role of RanBP3 on potential targets of its nuclear export activity. In transformed human cell lines, RanBP3 stimulated cessation of canonical Wnt signaling via nuclear export of active β-catenin (Hendriksen et al., 2005). Given that withdrawal of GSK3i is necessary for exiting naive pluripotency, a role for RanBP3 in cessation of Wnt/β-catenin activity was tested. We transiently transfected ESCs with the SUPERTOPFlash reporter and performed luciferase assays. Absence of RanBP3 increased Wnt activity both in 2iL conditions and after 6 h of GSK3i withdrawal (Figure 3A). The accumulation of Tcf7l1 in Ranbp3−/− cells indicated that the effect of RanBP3 is not caused by loss of Tcf7l1 repressor protein (Figure 3B). We reasoned that, if RanBP3 affects Wnt signaling by nuclear export of active β-catenin, then the ratio of active, non-phosphorylated to total β-catenin should increase in Ranbp3−/− cells. Indeed, the increased ratio of active-to-total β-catenin in Ranbp3−/− mutants was due to an overall decrease in total β-catenin (Figure 3B). This suggested that active β-catenin may be trapped in the nucleus, which was tested by measuring the nuclear localization of active, non-phosphorylated β-catenin in the presence or absence of proteasome inhibitor, MG132, and GSK3i. There was strong enrichment of nuclear, active, non-phosphorylated β-catenin in Ranbp3−/− cells withdrawn from GSK3i and treated with MG132 (Figure 3C). Nuclear β-catenin was not affected by absence of Tcf7l1 (Figure S4G) or the presence of the MEKi and LIF (Figure 3D) These results are consistent with RanBP3 stimulating nuclear export of β-catenin in ESCs, which identifies a mechanism that likely contributes to the delay of Ranbp3−/− cells in exiting the naive state.
Figure 3. Ranbp3 Prevents Prolonged Wnt/β-Catenin and Stat3 Activity during Naive Exit.

(A) ESCs of the indicated genotype were transfected with the Wnt-reporter construct pSuperTOPFLASH and maintained in 2iL or withdrawn from GSK3i for 6 h. The activity of the reporter was measured by relative luciferase activity. Data are presented as mean ± SEM for n = 4 transfections. * and # represent an FDR < 5% and <10% compared to wild-type, respectively.
(B) Western blot was used to detect of active, non-phosphorylated β-catenin, total β-catenin, and Tcf7l1 in the presence or absence of GSK3i for 24 h on gelatin for ESCs with the indicated genotypes. Levels of beta-tubulin were measured as a loading control. Numbers below each band are densitometric intensities rounded to two significant digits. The ratio is the intensity of non-phosphorylated β-catenin divided by intensity of total β-catenin bands. Blot image is representative of 2 independent experiments; n = 2.
(C) The localization of active, non-phosphorylated β-catenin was determined using immunofluorescence detection in wild-type and Ranbp3−/− ESCs in the presence or absence of GSK3i + MG132 for 6 h. Representative images of n = 2 experiments and >10 colonies. DAPI is used as a nuclear counterstain. Scale bars represent 10 μm. Yellow square regions are shown at right for detail with the nuclei indicated by the yellow arrowhead. Tcf7l1−/− cells are shown in Figure S4G.
(D) The localization of active, non-phosphorylated β-catenin was determined using immunofluorescence detection in wild-type and Ranbp3−/− cells in the presence or absence of 2iL for 24 h + MG132 for the final 6 h. Representative images of n = 2 experiments and >10 colonies. DAPI is used as a nuclear counterstain. Scale bars represent 7 μm. Yellow square regions are shown at right for detail with the nuclei indicated by the yellow arrowhead.
(E) Western blot was used to detect phospho-Stat3(Y706) in wild-type, Tcf7l1−/−, and Ranbp3−/− cells in the presence or absence of 2iL for 48 h. Levels of beta-tubulin were measured as a loading control. Numbers below each band are densitometric intensities rounded to two significant digits. Blot image is representative of 2 independent experiments; n = 2.(F) The presence and localization of phospho-Stat3(Y706) in the presence or absence of 2iL for 48 h was determined by immunofluorescence detection of phospho-Stat3(Y706) in wild-type, Tcf7l1−/−, and Ranbp3−/− cells. Representative images of n = 2 experiments and >10 colonies. DAPI is used as a nuclear counterstain. Scale bars represent 10 μm.
(G) The presence and localization of phospho-Stat3(Y706) was determined by immunofluorescence in wild-type and Ranbp3−/− cells withdrawn from LIF in the presence or absence of JAKi (1 μM or 5 μM) for 6 h. Representative images of n = 2 experiments and >10 colonies. DAPI is used as a nuclear counterstain. Scale bars represent 10 μm.
Additional potential targets of RanBP3 nuclear export were chosen to be tested based on previously identified roles of those proteins in pluripotency and nuclear localization. Because failure to export Tfe3 from the nucleus inhibits exit from the naive state (Betschinger et al., 2013; Villegas et al., 2019), we tested whether Ranbp3 mutations affected Tfe3 nuclear localization. The cellular localization of Tfe3 was determined by immunofluorescence in cells subjected to 2iL withdrawal and Fgf2 stimulation (Figure S4H). Tfe3 nuclear localization after 2iL withdrawal was not affected by Ranbp3 or Tcf7l1 mutations, as mutant cells appeared similar to wild-type controls (Figures S4H and S4I). Other mutations, including Tsc1−/− and Rragc−/−, retained more nuclear Tfe3 than wild-type cells (Figure S4I). Therefore, nuclear export of Tfe3 appears to be independent of RanBP3 function during exit from the naive state.
Cessation of nuclear Stat3 signaling is important for exit from the naive state of pluripotency (Martello et al., 2012, 2013; Stuart et al., 2014; Ying et al., 2008). Orthologs of Ranbp3 have been identified as inhibitors of Stat3 signaling activity in genetic screens in Drosophila (Baeg et al., 2005). Therefore, we tested whether RanBP3 stimulated cessation of Stat3 activity by nuclear export. Western blot analysis of total cell lysates showed that phosphorylation of Stat3 (Y705) was retained after 48 h without 2iL in Ranbp3−/− cells, but not in wild-type or Tcf7l1−/− cells (Figure 3E). Immunofluorescent staining of cells for phospho-Stat3 showed that the activated form of Stat3 is retained in nuclei of Ranbp3−/− cells, but not in wild-type or Tcf7l1−/− cells (Figure 3F). These changes in phospho-Stat3 occurred despite little to no change in expression and localization of total Stat3 by western blot and immunofluorescence after 48 h in −2iL/+Fgf2 media (Figures S4J and S4K). To directly distinguish between reduction of phosphorylation of Stat3 and export of nuclear phospho-Stat3, naive state cells in 2iL were switched to media without LIF and with the Jak inhibitor, pyridone 6 (JAKi). In these naive cells, nuclear export of phospho-Stat3 is reduced by absence of RanBP3 (Figure 3G), indicating importance of RanBP3 for nuclear export of phospho-Stat3. We suggest that these new roles of RanBP3 for cessation of both Wnt/β-catenin and Stat signaling support an early step in the transition out of naive pluripotency, wherein RanBP3-mediated nuclear export is required to halt nuclear activity of these signaling pathways.
Atp2b1 Reduction of Intracellular Ca2+ Is Required for Exit from the Naive State
Atp2b1 is a plasma membrane Ca2+ pump that is ubiquitously expressed and is genetically required for early mouse embryo-genesis (Okunade et al., 2004; Yanagida et al., 2004). Atp2b1 transports Ca2+ from the cytoplasm to extracellular space as a component of the normal cellular homeostatic mechanisms that maintain low intracellular Ca2+ concentrations (Strehler, 2015). The resistance to naive exit of Atp2b1−/− cells suggested the hypothesis that control of intracellular Ca2+ is important for the exit from naive pluripotency. To assess intracellular Ca2+ levels among pluripotent cells, including Atp2b1−/− mutants (Figure S5A), a Ca2+-sensitive, ratiometric dye (Fura-2 AM) was used (Grynkiewicz et al., 1985). Control treatments of ionomycin, a Ca2+-specific ionophore, and BAPTA-AM, an intracellular, Ca2+-specific chelator, caused the predicted effects of increasing and reducing intracellular Ca2+ levels in naive and primed cells (Figures S5B–S5D), demonstrating the utility of this method for measuring intracellular Ca2+. When 2iL was withdrawn from ESCs, intracellular Ca2+ levels were reduced in wild-type cells (Figures 4A and S5B–S5D). Mutation of Atp2b1 greatly increased intracellular Ca2+ in 2iL conditions, but mutation of other genes (Tcf7l1, Ranbp3, Rragc, Tsc1, Rab7, Vps11, Vps35, or Fbxw7) did not substantially affect intracellular Ca2+ (Figures 4A and S5B–S5D). Interestingly, the elevated intracellular Ca2+ in Atp2b1−/− mutants was decreased during naive exit, but it remained significantly elevated compared to other cells (Figures 4A and S5B–S5D).
Figure 4. Reduction of Intracellular Ca2+ Is Required for Exit from the Naive State.

(A) Intracellular free Ca2+ was measured with the ratiometric dye, Fura2-AM, over a 48-h time course following 2iL withdrawal in wild-type, Tcf7l1−/−, Atp2b1−/−, and Ranbp3−/− ESCs. Controls are shown in Figure S5D. Fura2 ratio is the 510-nm emission from excitation with 340 nm divided by the 510-nm emission from excitation with 380 nm. Mean represents the mean Fura2 ratio ± SD; n = 3.
