

Abstract
Exfoliation syndrome (XFS) is the commonest known risk factor for secondary glaucoma and a significant cause of blindness worldwide. Variants in two genes, LOXL1 and CACNA1A have been previously associated with XFS. To further elucidate the genetic basis of XFS, we collected a global sample of XFS cases to refine the association at LOXL1, which previously showed inconsistent results between populations, and to identify new variants associated with XFS. We identified a rare, protective allele at LOXL1 (p.407Phe, OR = 25, P =2.9 × 10−14) through deep resequencing of XFS cases and controls from 9 countries. This variant results in increased cellular adhesion strength compared to the wild-type (p.407Tyr) allele. A genome-wide association study (GWAS) of XFS cases and controls from 24 countries followed by replication in 18 countries identified seven genome-wide significant loci (P < 5 × 10−8). Index variants at the new loci map to chromosomes 13q12 (POMP), 11q23.3 (TMEM136), 6p21 (AGPAT1), 3p24 (RBMS3) and 5q23 (near SEMA6A). These findings provide biological insights into the pathology of XFS, and highlight a potential role for naturally occurring rare LOXL1 variants in disease biology.
Exfoliation syndrome (XFS) is an age-related systemic disorder involving the extracellular matrix (ECM). It is characterized by the excessive production and progressive accumulation of an abnormal extracellular material in various tissues1. Manifesting most conspicuously in the eye, XFS is the commonest cause of secondary glaucoma world-wide2 and is also a prognostic factor for progression of open-angle glaucoma3. This disease is common in many populations, with an estimated 60–70 million patients affected4–7. Exfoliation glaucoma (XFG) has a worse prognosis than other major types of glaucoma, and it is often resistant to intraocular pressure-lowering medical treatment, more often necessitating laser and surgical intervention3,8. Cumulatively, XFS/XFG is a significant cause of blindness globally.
The strong pattern of familial aggregation for XFS indicates a significant genetic contribution to disease pathology9,10 and LOXL1, a gene coding for lysyl oxidase homolog 1, was the first genetic locus reported to be associated with this disease11. Despite the overwhelming strength of the genetic association seen at polymorphisms mapping to LOXL1, the results are inconsistent due to risk alleles being ‘flipped’ in certain populations12,13. Such stark allele reversals imply that the genetic architecture underlying XFS disease biology is complex and worthy of further study.
Recently, CACNA1A was identified as the second locus associated with XFS14. Realizing that large and inclusive international collaborative efforts are essential in providing new biological leads in complex disease pathogenesis15–20, we report here a world-wide collaborative XFS study aimed at further understanding the genetic basis of the disorder. Firstly, due to the allele reversals seen at LOXL1 common polymorphisms led by rs3825942 G>A (p.153Gly>Asp) and to a lesser extent, rs1048661 T>G (p.141Leu>Arg)12,21–28(Supplementary Figure 1), we aimed to refine the LOXL1 genetic landscape by performing deep sequencing of the entire gene in 5,570 XFS and XFG cases and 6,279 controls from 9 countries (Supplementary Table 1). The previously reported CACNA1A locus was also sequenced to assess if rare non-synonymous amino acid substitutions within the gene could provide further insights 29–31. In our effort to identify additional genetic variants associated with XFS, we also conduct an expanded genome-wide association study (GWAS) of 13,838 cases and 110,275 controls from countries across six continents (Supplementary Table 2, Supplementary Figures 2 and 3).
Results
A LOXL1 rare missense variant protects against XFS.
We conducted deep re-sequencing of the entire LOXL1 and CACNA1A loci (see Methods) in 5,570 XFS cases and 6,279 controls. This sequencing effort confirmed previously reported strong allele reversals at key LOXL1 common variants and also at recently reported non-coding variants (Supplementary Tables 3, 4 and 5)13,21,23.