(B) Flow cytometry analysis of the naive state with Rex1-GFPd2 ESCs of wild-type (left panel) and Atp2b1−/− (right panel) cells after 40 h in the presence or absence of 2iL and/or BAPTA-AM. n = 2 biological replicates of >10,000 live-cell, singlet events were acquired for each treatment histogram.
(C) Flow cytometry analysis of the naive state with Rex1-GFPd2 ESC after 52 h in the presence or absence of 2iL with or without NCXi or carboxyeosin (50 μM or 5 μM). n = 2 biological replicates of >10,000 live-cell, singlet events were acquired for each treatment.
(D) Fluorescence microscopy of Atp2b1-mCH fusion ESCs. An in-frame fusion of mCherry to the C-terminal of Atp2b1 was engineered by CRISPR-mediated editing of the endogenous Atp2b1 gene. mCherry signal is shown in red. DAPI (blue) is used as a nuclear counterstain. Scale bar represents 7 mm.
(E) Intracellular free Ca2+ is visualized in wild-type or Atp2b1−/− cells transfected with the genetically encoded calcium indicator, GCaMP5G. Transfected cells are incubated in N2B27 in the presence or absence of 2iL for 24 h prior to live-cell imaging. Hoechst is used as a nuclear, live-cell counterstain. Scale bars represent 10 μm.
(F) Quantification of the nuclear-to-cytoplasmic ratio of GCaMP5G signal shown in (E) with the number of cells quantified shown.
To test whether the reduction of intracellular Ca2+ was necessary for exit from naive pluripotency, two types of experiments were performed. First, the elevated Ca2+ levels in Atp2b1−/− cells were reduced by BAPTA-AM, which reduced Rex1-GFPd2 levels in Atp2b1−/− cells after withdrawal of 2iL and addition of Fgf2 (Figures 4B and S5D). BAPTA-AM did not drive loss of Rex1-GFPd2 in the presence of 2iL culture conditions (Figure 4B), indicating a dominant role of 2iL in preventing exit. Second, intracellular Ca2+ levels were increased in wild-type ESCs to determine whether they were sufficient to inhibit naive exit. Treatment with SEA0400, a Na+/Ca2+ exchanger inhibitor (NCXi), and carboxeosin, a non-selective plasma membrane Ca2+ pump inhibitor, each increased intracellular Ca2+ (Figure S5E) and reduced loss of Rex1-GFPd2 in wild-type ESCs after withdrawal of 2iL (Figure 4C). Calcitonin, a peptide hormone, also increased intracellular Ca2+ (Figure S5F), but it did not affect Rex1-GFPd2 in the absence of GSK3i (Figure S5F). Although treatment with NCXi was slightly toxic to ESCs, it prevented loss of naive-like colony morphology (Figure S5G) and increased replating in naive conditions following withdrawal of 2iL and Fgf2 addition (Figure S5H). These results demonstrate that reduction of intracellular Ca2+ is important for exit from the naive state of pluripotency.
Previous experiments using mouse embryos and ESCs in serum and LIF conditions identified activation of the calcineurin-NFATc pathway for the differentiation of pluripotent cells (Li et al., 2011). To test whether this mechanism was directly related to the effects described above, we examined effects of inhibition of calcineurin phosphatase activity with cyclosporine A (CsA). Addition of CsA did not sustain self-renewal after LIF and MEKi were withdrawn (Figure S5I). Although CsA did not affect wild-type cells in 2iL or delay exit after 2iL was withdrawn, it did stimulate exit of Atp2b1−/− mutants and Tcf7l1−/− mutants during 2iL withdrawal (Figure S5J). Loss of Rex1-GFPd2 was also stimulated in Atp2b1−/− mutants by treatment with another calcineurin inhibitor, FK506, and by inhibition of the calcineurin-NFATc interaction with VIVIT (Figure S5J; Aramburu et al., 1999; Fruman et al., 1995). These results indicate a role for calcineurin-NFATc downstream of intracellular calcium for keeping cells in the naive state of pluripotency.
To better elucidate the cellular functions of Atp2b1 in ESCs, subcellular localization of Atp2b1 protein and activity was examined by fluorescence microscopy. An in-frame fusion between Atp2b1 and the mCherry fluorescent protein was engineered using CRISPR-Cas9 (Figure S5K) and confirmed at the Atp2b1 locus in ESCs (Figure S5L). Cells harboring the Atp2b1-mCh fusion gene displayed prominent fluorescence at their plasma membrane (Figure 4D), as expected from the characterization of Atp2b1 from previous research (Di Leva et al., 2008; Okunade et al., 2004). Subcellular localization of relative Ca2+ levels were examined with genetically encoded calcium indicator proteins GCaMP5G (Akerboom et al., 2012) and R-CEPIAer (Suzuki et al., 2014). Ca2+ signals were higher throughout Atp2b1−/− cells in 2iL, including the cytoplasm and nucleus (Figures 4E and 4F and 4F), but not the endoplasmic reticulum (Figure S5M). Upon withdrawal of 2iL, Ca2+ signal diminished for wild-type cells most substantially in the nucleus (Figures 4E and 4F); by contrast, Atp2b1−/− cells retained high nuclear Ca2+ signal (Figures 4E and 4F), suggesting a possible role for Ca2+ in the nucleus during the exit from naive pluripotency.
High Intracellular Ca2+ and Inhibition of Gsk3/Tcf7l1 Are Sufficient for Self-Renewal
RanBP3 and Atp2b1 were particularly interesting regulators of naive pluripotency because they appeared to affect basic cellular activities that had not been previously known to be important for pluripotency. In addition, the effects of each mutant were distinct from effects caused by the well-characterized inhibitor of pluripotency, Tcf7l1; Tcf7l1 expression was not diminished in Ranbp3−/− or Atp2b1−/− ESCs (Figure 3B), intra-cellular Ca2+ decreased in Tcf7l1−/− cells (Figure 4), and phospho-Stat3 and active β-catenin did not remain nuclear in Tcf7l1−/− cells (Figure 3). These observations prompted us to examine a potential epistatic relationship between Tcf7l1 and either Ranbp3 or Atp2b1. In particular, we were interested in whether combining effects of mutating Tcf7l1 with Atp2b1 or with Ranbp3 could enable ESC to self-renew without the inhibitors or LIF.
To begin to elucidate effects of mutations on self-renewal, long-term cell culture capabilities were first examined for Tcf7l1−/−, Atp2b1−/−, and Ranbp3−/− single-mutant ESCs. The stringent test was to determine whether mutant ESCs could be propagated in N2B27 alone, without LIF and inhibitors. Wild-type ESCs rapidly exit naive pluripotency and are incapable of self-renewal when switched to N2B27 alone (Ying et al., 2008), resulting in the failure of cultures to propagate (Figures 5A and S6A). Tcf7l1−/− and Atp2b1−/− mutants delayed their exit from naive pluripotency, but they each decline in ESC colony-forming ability within five passages (Figures 5A and S6A). Ranbp3−/− cells were less effective than either Tcf7l1−/− or Atp2b1−/−, and those single-mutant cells failed to self-renew after a single passage in N2B27 alone (Figure S6A). This result contrasted the strong effect of Ranbp3 mutation in short-term replating and Rex1-GFPd2 assays, where Ranbp3−/− cells displayed strong phenotypes in maintenance of naive state (Figures 2, S2, and S3), and it is consistent with Ranbp3−/− causing a highly penetrant but short-lived delay in the cessation of nuclear activity of proteins, such as phospho-Stat3 and β-catenin (Figure 3).
Figure 5. Atp2b1−/− Tcf7l1−/− Double-Mutant ESCs Self-Renew without Inhibitors or Exogenous Lif.

(A) Differential interference contrast (DIC) images displaying the morphology and density of wild-type, Tcf7l1−/−, Atp2b1−/−, and Atp2b1−/− Tcf7l1−/− double-mutant ESCs in N2B27 for indicated number of days and passage number. Scale bar represents 200 μm. See also Figure S6A.
(B) DIC and epifluorescence images (from Rex1-GFPd2) of Atp2b1−/− Tcf7l1−/− double-mutant ESCs in N2B27 for 10 passages compared to wild-type cells grown in 2iL. Scale bars represent 200 μm.
(C) Flow cytometry analysis of the naive state with Rex1-GFPd2 for wild-type cells cultured in 2iL or serum/LIF/GSK3i or Atp2b1−/− Tcf7l1−/− double-mutant cells cultured continuously either in 2iL or in N2B27 alone for 15 passages (Inh. Ind, p15). n = 2 biological replicates of >10,000 live-cell, singlet events were acquired for each treatment. Negative control reference (gray histogram) is wild-type ESCs in N2B27 on fibronectin for 48 h and is identical in each panel.
(D) The relative mRNA expression (ΔΔCt-method) of naive, formative, and primed state marker genes in inhibitor independent, Atp2b1−/− Tcf7l1−/− double-mutant cells maintained in N2B27 as measured by qRT-PCR. Bars represent mean ± SD for 3 biological replicates. Gapdh expression is used as a loading control. * represents FDR < 5% for indicated comparisons except Rex1 and Fgf5, where it represents FDR < 5% for all comparisons.
(E) The presence and localization of Nanog or Klf4 were determined by immunofluorescence detection in wild-type, Tcf7l1−/−, Atp2b1−/−, Atp2b1−/− Tcf7l1−/− double-mutant ESCs in 2iL, and inhibitor-independent Atp2b1−/− Tcf7l1−/− double-mutant ESCs in N2B27. Representative images of n = 2 experiments and >25 colonies. DAPI is used as a nuclear counterstain. Scale bars represent 10 μm.
Given the ability of Tcf7l1−/− and Atp2b1−/− mutations to extend ESC self-renewal in single mutants, we generated Tcf7l1−/− Atp2b1−/− double-mutant ESCs (Figure S6B). Atp2b1−/− Tcf7l1−/− double mutants did not display apparent defects in 2iL media and continued to proliferate and form colonies with naive-like morphology after withdrawal of 2iL (Figure 5A). Cultures of Atp2b1−/− Tcf7l1−/− double-mutant cells did not experience a significant bottleneck during passaging in N2B27 alone, and they retained the ability to form colonies when replated onto 2iL conditions (Figure S6C). In contrast, the Atp2b1−/− and Tcf7l1−/− single mutants only retained the initial number of naive-competent colony-forming units without expansion through passaging (Figure S6C). Through passage 10, Atp2b1−/− Tcf7l1−/− cells continued to proliferate and express the naive marker Rex1-GFPd2 (Figures 5B and 5C). In long-term N2B27 alone conditions (10 or more passages), Atp2b1−/− Tcf7l1−/− cells continued to express naive genes (Esrrb, Nanog, Tfcp2l1, Rex1, Klf2, Tcl1, Klf4, and Tbx3), albeit at lower levels than wild-type cells in 2iL (Figure 5D). Expression of formative stage genes (Otx2 and Dnmt3a) remained at lower levels in Atp2b1−/− Tcf7l1−/− cells compared to wild-type cells 24 h after withdrawal of 2iL (Figure 5D). Primed state genes (Fgf5 and Oct6) were expressed at relatively low levels in Atp2b1−/− Tcf7l1−/− cells (Figure 5D). Nanog levels fluctuated similarly in Atp2b1−/− Tcf7l1−/− colonies in N2B27 alone and wild-type colonies in 2iL (Figure 5E); however, immunofluorescence signal was undetectable in the weakest cells in double mutants (Figure S6D; Abranches et al., 2014). Sox2 and Klf4 immunofluorescence was maintained throughout colonies of Atp2b1−/−Tcf7l1−/− cells (Figures 5E and S6E). These results are consistent with the self-renewal of ESCs in N2B27 alone, albeit with a reduced level of naive specific gene expression. We suggest that the naive state is maintained at a substantial frequency of Atp2b1−/− Tcf7l1−/− cells in N2B27 alone, and this maintenance is sufficient to support self-renewal of these ESCs without inhibitors or LIF.
Assessing the pluripotency of inhibitor-independent Atp2b1−/− Tcf7l1−/− cells is complicated by the roles of Tcf7l1-mediated repression and reduction of intracellular Ca2+ during the generation of lineage-specific cell types. To test whether the combined effects of Tcf7l1 and Atp2b1 ablation kept cells in a pluripotent state, combinations of small molecules were tested to conditionally increase intracellular calcium and inhibit the Gsk3/Tcf7l1 axis in wild-type ESCs (Figure S6F). The combination of calcitonin paired with GSK3i (CTCH) was effective for long-term proliferation of ESCs in N2B27 media (Figure 6A). Atp2b1−/− cells and Tcf7l1−/− cells in these CTCH media (N2B27; calcitonin 100 nM; CH99021 3 μM) were maintained similar to wild-type cells in CTCH media (Figure S6G). The combination of GSK3i alone with Atp2b1 ablation was sufficient to maintain ESCs; however, calcitonin alone did not enable long-term self-renewal of Tcf7l1−/− cells (Figure S6G). For wild-type ESCs, CTCH maintained high levels of Rex1-GFPd2 (Figure 6B), high proliferation (Figure S6H), and high intracellular Ca2+ (Figure 6C). Expression of naive genes (Esrrb, Nanog, Tfcp2l1, Rex1, Klf2, Tcl1, Klf4, and Tbx3) remained high, and formative genes (Otx2 and Dnmt3a) and primed genes (Fgf5 and Oct6) remained low in wild-type cells passaged in CTCH media (Figure 6D). Cells that had been maintained in CTCH media for more than 10 passages formed embryoid bodies similarly to cells that had been maintained in 2iL (Figure 6E). Embryoid bodies from the CTCH media cells reduced naive gene expression (Rex1 and Nanog) and induced markers of ectoderm (Fgf5 and Sox1), mesoderm (Mixl1), and endoderm (Sox17, GATA4, and aFP) cell line-ages (Figure 6E). Immunofluorescent staining for marker protein expression in embryoid bodies from CTCH media were similar to those from 2iL; embryoid body cells displayed staining for Nanog and for markers of mesoderm (Mixl1 and Eomes), endoderm (Eomes, FoxA2, and CK19), and ectoderm (Sox1, Nestin, and βIII-tubulin; Figure S6I). Although long-term passaging in CTCH media stimulated activation of endoderm markers (Sox17, GATA4, and aFP) and low brachyury (T) levels in embryoid bodies compared to those generated from 2iL cells (Figure 6E), these data suggest that combining the inhibition of Gsk3 with high intracellular Ca2+ by calcitonin is sufficient for ESCs to retain pluripotency and capacity for specification into each primary germ layer.
Figure 6. Combination of Calcitonin and GSK3i (CTCH) in N2B27 Media Supports Self-Renewal of Wild-Type ESCs.

(A) DIC images displaying the morphology and density of wild-type and Atp2b1−/− Tcf7l1−/− double-mutant ESCs in 2iL, N2B27 alone, or CTCH media for indicated number of passages (P) and days (d). Scale bar represents 200 mm. See also Figures S6F and S6G.
(B) (Left) DIC and epifluorescence images (from Rex1-GFPd2) of wild-type cells in CTCH media for 26 passages. (Right) Flow cytometry analysis of the naive state with Rex1-GFPd2 of wild-type cells in CTCH media (green) or 2iL (red) for 26 passages. n = 2 biological replicates of >10,000 live-cell, singlet events were acquired for each sample.
(C) Intracellular free Ca2+ levels are localized in wild-type or Atp2b1−/− cells transfected with the genetically encoded calcium indicator, GCaMP5G. Transfected cells are treated with 2iL, GSK3i only, calcitonin (CT) only, or combinations thereof. Scale bar represents 14 μm.
(D) The relative mRNA expression measured by qRT-PCR (ΔΔCt-method) of naive, formative, and primed state marker genes in wild-type cells maintained in CTCH media. Data represent mean ± SD for 3 biological replicates. Gapdh expression is used as a loading control. * represents FDR < 5% for indicated comparisons.
(E) The relative mRNA expression measured by qRT-PCR (ΔΔCt-method) of pluripotency genes and germ layer (ectoderm, mesoderm, and endoderm) marker genes during embryoid body differentiation from wild-type cells previously maintained in either 2iL or CTCH media. Data represent individual measurements from biological duplicates. See also Figure S7.
DISCUSSION
In this study, we used a CRISPR mutagenesis screen to identify genes required for exit from naive pluripotency. We focused on two of the screen hits that displayed the strongest phenotypes in assays for naive exit. Examination of mutants showed that nuclear export requiring RanBP3 is important for rapidly exiting the naive state by removing nuclear activities, including active β-catenin and phospho-Stat3. Whereas the effect of Ranbp3−/− mutation was transient and did not contribute to long-term self-renewal in N2B27 alone, increased levels of intracellular Ca2+ caused by ablation of Atp2b1 resulted in a more sustained effect on ESCs. Atp2b1−/− Tcf7l1−/− double-mutant ESCs were able to self-renew in N2B27 alone, thus identifying a two-gene combination necessary for exiting naive pluripotency. To the best of our knowledge, this finding represents the only duo of mutations capable of supporting self-renewal in this manner.
In comparison to recently published CRISPR screens for regulators of naive pluripotency, a highly overlapping set of genes was identified despite different procedures being used by each (Hackett et al., 2018; Li et al., 2018). Genes contributing to extra-cellular signaling pathways (Wnt/β-catenin, Lif/Jak/Stat, and Fgf/Mek/Erk) and the lysosome-associated amino-acid-sensing pathway were identified in each screen, supporting a central role for each in the exit from naive pluripotency. Whereas these other studies provided new insights by focusing mechanistic examination on biological processes highly represented by multiple gene hits in CRISPR screens (lysosome activity and mTorc1 pathway; Hackett et al., 2018; Li et al., 2018), we focused on genes that caused the strongest phenotypes when mutated. As a result, the major discoveries of processes from this study (nuclear export and intracellular Ca2+) are distinct from previous studies using CRISPR screens. Based on the wealth of genes identified from the CRISPR screens, we anticipate that additional curation and secondary screening with the known gene hits will provide additional novel insights into the exit from naive pluripotency.
Despite the importance of Ca2+ in many mechanisms essential for embryonic development and cellular functions, the effect of Ca2+ on pluripotency has been poorly understood. Several experiments manipulating intracellular Ca2+ levels have been performed on ESCs using epidermal growth factor (EGF), GABA agonists, estrogen, or lysophosphatidic acid; however, these experiments were performed on ESCs in serum + LIF media and did not examine state of pluripotency (Hao et al., 2016). Similarly, serum + LIF conditions were also used to elucidate calcineurin stimulation of NFATc3, leading to differentiation of ESCs (Li et al., 2011). By contrast to substantial effects caused by manipulating Ca2+ or calcineurin in serum + LIF conditions, the manipulation of Ca2+ homeostasis proteins themselves (STIM1, 1,4,5-triphosphate receptors, TPC2, and CD38) has caused remarkably few effects on ESCs and pluripotency (Hao et al., 2014; Liang et al., 2010; Wei et al., 2012, 2015; Zhang et al., 2013). Atp2b1 mutation appears to be unique in its effects on pluripotency, which function through elevated intracellular Ca2+. Although calcitonin was able to replace Atp2b1 ablation in combination with GSK3i, it could not maintain self-renewal of Tcf7l1−/− cells without GSK3i. Given GSK3 has many roles in addition to regulating Tcf7l1/β-catenin, Ca2+-independent effects of calcitonin may cause ESCs to depend on Tcf7l1-independent effects of GSK3i.