We first analyzed the sequencing data to find unifying consistent common variants associated with XFS across ethnic groups that could have been missed by previous efforts. Single variant analysis showed that for all common variants polymorphic across all collections studied, rs3825942 G>A (encoding for LOXL1 p.153Gly>Asp) remains the most significantly associated variant (Pfixed-effects = 4.14 × 10−62), but with very high heterogeneity across study groups (Prandom effects = 0.0039). No LOXL1 common variant was consistently associated across all collections, and no LOXL1 common variant surpassed genome-wide significance on random effects analysis (Supplementary Table 6). Conditioning for allele dosage at rs3825942 G>A abolished all residual evidence of association across the collections (Supplementary Table 6). The commonly reported rs1048661 T>G (p.141Leu>Arg) polymorphism was not significant in the meta-analysis of sequencing data either before (P=0.25) or after (P=0.53) conditioning for rs3825942 G>A (Pfor heterogeneity < 1×10−10; l2 = 98.3%). Recognizing that single variant analysis could have missed a consistent LOXL1 haplotypic association which is unreversed across populations, we followed up our search by phasing haplotypes of 57 SNPs across the entire LOXL1 sequenced locus in 20-SNP sliding windows. All analyzed haplotypes showed reversal of effect across the locus, with no exceptions (Supplementary Dataset 1). It is thus unlikely that we may have missed an “unflipped” common variant which shows consistent association with XFS across our world-wide sample.
The re-sequencing of LOXL1 revealed a total of 63 unique non-synonymous variants across the nine countries studied (Supplementary Table 7 and Supplementary Figure 4). Due to the limited insights shown by all the reversed LOXL1 common haplotypes, we next evaluated the hypothesis that rare alleles collectively within LOXL1 (MAF<1%) could contribute to XFS risk. We observed a broad enrichment of rare LOXL1 non-synonymous variants in the normal controls compared to the XFS patients (OR = 0.46, P = 4.2 × 10−7; Table 1). As the vast majority of non-synonymous variants do not exert functional effects31–34, we performed a second test restricting the analysis to aggregate only rare, non-synonymous variants conservatively predicted to be deleterious by all five functional effect prediction algorithms (SIFT, Polyphen 2-HumDiv, LRT score, MutationTaster, and Condel)33. In so doing, we observed a substantially larger protective effect size conferred by rare variant burden (OR = 0.18, P = 4.23 × 10−11; Table 2). This protective burden of alleles conservatively predicted to affect LOXL1 function remained significant even after accounting for co-segregation at the sentinel rs3825942 G>A SNP (Supplementary Table 8).
Table 1.
Association of all rare, non-synonymous variants at LOXL1 and risk of exfoliation syndrome. No additional filters by functional effect prediction algorithms were applied. Frequencies for rare variant carriers are given in percentages.
| Collection | N cases | N controls | Allele burden cases |
Allele burden controls |
Carrier freq cases |
Carrier freq controls |
Allele OR | L95 | U95 | P |
|---|---|---|---|---|---|---|---|---|---|---|
| Japanese | 2827 | 3013 | 34 | 100 | 1.20 | 3.32 | 0.36 | 0.24 | 0.53 | 8.03 × 10−8 |
| Greece | 355 | 1075 | 3 | 17 | 0.85 | 1.58 | 0.53 | 0.16 | 1.82 | 0.44 |
| Italy | 454 | 267 | 10 | 3 | 2.20 | 1.12 | 1.97 | 0.54 | 7.19 | 0.39 |
| Russia | 476 | 859 | 2 | 5 | 0.42 | 0.58 | 0.72 | 0.14 | 3.72 | 1 |
| USA | 212 | 161 | 2 | 2 | 0.94 | 1.24 | 0.76 | 0.11 | 5.41 | 1 |
| Mexico | 116 | 205 | 2 | 9 | 1.72 | 4.39 | 0.39 | 0.083 | 1.81 | 0.34 |
| South Africa | 95 | 250 | 1 | 21 | 1.05 | 8.40 | 0.12 | 0.016 | 0.90 | 0.014 |
| India | 648 | 263 | 12 | 8 | 1.85 | 3.04 | 0.61 | 0.25 | 1.49 | 0.32 |
| Pakistan | 383 | 186 | 7 | 4 | 1.83 | 2.15 | 0.85 | 0.25 | 2.92 | 0.76 |
| Stratified meta-analysis for all sequenced collections | 0.46 | 0.34 | 0.62 | 4.2 × 10−7 | ||||||
Table 2.