It is interesting to consider a role for intracellular Ca2+ in coordinating the exit from naive pluripotency in epiblast cells to occur relative to implantation of the embryo in the uterus. A careful analysis of intracellular Ca2+ concentrations with fluorescent dyes showed that Ca2+ levels spontaneously oscillate in individual ESCs as they progress through the cell cycle in LIF + serum media (Kapur et al., 2007). Thus, intracellular Ca2+ is dynamically regulated in pluripotent cells. Our results indicate that elevated intracellular Ca2+ protects pluripotent cells from exiting the naive state. By contrast, an influx of intracellular Ca2+ in embryonic trophectoderm stimulates progression of preimplantation embryonic development for those cells (Stachecki and Armant, 1996; Wang et al., 1998, 2013). The uterine endometrium secretes calcitonin within a so-called window of implantation, and embryonic cells respond by increasing intracellular Ca2+ (Ding et al., 1994; Wang et al., 1998, 2013). This paracrine signaling involving Ca2+ has been shown to be important for coordinating embryo-uterine interaction during implantation (Armant, 2005; Stachecki and Armant, 1996; Wang et al., 2013). Interestingly, LIF/Jak/Stat and Wnt/β-catenin signaling are similarly coordinated by paracrine signals from the endometrium to the embryonic trophoblast important for successful implantation (Armant, 2005; Fritz et al., 2014). It is important to consider that the same signals (LIF/Jak/Stat, Wnt/β-catenin, and high Ca2+) all stimulate maintenance of naive pluripotency and are all activated by the endometrium during implantation. We suggest that maintenance of the naive state by the combinations of these signals enables pluripotency to be maintained in the epiblast during the dynamic process of implantation, and intracellular Ca2+ plays a fundamental role in the process. The development of CTCH media may provide a method of better elucidating pathway-specific effects during the maintenance of the naive state of pluripotency.
In summary, the CRISPR screen confirmed known processes (Wnt/β-catenin/Tcf7l1, Fgf/Mek/Erk, LIF/Jak/Stat, and mTorc1/TSC) and discovered new ones (nuclear export, Ca2+ homeostasis, and endosome-lysosome trafficking) important for the exit out of the naive state of pluripotency. Elevated intracellular Ca2+ inhibited exit from naive pluripotency, and Atp2b1−/− Tcf7l1−/− ESCs were able to self-renew without exogenous LIF or inhibition of Mek or Gsk3 kinases. We suggest that intracellular Ca2+ plays a central role in regulating pluripotency and coordinating the state of pluripotency with embryonic development.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents can be directed to, and will be fulfilled, by the Lead Contact, Bradley J. Merrill (merrillb@uic.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
Mouse embryonic stem cells utilized for the screen are male, 129/Sv (Pereira et al., 2006). These cells were isolated under a protocol approved by the University of Illinois at Chicago Animal Care Committee. Male, 129/Sv ESC that harbor Rex1::GFPd2 knock-in (A gift from Dr. Austin Smith) (Wray et al., 2011) were used for validation and clone construction.
METHOD DETAILS
Culture Methods
Both lines of ESC are passaged every 2–4d gelatin in N2B27 supplemented with 1 μM PD0325901 (MEKi, Stemgent), 3 μM CHIR99021 (GSK3i, Sigma), and 1000 units/mL LIF (Millipore) (2iL). For the screen, cells were passaged on tissue culture plates pre-coated with 7.5 μg/ml poly-ornithine (Advanced Biomatrix) and 5 μg/ml ultrapure laminin (Corning; Novus), as described previously (Buecker et al., 2014). For the validation and clone construction, Rex1-GFPd2 cells were passaged on tissue culture plates pre-coated with 0.1% gelatin (Millipore).
The conditions for exit from naive pluripotency involve plating cells on plates coated with 15 μg/mL fibronectin (Millipore) in N2B27 without 2iL(−2iL). Where indicated, Fgf2 12 ng/mL Fgf2 (R&D) was included after 24h in N2B27. Media was changed daily.
To differentiate cells to embryoid bodies, cells were first transitioned Knockout DMEM (GIBCO #10829–018) supplemented with the following: 15% EmbryoMax ES Cell Qualified Fetal Bovine Serum (Millipore), 2 mM L-Glutamine (ThermoFisher), 1000 U/ml Pen-Strep (ThermoFisher), 1 mM HEPES, 1 × MEM NEAA (ThermoFisher) 55 uM 2-mercaptoethanol (ThermoFisher), 100 U/ml LIF. After several passages, 1 million cells were plated on bacterial Petri dishes in the above media without LIF. Embryoid bodies were prevented from attaching by pipette. 1mL samples of embryoid bodies were taken daily for RNA isolation and media was changed every other day by allowing embryoid bodies to settle by gravity in a 15mL conical tube. For immunofluorescence experiments, entire plates of embryoid bodies were taken at indicated times, allowed to settle by gravity, and fixed in 4% PFA for 2h (Baillie-Johnson et al., 2015).
For clonogenicity assays, 3000 cells were plated per well of 6-well plate coated with 0.1% gelatin in duplicate. At day 7–10, colony counts were performed by hand or by automated whole-well imaging on a Nexcelom Celigo Imaging Cytometer. For proliferation assay, cells were plated and stained with Hoechst 33342 and propidium iodide and counted on the Nexcelom Celigo Imaging Cytometer to determine the number of viable cells.
Treatment concentrations for various drugs are indicated as follows. For Jak inhibition, the previously characterized Pyridone 6(1uM or 5uM, Calbiochem) was used(Yi and Merrill, 2010). For manipulation of intracellular calcium concentration: Cyclosporin A (1uM, Sigma), FK506(Tocris, 25uM), VIVIT (Tocris, 15uM), Ionomycin(5uM, Sigma), BAPTA-AM (1uM, Invitrogen), SEA-0400(100uM, Tocris), human calcitonin(10–100nM, Anaspec), and Carboxyeosin(5–50uM, MGTi). For proteasome inhibition, MG-132 was used (5uM, Sigma).
Genome-wide Knockout Screen
First, stable Cas9 expressing mESCs were made by transducing mESCs with lentivirus containing Lenti-Cas9 Blast (A gift from Feng Zhang, Addgene:52962) followed by selection with 2ug/ml blasticidin (ThermoFisher) for 4 days. Clones were picked and screened for cas9 activity by transfecting an pKLV-U6-Lef1-PGKpuro2ABFP (empty, pKLV-U6-Lef1-PGKpure2ABFP was a gift from using Surveyor mutation detection kit (IDT).
The genome-wide sgRNA library was purchased from Addgene and utilized as previously published with a few modifications (Koike-Yusa et al., 2014). Library amplification was performed in liquid culture with selection. Virus construction was performed in HEK293FT cells as previously described. 3×107 mESC-Cas9 cells were transduced with library virus, selected with 2ug/ml puromycin (ThermoFisher)
Library preparation made use of amplicons with CS1 and CS2 linkers for Illumina sequencing. Plasmid, mutant, and enriched mutant libraries were prepared concurrently by initial 30 cycle sgRNA specific PCR. 5ng of the sgRNA libraries were barcoded with Fluidigm Access Array barcodes using AccuPrime Supermix II (ThermoFisher) PCR mix (95°C for 5 m, 8 cycles of 95°C for 30 s, 60°C for 30 s and 72°C for 30 s, and one cycle of 72°C for 7 m). Barcoded PCR products were analyzed on a 2200 TapeStation (Agilent) before and after 2 rounds of 0.6x AMPure bead (Beckman Coulter) purification to exclude primer dimers. Sequencing of the libraries was performed on an Illumina HiSeq.
Arrayed Validation Screen
sgRNA design –
Where possible, independent sgRNAs from the screen library were designed for validation of the top 40 candidates (Table S6) and cloned into pSP-sgRNA (Addgene).
Transfection and Selection –
Within 2 hr of transfections, 2.5 × 105 mESCs were freshly plated in each well of 24 wells dishes. For each well, 2.5 μL of Lipofectamine 2000(ThermoFisher) and relevant DNAs were incubated in 125 μL OPTI-MEM (ThermoFisher) before adding to wells. For the Cas9 mutagenesis of 40 distinct genes in ES cells, transfections included 175ng pPGKpuro (Addgene), 175ng pX330 (lacking sgRNA insert), and 175ng of the relevant pSPgRNA plasmid. To assess background mutation rate due to possible deep sequencing or amplification errors, a transfection containing pSPgRNA with empty sgRNA site was assessed alongside the other sgRNA-containing transfections. Two days after transfection, cells were split into 2 μg/ml puromycin and selection was applied for 48hrs before seeding for 2 sequential rounds of replating selection. For each round of replating selection, cells were subjected to 4 days in absence of 2iL on fibronectin followed by 4 days in 2iL on gelatin. Genomic DNA was isolated at the end of puromycin selection and after each round of replating selection by overnight lysis with Bradley Lysis buffer (10mM Tris-HCl, 10mM EDTA, 0.5% SDS, 10mM NaCl) containing 1mg/ml Proteinase K, followed with EtOH/NaCl precipitation, two 70% EtOH washes, and eluted in 50 μL of ddH2O or TE.
Targeted deep-sequencing preparation –
Genomic DNA was harvested four days after transfection and approximately 100ng of DNA was used in PCR to amplify respective target sites while attaching adaptor sequences for subsequent barcoding steps (Table S6). PCR products were analyzed via agarose gel. Amplicons were normalized with SequalPrep (ThermoFisher) plates followed by pico green quantification and pooled robotically by pipetting with the Eppendorf EPmotion. Pooled PCR products were purified with AMPure XP beads (Beckman Coulter), and 5ng of the purified pools was barcoded with Fluidigm Access Array barcodes using AccuPrime Supermix II (ThermoFisher) (95°C for 5 m, 8 cycles of 95°C for 30 s, 60°C for 30 s and 72°C for 30 s, and one cycle of 72°C for 7 m). Barcoded PCR products were analyzed on a 2200 TapeStation (Agilent) before and after 2 rounds of 0.6x AMPure XP bead purification to exclude primer dimers. A final pool of amplicons was created and loaded onto an Illumina MiniSeq generating 150bp paired-end reads. Sequencing data analysis for indel frequency determination is described below.
Creation of Knockout ESCs
Using the sgRNAs from the arrayed validation for chosen candidates, transfections were repeated as described above to create a pool of mutants. 12–24 clones were then picked based on the editing frequencies observed in the arrayed validation. Target specific amplicons were created and sequenced by NGS, as above, to identify bi-allelic frameshifted mutants.
Immunofluorescence
Fifty thousand cells were plated on μ-Slide 8 Well, polymer coverslip (Ibidi). After indicated culture times, cells were fixed in 4% paraformaldehyde for 15–20min followed by blocking and permeabilized in blocking buffer (0.3% Triton X-100 and 5% FBS in PBS). For P-Stat3, an additional permeabilization step of incubation in 100% ice cold methanol at −20°C for 10 minutes was required prior to blocking. The following antibodies were diluted as indicated in Antibody Diluent Buffer (0.3% Triton X-100, 5% BSA in PBS) and incubated overnight: RAN (1:100, BD), Tfe3(1:300, Sigma), P-Stat3(1:100, Cell Signaling), Non-phospho (Active) β-Catenin (Ser33/37/Thr41) (D13A1) (1:800, Cell Signaling), Stat3(1:100, Cell Signaling), Nanog (1:1000, Cell Signaling), Sox2(1:200, Cell Signaling), Klf4(1:100, R&D). No primary antibody was used as a negative control. In preparation for imaging, cells were washed three times with PBS, then incubated for 1–2h in secondary antibody dilution (1:100, Cy5- or FITC- conjugated, Jackson ImmunoResearch) Cells were washed, counterstained with DAPI for 15 min, and mounted in mounting medium (Ibidi).
Fixed embryoid bodies were washed, blocked, and permeabilized in PBSFT (0.2% Triton X-100 and 10% FBS in PBS). The following antibodies were diluted as indicated in blocking buffer and inbubated overnight: Nanog (1:1000, Cell Signaling), Mixl1(1:150, R&D), Eomes(1:500, Abcam), FoxA2(1:100, Santa Cruz), CK19(1:100, DSHB), Sox1(1:100, R&D), Nestin(1:300, BD), βIII-tubulin(1:1000, Promega). No primary antibody was used as a negative control. In preparation for imaging, cells were washed five times with PBSFT, then incubated for overnight in secondary antibody (1:100, Cy5- conjugated, Jackson ImmunoResearch) and Hoechst 33342. Cells were washed with PBT (0.2% Triton X-100, 0.2% FBS in PBS) and mounted in mounting medium. When ready for imaging, embryoid bodies were pipetted with cut pipette tips onto μ-Slide 18 Well - Flat, polymer coverslips (Ibidi).
Cells were imaged immediately on a Zeiss Laser Scanning Confocal Microscope (LSM) 700 confocal microscope system. Different sample images of the same antigen were acquired under constant acquisition settings.
Immunoblotting
Protein for immunoblotting was isolated using either heated SDS-Loading buffer or actively by freeze-thaw in ice-cold lysis buffer (20 mM HEPES, 1% Triton X-100, 150 mM NaCl, 1 mM EDTA, 10 mM sodium pyrophosphate, 100 mM NaF, 5 mM iodo-acetic acid, 20 nM okadaic acid, 0.2 mM phenylmethylsulfonyl fluoride and a complete protease inhibitor cocktail tablet (ThermoFisher)). When the latter lysis buffer method was used, protein samples were quantified and normalized by Bradford prior to loading for polyacrylamide gel electrophoresis (PAGE) in 8% SDS-PAGE gels. Protein was transferred to PVDF, blocked in 5% Milk in 0.1% Tween-20 in TBS(TBST) for at least 1h and incubated overnight in either 5% Milk or 5% BSA in TBST with one of the following primary antibodies: Non-P, Active B-catenin(1:1000, Cell Signaling, Milk), Total B-catenin(1:1000, Cell Signaling, Milk), Tcf7l1(1:2000, In-house, Milk), Tubulin-E7(1:3000,DSHB, Milk), P-Stat3-Y705(1:1000, Cell Signaling, BSA), RanBP3(1:1000, Cell Signaling, Milk), P-RanBP3-S58(1:1000, Cell Signaling, BSA), and Anti-HA(1:4000, Millipore, Milk).
Luciferase Assay
Luciferase assay was performed as previously described (Shy et al., 2013; Yi et al., 2011). Briefly, one hundred thousand cells were transfected at the time of plating on 24-well plates using 400ng of a mix of pSuperTOPflash and pRL-CMV (10:1) and Lipofectamine 2000 overnight in 2iL. Approximately 18h after transfection, cells were washed and placed in either 2iL or 1iL-GSK3i. Cells were harvested 6h later and lysed in 1x passive lysis buffer (Promega). Luciferase activity was measured using a Clarity luminometer (Bio-Tek) and a dual luciferase reporter assay system (Promega). Each transfection was carried out in duplicate, and each experiment was repeated at least twice.
Intracellular Calcium Measurement and Visualization
Measurement of intracellular calcium was performed through the use of Fura-2-AM (ThermoFisher). Staining was performed in phenol free red media containing 1uM of Fura-2-AM for 30min at 37°C. Following staining, de-esterification was allowed to occur in Ca-free Hank’s Buffered Saline Solution (HBSS) for 20min. For controls, treatment with Ionomycin (5uM, ThermoFisher) in Ca+HBSS or BAPTA-AM (1uM, ThermoFisher) in Ca-free HBSS was performed for 10min, all other experimental samples were washed with Ca-free HBSS. Whole well, resting intracellular calcium concentration was measured by exciting with 340nm and 380nm followed by measuring emissions at 510nm on a Tecan Infinite M200, as previously described (Martinez et al., 2016, 2017). The Fura2 ratio is the ratio of emission at 510nm from excitation at 340nm and 380nm(340nm/380nm). For timed experiments, cells were plated at the same time and density in 2iL on fibronectin and allowed to exit the naive state by placement in N2B27–2iL at differing times that allowed for acquisition all at once.
To visualize intracellular calcium, cells were transfected in 24-well plates using 500ng of either pCMV-GCaMP5G or pCMV R-CEPIA1er as described above. 24h later, fifty thousand cells were plated on m-Slide 8 Well, glass coverslip (Ibidi) coated with fibronectin in indicated treatments for 24h. Cells were then counterstained with Hoechst 33342 and imaged live on the Zeiss LSM 710 BiG system.
Flow Cytometry
Cells are prepared for flow cytometry by trypsinization and resuspension in PBS + 5%FBS and filtered. Flow cytometry is performed using the BD LSRFortessa. Unless otherwise noted, 10,000 live, single cells were collected prior to the measurement of Rex1-GFPd2.
Atp2b1-mCh Construction
An sgRNA was designed to target the C terminus of mouse Atp2b1 and cloned into px330. A repair template donor was created by PCR with ultramers to mCh with homology to the regions surrounding the stop codon of Atp2b1. mESC cells were transfected with 250ng each of px330-Atp2b1-Cterm sgRNA and donor. Red positive cells were sorted and expanded. The heterogeneous population was expanded, genotyped to confirm correct insertion, and then imaged by confocal microscopy.
RanBP3 Complementation and Ran Transfections
mRanBP3 cDNA was amplified using target specific primers based on annotations in Ensembl from wild-type mESC cDNA. mRanBP3 was cloned Gibson cloning into pKH3-HA-hRanBP3(A gift from Yoon, S.O.) (Yoon et al., 2008) linearized by PCR to remove hRanBP3. Cells were transfected as described above but with 250ng of pKH3-HA-mRanBP3 or pcDNA3-empty and 250ng pmCherry-C1(Clontech). 24h after transfection cells plated for 48h on fibronectin in N2B27 without 2iL then analyzed by flow cytometry. Live, singlet, mCherry+ cells were analyzed for Rex1-GFPd2 expression. pmCherry-C1-RanQ69L was corrected to WT by site directed mutagenesis and then used as a template to make RanT24N and Ran G19V. Cells were transfected and analyzed as in the RanBP3 complementation above.
RNA Isolation and RT-qPCR
RNA was isolated from cells using TRIzol reagent (ThermoFisher) followed by column purification with Direct-zol Rna MiniPrep Plus Kit (Zymo) with on-column DNase digestion. 3 μg of RNA was converted to cDNA using Superscript III (ThermoFisher) and quantitative real time PCR was performed using primers indicated in Table S6 combined with 100ng of cDNA from each sample and with Perfecta SYBR Green Supermix (QuantaBio). qPCR was performed on a C1000 thermal cycler and CFX96 Real Time System (Bio-Rad). The ΔΔCt method was used for quantification and plotting.