Association of rare, non-synonymous variants at LOXL1 and risk of exfoliation syndrome tagged as deleterious by five functional prediction algorithms (SIFT, Polyphen2-HumDiv, LRT score, MutationTaster, and CONDEL). Frequencies for rare variant carriers are given in percentages.
| Collection | N cases | N controls | Allele burden cases |
Allele burden controls |
Carrier freq cases |
Carrier freq controls |
OR | L95 | U95 | P |
|---|---|---|---|---|---|---|---|---|---|---|
| Japanese | 2827 | 3013 | 11 | 85 | 0.39 | 2.82 | 0.14 | 0.073 | 0.26 | 3.49 × 10−13 |
| Greece | 355 | 1075 | 1 | 6 | 0.28 | 0.56 | 0.50 | 0.061 | 4.19 | 1 |
| Italy | 454 | 267 | 1 | 3 | 0.22 | 1.12 | 0.20 | 0.020 | 1.88 | 0.15 |
| Russia | 476 | 859 | 0 | 4 | 0 | 0.47 | 0 | N/A | N/A | 0.56 |
| USA | 212 | 161 | 1 | 1 | 0.47 | 0.62 | 0.76 | 0.047 | 12.18 | 1 |
| Mexico | 116 | 205 | 0 | 0 | 0 | 0 | 0 | NA | NA | 1 |
| South Africa | 95 | 250 | 0 | 2 | 0 | 0.80 | 0 | NA | NA | 0.38 |
| India | 648 | 263 | 5 | 7 | 0.77 | 2.66 | 0.29 | 0.091 | 0.91 | 0.047 |
| Pakistan | 383 | 186 | 0 | 3 | 0 | 1.61 | 0 | NA | NA | 0.035 |
| Stratified meta-analysis for all sequenced collections | 0.18 | 0.11 | 0.30 | 4.23 × 10−11 | ||||||
| Meta-analysis excluding Russia, Mexico, South Africa, and Pakistan* | 0.19 | 0.11 | 0.31 | 1.41 × 10−10 | ||||||
Excluding collections where allele zero in either cases or controls
One of the rare, non-synonymous variants, rs201011613 A>T encoding for LOXL1 p.407Tyr>Phe, showed genome-wide significance on single-variant analysis. This variant was conservatively predicted by all five protein functional predictive algorithms to affect LOXL1 function, and is found exclusively in the Japanese (Table 3,Supplementary Figure 4a and 4b, Supplementary Table 7). The rare rs201011613-T (LOXL1 p.407Phe) allele was observed in only 2 XFS cases (N= 3,909, 0.026%) but was observed in 68 out of 5,338 (0.64%) age- and hospital-matched controls (N=5,338, 0.64%) with no eye disorders (Table 3), conferring a 25-fold resistance to XFS (PFisher’s exact =2.9 × 10−14). We examined the individuals carrying the rare p.407Phe allele who had also undergone genome-wide genotyping for evidence of population substructure, but found no evidence that these carriers clustered along the major axes of population stratification (Supplementary Figure 5)35,36.
Table 3.
Association between LOXL1 p.Y407F (rs201011613, A>T base change) and exfoliation syndrome.
| p.Y407F | N cases |
N controls |
Allele count in cases (%) |
Allele count controls |
Freq. cases (%) |
Freq. controls (%) |
OR | L95 | U95 | p |
|---|---|---|---|---|---|---|---|---|---|---|
| Japan sequencing | 2827 | 3013 | 1 | 36 | 0.018 | 0.60 | 0.029 | 0.0040 | 0.21 | 8.3 × 10−10 |
| Japan replication | 1082 | 2325 | 1 | 32 | 0.046 | 0.69 | 0.067 | 0.0091 | 0.49 | 8.9 × 10−5 |
| Japan Combined | 3909 | 5338 | 2 | 68 | 0.026 | 0.64 | 0.040 | 0.0098 | 0.16 | 2.9 × 10−14 |
| European sequencing | 1613 | 2567 | 0 | 0 | 0.0 | 0.0 | - | - | - | - |
| South Africa sequencing | 95 | 250 | 0 | 0 | 0.0 | 0.0 | - | - | - | - |
| South Asia sequencing | 1031 | 449 | 0 | 0 | 0.0 | 0.0 | - | - | - | - |
This rare variant was found exclusively in the Japanese, and was not polymorphic in Europe, Africa, and South Asia. P-values are by Fisher’s exact test.
We next examined the haplotype background for all 37 p.407Phe allele carrying Japanese individuals who underwent sequencing for the LOXL1 locus (Table 3), and found that in 35 of the 37 individuals p.407Phe segregated with the common rs3825942-A (p.153Asp) haplotype. As previously discussed, this common rs3825942-A haplotype was associated with protection against XFS in Japan but conferred susceptibility to XFS in Black Africans and was thus only nominally significant in the meta-analysis of all collections which underwent resequencing for LOXL1 (Prandom effects = 0.0039; Supplementary Table 3).