QUANTIFICATION AND STATISTICAL ANALYSIS
Screen Analysis
The data processing and analysis of the screen was performed using the Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout(MAGeCK) command line tool (Li et al., 2014). MAGeCK analysis takes all the raw sequencing data and an sgRNA reference list as input and outputs sgRNA counts/statistics (Table S1) as well as gene-level (Table S2) and GO-term hit data (Table S3). The sgRNA count table and gene hit information are used as raw data to construct the plots shown in Figure S1 with R scripts.
Targeted deep-sequencing analysis
Determination of indel frequencies made use of CRISPResso command line tools that demultiplexed by amplicon, where appropriate, and determined indel frequency by alignment to reference amplicon files (Pinello et al., 2016)(Table S5). Outputs were assembled and analyzed using custom command-line, python, and R scripts which are available upon request. Demultiplexed FASTQ files were also aligned to the mouse genome to create BAM files for input into CrispRVariants for cut-site and allele visualization (Lindsay et al., 2016).
Image Analysis
Immunofluorescence images were processed using a custom Fiji script for batch conversion to RGB and montaging for publication(Schindelin et al., 2012). No other manipulations were applied to images. Scale bar lengths are indicated in figure legends. For Tfe3 quantification, DAPI-labeled nuclei were identified and the average Tfe3 nuclear fluorescence intensity was quantified using CellProfiler(Broad Institute) (Carpenter et al., 2006).
Flow cytometry analysis
Analysis and visualization of flow cytometry data was performed using FlowJo (v9.3.2).
Data Visualization and Statistical Analysis
Where applicable, most data visualization and statistical analysis was performed use R (v3.3.1), RStudio(v1.1.383), and packages: ggplot2, dplyr, magrittr, multcomp, crispRvariants. In some cases, data visualization was performed using Microsoft Excel. Schematics and figures were prepared using Adobe Illustrator (v22.1). In barplots throughout figures, data are represented as mean ± standard deviation or standard error of the mean, as indicated. Statistics were performed in R with the package multcomp by performing one-way ANOVA with Tukey contrasts followed by p value correction for multiple testing with Benjamini–Hochberg procedure (false discovery rate, FDR).
DATA AND SOFTWARE AVAILABILITY
All raw data and custom scripts are either provided as a supplemental table (Tables S1, S2, S3, S4, S5, and S6) or are made available on Mendeley Data with https://doi.org/10.17632/9mfp9n3j6p.1. The accession number for the all sequencing in this paper is SRANCBI: SRP151214.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Non-phospho (Active) β-Catenin (Ser45) (D2U8Y) XP®Rabbit mAb | Cell Signaling Technology | Cat#19807; RRID:AM_2650576 |
| β-Catenin (D10A8) XP® Rabbit mAb | Cell Signaling Technology | Cat# 8480; RRID:AB_11127855 |
| Phospho-Stat3 (Tyr705) (D3A7) XP® Rabbit mAb | Cell Signaling Technology | Cat# 9145; RRID:AB_2491009 |
| Tcf7l1 Rabbit pAb | This Paper | N/A |
| Stat3 (D3Z2G) Rabbit mAb | Cell Signaling Technology | Cat# 12640; RRID:AB_2629499 |
| Tubulin, Beta Mouse mAb | Developmental Studies Hybridoma Bank | Cat# E7; RRID:AB_528499 |
| Ran Mouse mAb | BD Biosciences | Cat# 610340; RRID:AB_397730 |
| Tfe3 Rabbit pAb | Sigma-Aldrich | Cat# HPA023881; RRID:AB_1857931 |
| Nanog (D2A3) XP® Rabbit mAb (Mouse Specific) | Cell Signaling Technology | Cat# 8822P; RRID:AB_11217637 |
| Klf4 pAb | R&D | Cat# AF3158; RRID:AB_2130245 |
| Sox2 (L1D6A2) Mouse mAb | Cell Signaling Technology | Cat# 4900S; RRID:AB_10560516 |
| RanBP3 Rabbit pAb | Cell Signaling Technology | Cat# 93706; RRID:AB_2800209 |
| Anti-HA Tag Antibody | Millipore | Cat#05–904; RRID:AB_417380 |
| Mixl1 Rat mAb | R&D | Cat# MAB8099 |
| Eomes Rabbit pAb | Abcam | Cat# ab23345; RRID:AB_778267 |
| FoxA2 Mouse mAb | Santa Cruz | Cat# sc374376; RRID:AB_10989742 |
| CK19 Rat mAb | Developmental Studies Hybridoma Bank | Cat# TROMA-III; RRID:AB_2133570 |
| Sox1 Goat pAb | R&D | Cat# AF3369; RRID:AB_2239879 |
| Nestin Mouse mAb | BD Biosciences | Cat# 556309; RRID:AB_396354 |
| βIII-tubulin Mouse mAb | Promega | Cat# G7121; RRID:AB_430874 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PD0325901 | Stemgent | Cat#38808 |
| CHIR99021 | Sigma | Cat#SML1046 |
| ESGRO (LIF) | Millipore | Cat#ESG1107 |
| Poly-L-Ornithine, Solution, 0.1mg/ml | Advanced Biomatrix | Cat#5058 |
| Laminin ultrapure mouse | Corning | Cat#354239 |
| EmbryoMax 0.1% Gelatin Solution | Millipore | Cat#ES006B |
| Fibronectin | Millipore | Cat#FC010 |
| Pyridone 6 | Calbiochem | Cat#420097 |
| Cyclosporin A | Sigma | Cat#30024 |
| FK506 | Tocris | Cat#3631 |
| VIVIT | Tocris | Cat#3930 |
| Ionomycin | ThermoFisher | Cat#I24222 |
| BAPTA-AM | Invitrogen | Cat#B6769 |
| SEA-0400 | Tocris | Cat#6164 |
| 5(6)-Carboxyeosin | MGTi | Cat#M1300 |
| Human Calcitonin | Anaspec | Cat#AS-20673 |
| Fgf2 | R&D | Cat#233-FB-025 |
| Blasticidin S HCl | ThermoFisher | Cat#A11139–03 |
| Puromycin Dihydrochloride | ThermoFisher | Cat#A1113802 |
| AccuPrime Supermix II | ThermoFisher | Cat#12341012 |
| AMPure XP Beads | Beckman Coulter | Cat#A63880 |
| Lipofectamine 2000 | ThermoFisher | Cat#11668–019 |
| Fura2-AM | ThermoFisher | Cat#F1221 |
| TRIzol Reagent | ThermoFisher | Cat#1556026 |
| Perfecta SYBR Green Supermix for iQ | QuantaBio | Cat#95053 |
| Knockout DMEM | ThermoFisher | Cat#10829–018 |
| EmbryoMax ES Cell Qualified FBS | Millipore | Cat#ES-009-B |
| L-Glutamine | ThermoFisher | Cat#25030–081 |
| Penicillin-Streptomycin | ThermoFisher | Cat#15140–163 |
| HyClone HEPES Solution | ThermoFisher | Cat#SH3023701 |
| MEM Non-Essential Amino Acids Solution | ThermoFisher | Cat#11140–076 |
| 2-mercaptoethanol | ThermoFisher | Cat#21985023 |
| Hoechst 33342 | ThermoFisher | Cat#H3570 |
| Critical Commercial Assays | ||
| Surveyor Mutation Detection Kit | IDT | Cat#706020 |
| SequalPrep Normalization Plate Kit, 96-well | ThermoFisher | Cat#A1051001 |
| Direct-zol RNA Miniprep Plus Kit | Zymo | Cat#R2072 |
| Experimental Models: Cell Lines | ||
| mESC 129/sv Male | Pereira et al., 2006 | N/A |
| mESC harboring Rex1::GFPd2 129/sv Male | Smith, AS | Wray et al., 2011 |
| mESC-Cas9(derived from mESC 129/sv Male) | This Paper | N/A |
| Deposited Data | ||
| Screen Sequencing Data | SRA-NCBI | SRP151214 |
| Amplicon Sequencing Data for Validation and Clone Construction | SRA-NCBI | SRP151214 |
| Raw data including: Uncropped blots, scripts for analysis and visualization, etc. | Mendeley | https://doi.org/10.17632/9mfp9n3j6p.1 |
| Oligonucleotides | ||
| See Table S6 for Oligos Used in this Study | N/A | |
| Recombinant DNA | ||
| Genome-wide Mouse Lentiviral CRISPR gRNA Library v1 | Addgene | Addgene:50947 |
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | Addgene | Addgene:42230 |
| pSPgRNA | Addgene | Addgene:47108 |
| lentiCas9-Blast | Addgene | Addgene:52962 |
| pKLV-U6gRNA(BbsI)-PGKpuro2ABFP | Addgene | Addgene:50946 |
| pKLV-U6-Lef1-PGKpuro2ABFP | This Paper | N/A |
| pSPgRNAs Used for Validation, Clone Construction, Atp2b1-mCh See Table S6 | This Paper | N/A |
| pPGKpuro | Addgene | Addgene:11349 |
| pKH3-HA-mRanBP3 | This Paper | N/A |
| pmCherry-C1 | Clontech | Cat#632524 |
| pmCherry-C1-RanQ69L | Addgene | Addgene:30309 |
| pmCherry-C1-RanWT | This Paper | N/A |
| pmCherry-C1-RanG19V | This Paper | N/A |
| pmCherry-C1-RanT24N | This Paper | N/A |
| pCMV R-CEPIA1er | Addgene | Addgene:58216 |
| pCMV-GCaMP5G | Addgene | Addgene:31788 |
| Software and Algorithms | ||
| MAGeCK | Liu, S | Li et al., 2014 |
| R | R Project | v3.3.1 |
| RStudio | RStudio | v1.1.383 |
| CRISPResso | Pinello, L | Pinello et al., 2016 |
| CrispRVariants | Lindsay, H, Bioconductor | Lindsay et al., 2016 |
| Fiji | NIH | Schindelin et al., 2012 |
| CellProfiler | Broad Institute | Carpenter et al., 2006 |
| FlowJo | FlowJo | V9.3.2 |
| Other | ||
| μ-Slide 8 Well, polymer coverslip | Ibidi | Cat#80826 |
| μ-Slide 8 Well, glass coverslip | Ibidi | Cat#80827 |
| μ-Slide 18 Well - Flat Uncoated, polymer coverslip | Ibidi | Cat#81821 |
Highlights.
A CRISPR screen identifies Ranbp3 and Atp2b1 as regulators of naive pluripotency
Nuclear export of β-catenin and phospo-Stat3 is reduced in Ranbp3−/− cells
Atp2b1−/− Tcf7l1−/− double-knockout ESCs sustain self-renewal in N2B27 alone
Wild-type ESCs self-renew in N2B27 with the combination of calcitonin and GSK3i
ACKNOWLEDGMENTS
We thank Maureen Regan of the UIC Genome Editing Core for her helpful discussions regarding screens and large-scale cloning of sgRNAs; Jenny Zhang and Hannah Pennington for helpful discussions on several aspects of this study; the Sequencing, Fluorescence Imaging, and Flow Cytometry core facilities within UIC Research Resources Center; and Alex Yemelyanov of the viral core at Northwestern University. This work was supported by grant and fellowship funding from the NIH (R01-HD081534 to B.J.M.; F30-HD090938 to M.S.M.) and the UIC Center for Clinical and Translation Sciences (to M.S.M.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.stem.2019.04.015.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abranches E, Guedes AM, Moravec M, Maamar H, Svoboda P, Raj A, and Henrique D (2014). Stochastic NANOG fluctuations allow mouse embryonic stem cells to explore pluripotency. Development 141, 2770–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, Calderón NC, Esposti F, Borghuis BG, Sun XR, et al. (2012). Optimization of a GCaMP calcium indicator for neural activity imaging.J. Neurosci 32, 13819–13840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramburu J, Yaffe MB, López-Rodríguez C, Cantley LC, Hogan PG, and Rao A (1999). Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science 285, 2129–2133. [DOI] [PubMed] [Google Scholar]
- Armant DR (2005). Blastocysts don’t go it alone. Extrinsic signals fine-tune the intrinsic developmental program of trophoblast cells. Dev. Biol 280, 260–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeg GH, Zhou R, and Perrimon N (2005). Genome-wide RNAi analysis of JAK/STAT signaling components in Drosophila. Genes Dev 19, 1861–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie-Johnson P, van den Brink SC, Balayo T, Turner DA, and Martinez Arias A (2015). Generation of aggregates of mouse embryonic stem cells that show symmetry breaking, polarization and emergent collective behaviour in vitro. J. Vis. Exp 53252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betschinger J, Nichols J, Dietmann S, Corrin PD, Paddison PJ, and Smith A (2013). Exit from pluripotency is gated by intracellular redistribution of the bHLH transcription factor Tfe3. Cell 153, 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowling S, Di Gregorio A, Sancho M, Pozzi S, Aarts M, Signore M, D Schneider M, Martinez-Barbera JP, Gil J, and Rodriguez TA (2018). P53 and mTOR signalling determine fitness selection through cell competition during early mouse embryonic development. Nat. Commun 9, 1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brons IG, Smithers LE, Trotter MW, Rugg-Gunn P, Sun B, Chuva de Sousa Lopes SM, Howlett SK, Clarkson A, Ahrlund-Richter L, Pedersen RA, and Vallier L (2007). Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 448, 191–195. [DOI] [PubMed] [Google Scholar]
- Buecker C, Srinivasan R, Wu Z, Calo E, Acampora D, Faial T, Simeone A, Tan M, Swigut T, and Wysocka J (2014). Reorganization of enhancer patterns in transition from naive to primed pluripotency. Cell Stem Cell 14, 838–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. (2006). CellProfiler: image analysis software for identifying and quantifying cell pheno-types. Genome Biol 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke R, Heler R, MacDougall MS, Yeo NC, Chavez A, Regan M, Hanakahi L, Church GM, Marraffini LA, and Merrill BJ (2018). Enhanced bacterial immunity and mammalian genome editing via RNA-poly merase-mediated dislodging of Cas9 from double-strand DNA breaks. Mol. Cell 71, 42–55.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Leva F, Domi T, Fedrizzi L, Lim D, and Carafoli E (2008). The plasma membrane Ca2+ ATPase of animal cells: structure, function and regulation. Arch. Biochem. Biophys 476, 65–74. [DOI] [PubMed] [Google Scholar]
- Ding YQ, Zhu LJ, Bagchi MK, and Bagchi IC (1994). Progesterone stimulates calcitonin gene expression in the uterus during implantation. Endocrinology 135, 2265–2274. [DOI] [PubMed] [Google Scholar]
- Du P, Pirouz M, Choi J, Huebner AJ, Clement K, Meissner A, Hochedlinger K, and Gregory RI (2018). An intermediate pluripotent state controlled by microRNAs is required for the naive-to-primed stem cell transition. Cell Stem Cell 22, 851–864.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidalgo M, Huang X, Guallar D, Sanchez-Priego C, Valdes VJ, Saunders A, Ding J, Wu WS, Clavel C, and Wang J (2016). Zfp281 coordinates opposing functions of Tet1 and Tet2 in pluripotent states. Cell Stem Cell 19, 355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley LWS, Vardhana SA, Carey BW, Alonso-Curbelo D, Koche R, Chen Y, Wen D, King B, Radler MR, Rafii S, et al. (2018). Pluripotency transcription factors and Tet1/2 maintain Brd4-independent stem cell identity. Nat. Cell Biol 20, 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz R, Jain C, and Armant DR (2014). Cell signaling in trophoblast-uterine communication. Int. J. Dev. Biol 58, 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman DA, Bierer BE, Benes JE, Burakoff SJ, Austen KF, and Katz HR (1995). The complex of FK506-binding protein 12 and FK506 inhibits calcineurin phosphatase activity and IgE activation-induced cytokine transcripts, but not exocytosis, in mouse mast cells. J. Immunol 154, 1846–1851. [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, and Tsien RY (1985). A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem 260, 3440–3450. [PubMed] [Google Scholar]
- Hackett JA, Huang Y, Günesdogan U, Gretarsson KA, Kobayashi T, and Surani MA (2018). Tracing the transitions from pluripotency to germ cell fate with CRISPR screening. Nat. Commun 9, 4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao B, Lu Y, Wang Q, Guo W, Cheung KH, and Yue J (2014). Role of STIM1 in survival and neural differentiation of mouse embryonic stem cells independent of Orai1-mediated Ca2+ entry. Stem Cell Res. (Amst.) 12, 452–466. [DOI] [PubMed] [Google Scholar]
- Hao B, Webb SE, Miller AL, and Yue J (2016). The role of Ca(2+) signaling on the self-renewal and neural differentiation of embryonic stem cells (ESCs). Cell Calcium 59, 67–74. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Ohta H, Kurimoto K, Aramaki S, and Saitou M (2011). Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 146, 519–532. [DOI] [PubMed] [Google Scholar]
- Hendriksen J, Fagotto F, van der Velde H, van Schie M, Noordermeer J, and Fornerod M (2005). RanBP3 enhances nuclear export of active (beta)-catenin independently of CRM1. J. Cell Biol 171, 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman JA, Wu CI, and Merrill BJ (2013). Tcf7l1 prepares epiblast cells in the gastrulating mouse embryo for lineage specification. Development 140, 1665–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izaurralde E, Kutay U, von Kobbe C, Mattaj IW, and Görlich D (1997). The asymmetric distribution of the constituents of the Ran system is essential for transport into and out of the nucleus. EMBO J 16, 6535–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur N, Mignery GA, and Banach K (2007). Cell cycle-dependent calcium oscillations in mouse embryonic stem cells. Am. J. Physiol. Cell Physiol 292, C1510–C1518. [DOI] [PubMed] [Google Scholar]
- Kinoshita M, and Smith A (2018). Pluripotency deconstructed. Dev. Growth Differ 60, 44–52. [DOI] [PubMed] [Google Scholar]
- Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera Mdel.C., and Yusa K (2014). Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol 32, 267–273. [DOI] [PubMed] [Google Scholar]
- Koyama M, Shirai N, and Matsuura Y (2014). Structural insights into how Yrb2p accelerates the assembly of the Xpo1p nuclear export complex. Cell Rep 9, 983–995. [DOI] [PubMed] [Google Scholar]
- Li X, Zhu L, Yang A, Lin J, Tang F, Jin S, Wei Z, Li J, and Jin Y (2011). Calcineurin-NFAT signaling critically regulates early lineage specification in mouse embryonic stem cells and embryos. Cell Stem Cell 8, 46–58. [DOI] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Köster J, Xu H, Chen CH, Xiao T, Liu JS, Brown M, and Liu XS (2015). Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol 16, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Yu JSL, Tilgner K, Ong SH, Koike-Yusa H, and Yusa K (2018). Genome-wide CRISPR-KO screen uncovers mTORC1-mediated Gsk3 regulation in naive pluripotency maintenance and dissolution. Cell Rep 24, 489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Wang YJ, Tang Y, Cao N, Wang J, and Yang HT (2010). Type 3 inositol 1,4,5-trisphosphate receptor negatively regulates apoptosis during mouse embryonic stem cell differentiation. Cell Death Differ 17, 1141–1154. [DOI] [PubMed] [Google Scholar]