Although the rare protective p.407Phe allele does not segregate with the common p.Arg141Leu polymorphism, functional biological testing would be needed to assess the relative impact of all three (at positions 141, 153, and 407) non-synonymous variants to LOXL1 function.
At the CACNA1A locus, a total of 200 unique rare, non-synonymous amino acid substitutions were observed in the coding frame of CACNA1A after re-sequencing in East Asians, Europeans, South Africa, and South Asians. In contrast to LOXL1, we did not observe any consistent evidence of association between rare variant burden at CACNA1A and susceptibility to exfoliation syndrome (Supplementary Table 9). This is not surprising, as only some common variant GWAS loci harbor additional rare variant burden37.
Biological relevance of LOXL1 p.Y407F.
XFS is characterized by excessive production and progressive accumulation of an abnormal fibrillar material, termed exfoliation material, containing ECM components such as elastin, fibrillin-1, and fibronectin localized to the surface of various cell types including lens epithelial cells1,38,39. LOXL1 has been reported to modulate ECM biogenesis by cross-linking elastin and collagen in connective tissues40–42. We thus performed experiments assaying the effects of LOXL1 variants on ECM metabolism, with elastin, fibrillin-1, collagen type IV, and fibronectin as cellular biochemical readouts, and overall relative cell adhesion as a cellular physiological readout.
We assessed functional effects for the common, flipped p.Arg141Leu and p.Gly153Asp polymorphisms as well as the rare protective p.Tyr407Phe variant using four constructs carrying these three variants in naturally occurring haplotypes (Figure 1a). This experimental design also allowed for measurement of the effect of p.Tyr407Phe while conditioning for the effect of all p.Arg141Leu - p.Gly153Asp haplotype combinations. The ability to condition against and account for the effect of p.Gly153Asp is particularly important as the rare protective p.407Phe segregates with the p.153Asp allele. Using human lens epithelial cell (HLEC) 3D cell cultures that were transiently expressing the four haplotypes, we observed that the LOXL1 protein was secreted at detectable levels, and no significant difference in LOXL1 secretion could be observed between any of the four haplotypes (Supplementary Figure 6a).
Figure 1.
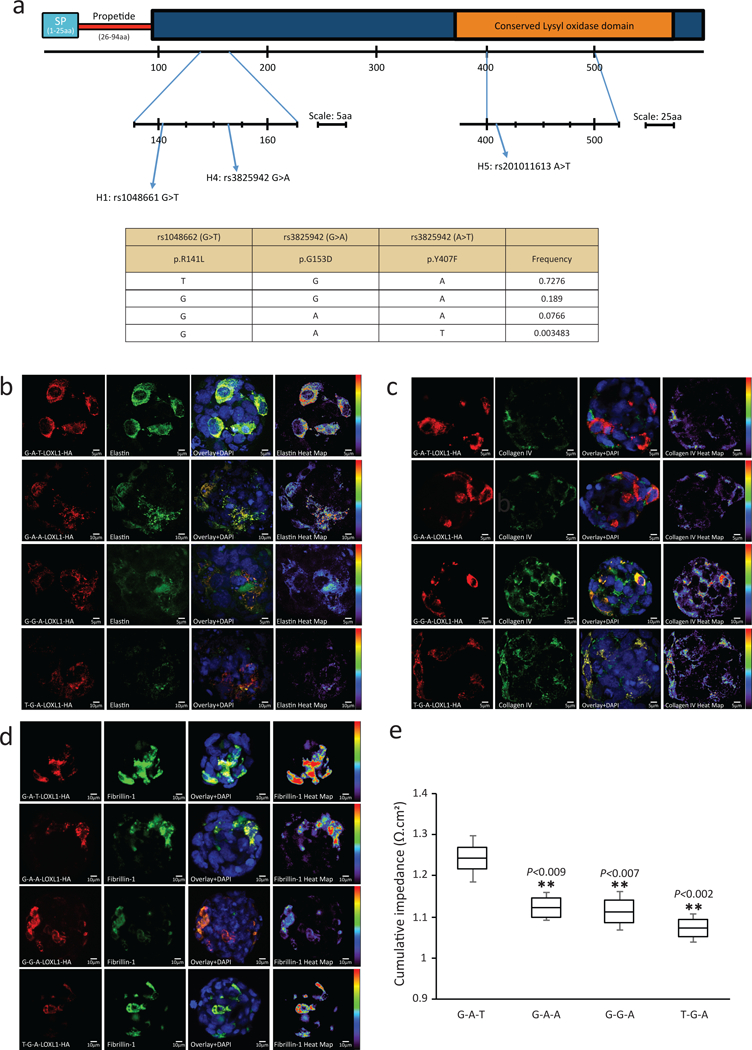
LOXL1 p.Y407F regulates ECM synthesis and improves cellular adhesion.