- Lindsay ME, Holaska JM, Welch K, Paschal BM, and Macara IG (2001). Ran-binding protein 3 is a cofactor for Crm1-mediated nuclear protein export. J. Cell Biol 153, 1391–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay H, Burger A, Biyong B, Felker A, Hess C, Zaugg J, Chiavacci E, Anders C, Jinek M, Mosimann C, et al. (2016). CrispRVariants charts the mutation spectrum of genome engineering experiments. Nat. Biotechnol 34, 701–702. [DOI] [PubMed] [Google Scholar]
- Lounsbury KM, Richards SA, Carey KL, and Macara IG (1996). Mutations within the Ran/TC4 GTPase. Effects on regulatory factor interactions and subcellular localization. J. Biol. Chem 271, 32834–32841. [DOI] [PubMed] [Google Scholar]
- Martello G, Sugimoto T, Diamanti E, Joshi A, Hannah R, Ohtsuka S, Göttgens B, Niwa H, and Smith A (2012). Esrrb is a pivotal target of the Gsk3/Tcf3 axis regulating embryonic stem cell self-renewal. Cell Stem Cell 11, 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martello G, Bertone P, and Smith A (2013). Identification of the missing pluripotency mediator downstream of leukaemia inhibitory factor. EMBO J 32, 2561–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez NA, Ayala AM, Martinez M, Martinez-Rivera FJ, Miranda JD, and Silva WI (2016). Caveolin-1 regulates the P2Y2 receptor signaling in human 1321N1 astrocytoma cells. J. Biol. Chem 291, 12208–12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez M, Martínez NA, and Silva WI (2017). Measurement of the intra-cellular calcium concentration with Fura-2 AM using a fluorescence plate reader. Bio-protocol 7, e2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgani S, Nichols J, and Hadjantonakis AK (2017). The many faces of pluripotency: in vitro adaptations of a continuum of in vivo states. BMC Dev. Biol 17, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okunade GW, Miller ML, Pyne GJ, Sutliff RL, O’Connor KT, Neumann JC, Andringa A, Miller DA, Prasad V, Doetschman T, et al. (2004). Targeted ablation of plasma membrane Ca2+-ATPase (PMCA) 1 and 4 indicates a major housekeeping function for PMCA1 and a critical role in hyperactivated sperm motility and male fertility for PMCA4. J. Biol. Chem 279, 33742–33750. [DOI] [PubMed] [Google Scholar]
- Pereira L, Yi F, and Merrill BJ (2006). Repression of Nanog gene transcription by Tcf3 limits embryonic stem cell self-renewal. Mol. Cell. Biol 26, 7479–7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinello L, Canver MC, Hoban MD, Orkin SH, Kohn DB, Bauer DE, and Yuan GC (2016). Analyzing CRISPR genome-editing experiments with CRISPResso. Nat. Biotechnol 34, 695–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren M, Villamarin A, Shih A, Coutavas E, Moore MS, LoCurcio M, Clarke V, Oppenheim JD, D’Eustachio P, and Rush MG (1995). Separate domains of the Ran GTPase interact with different factors to regulate nuclear protein import and RNA processing. Mol. Cell. Biol 15, 2117–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazi MN, Scialdone A, Skorupska N, Weberling A, Recher G, Zhu M, Jedrusik A, Devito LG, Noli L, Macaulay IC, et al. (2017). Pluripotent state transitions coordinate morphogenesis in mouse and human embryos. Nature 552, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shy BR, Wu CI, Khramtsova GF, Zhang JY, Olopade OI, Goss KH, and Merrill BJ (2013). Regulation of Tcf7l1 DNA binding and protein stability as principal mechanisms of Wnt/β-catenin signaling. Cell Rep 4, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A (2017). Formative pluripotency: the executive phase in a developmental continuum. Development 144, 365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AG, Heath JK, Donaldson DD, Wong GG, Moreau J, Stahl M, and Rogers D (1988). Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature 336, 688–690. [DOI] [PubMed] [Google Scholar]
- Stachecki JJ, and Armant DR (1996). Transient release of calcium from inositol 1,4,5-trisphosphate-specific stores regulates mouse preimplantation development. Development 122, 2485–2496. [DOI] [PubMed] [Google Scholar]
- Strehler EE (2015). Plasma membrane calcium ATPases: from generic Ca(2+) sump pumps to versatile systems for fine-tuning cellular Ca(2.). Biochem. Biophys. Res. Commun 460, 26–33. [DOI] [PubMed] [Google Scholar]
- Stuart HT, van Oosten AL, Radzisheuskaya A, Martello G, Miller A, Dietmann S, Nichols J, and Silva JC (2014). NANOG amplifies STAT3 activation and they synergistically induce the naive pluripotent program. Curr. Biol 24, 340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J, Kanemaru K, Ishii K, Ohkura M, Okubo Y, and Iino M (2014). Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun 5, 4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesar PJ, Chenoweth JG, Brook FA, Davies TJ, Evans EP, Mack DL, Gardner RL, and McKay RD (2007). New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 448, 196–199. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, and Jones JM (1998). Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147. [DOI] [PubMed] [Google Scholar]
- Tosti E, Boni R, and Gallo A (2016). Ion currents in embryo development. Birth Defects Res. C Embryo Today 108, 6–18. [DOI] [PubMed] [Google Scholar]
- Tsukiyama T, and Ohinata Y (2014). A modified EpiSC culture condition containing a GSK3 inhibitor can support germline-competent pluripotency in mice. PLoS ONE 9, e95329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villegas F, Lehalle D, Mayer D, Rittirsch M, Stadler MB, Zinner M, Olivieri D, Vabres P, Duplomb-Jego L, De Bont ESJM, et al. (2019). Lysosomal signaling licenses embryonic stem cell differentiation via inactivation of Tfe3. Cell Stem Cell 24, 257–270.e8. [DOI] [PubMed] [Google Scholar]
- Wang J, Rout UK, Bagchi IC, and Armant DR (1998). Expression of calcitonin receptors in mouse preimplantation embryos and their function in the regulation of blastocyst differentiation by calcitonin. Development 125, 4293–4302. [DOI] [PubMed] [Google Scholar]
- Wang FW, Zhang YM, Wang Z, Liu SM, Wang LY, Zhang XL, Jia DY, Hao AJ, and Wu YL (2013). Calcitonin promotes mouse pre-implantation development: involvement of calcium mobilization and P38 mitogen-activated protein kinase activation. Reprod. Domest. Anim 48, 382–389. [DOI] [PubMed] [Google Scholar]
- Wei WJ, Sun HY, Ting KY, Zhang LH, Lee HC, Li GR, and Yue J (2012). Inhibition of cardiomyocytes differentiation of mouse embryonic stem cells by CD38/cADPR/Ca2+ signaling pathway. J. Biol. Chem 287, 35599–35611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Lu Y, Hao B, Zhang K, Wang Q, Miller AL, Zhang LR, Zhang LH, and Yue J (2015). CD38 is required for neural differentiation of mouse embryonic stem cells by modulating reactive oxygen species. Stem Cells 33, 2664–2673. [DOI] [PubMed] [Google Scholar]
- Williams RL, Hilton DJ, Pease S, Willson TA, Stewart CL, Gearing DP, Wagner EF, Metcalf D, Nicola NA, and Gough NM (1988). Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature 336, 684–687. [DOI] [PubMed] [Google Scholar]
- Wray J, Kalkan T, Gomez-Lopez S, Eckardt D, Cook A, Kemler R, and Smith A (2011). Inhibition of glycogen synthase kinase-3 alleviates Tcf3 repression of the pluripotency network and increases embryonic stem cell resistance to differentiation. Nat. Cell Biol 13, 838–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagida E, Shoji S, Hirayama Y, Yoshikawa F, Otsu K, Uematsu H, Hiraoka M, Furuichi T, and Kawano S (2004). Functional expression of Ca2+ signaling pathways in mouse embryonic stem cells. Cell Calcium 36, 135–146. [DOI] [PubMed] [Google Scholar]
- Yang S-H, Kalkan T, Morrisroe C, Smith A, and Sharrocks AD (2012). A genome-wide RNAi screen reveals MAP kinase phosphatases as key ERK pathway regulators during embryonic stem cell differentiation. PLoS Genet 8, e1003112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F, and Merrill BJ (2010). Non-cell-autonomous stimulation of stem cell proliferation following ablation of Tcf3. Exp. Cell Res 316, 1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F, Pereira L, Hoffman JA, Shy BR, Yuen CM, Liu DR, and Merrill BJ (2011). Opposing effects of Tcf3 and Tcf1 control Wnt stimulation of embryonic stem cell self-renewal. Nat. Cell Biol 13, 762–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, and Smith A (2008). The ground state of embryonic stem cell self-renewal. Nature 453, 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SO, Shin S, Liu Y, Ballif BA, Woo MS, Gygi SP, and Blenis J (2008). Ran-binding protein 3 phosphorylation links the Ras and PI3-kinase pathways to nucleocytoplasmic transport. Mol. Cell 29, 362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZH, Lu YY, and Yue J (2013). Two pore channel 2 differentially modulates neural differentiation of mouse embryonic stem cells. PLoS ONE 8, e66077. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data and custom scripts are either provided as a supplemental table (Tables S1, S2, S3, S4, S5, and S6) or are made available on Mendeley Data with https://doi.org/10.17632/9mfp9n3j6p.1. The accession number for the all sequencing in this paper is SRANCBI: SRP151214.