(a) Schematic diagram of LOXL1 indicating the protein domain positions for the variants evaluated in this study.
b) Immunofluorescent staining of HA-tagged LOXL1 variants overexpressed in HLEC cells labelled with anti-HA for detection of overexpressed forms of LOXL1 (red) and elastin (green). Cell nuclei are stained in blue. The heat map for elastin indicates the intensity of elastin staining from red (increased expression) to purple (decreased expression).
c) Immunofluorescent staining of HA-tagged LOXL1 variants overexpressed in HLEC cells labelled with anti-HA for detection of overexpressed forms of LOXL1 (red) and collagen IV (green). Cell nuclei are stained in blue.. The heat map for collagen IV indicates the intensity of collagen IV staining from red (increased expression) to purple (decreased expression).
d) Immunofluorescent staining of HA-tagged LOXL1 variants overexpressed in HLEC cells labelled with anti-HA for detection of overexpressed forms of LOXL1 (red) and fibrillin 1 (green). Cell nuclei are stained in blue. The heat map for fibrillin 1 indicates the intensity of fibrillin 1 staining from red (increased expression) to purple (decreased expression).
e) Cumulative average of impedance values (as a surrogate for cellular adhesion strength) measured over 35h post nucleofection of HLECs overexpressing the four tested LOXL1 haplotypes. Data represent mean ± s.e.m. of four independent experiments. ** represents P<0.01 when compared against the rare, protective LOXL1 p.407F-carrying G-A-T haplotype. The four haplotypes tested were LOXL1-(G-A-T), - (G-A-A), -(T-G-A) and -(G-G-A). This experiment was further validated in Supplementary Figure 7.
In contrast, when we overexpressed the rare p.407Phe-carrying LOXL1–141Arg-153Asp-407Phe (G-A-T) haplotype in HLEC cultures, we observed a dose-dependent increase in elastin on Western blot (Supplementary Figure 6b), with the increase in elastin reiterated by 3D spheroid culture immunofluorescence analysis (Figure 1b). Overexpression of this rare p.407Phe-carrying haplotype also resulted in a decrease in collagen IV (Figure 1c) and an increase in fibrillin-1 (Figure 1d) levels when compared to the other three haplotypes that included the wild-type p.407Tyr allele. This result suggests that the rare p.407Phe allele had an overall upregulating effect on ECM components such as elastin and fibrillin.
As the ECM is important for cellular adhesion, we next asked whether these observed in-vitro functional biochemical effects of the rare LOXL1 p.407Phe allele on ECM components would translate to a physiological outcome affecting cell-cell adhesion. To this end, HLEC 3D spheroids overexpressing the four haplotypes were analyzed for their relative cellular adhesion strength using the microelectrodes of the Roche xCelligence real-time cell analysis system. Using this previously described methodology, the change in cellular impedance readout is directly proportional to the quality of cell attachment43–45. We observed that the rare, protective LOXL1 p.407Phe-carrying G-A-T haplotype conferred a significant increase in cellular adhesion strength in comparison to the remaining three haplotypes carrying the wild-type p.407Tyr allele (P<0.01 for all comparisons; Figure 1e). We observed no significant difference in relative cellular adhesion strength when the remaining three haplotypes carrying p.407Tyr (but with different combinations of the p.Arg141Leu and p.Gly153Asp alleles) were compared to one another (Figure 1e), suggesting that the common p.Arg141Leu and p.Gly153Asp polymorphisms have no significant effect on the strength of cellular adhesion in this assay. To ensure that the increase in cell-cell adhesion was unique to the rare protective p.407Phe (rs201011613-T) allele, two additional haplotype constructs covering the G-G-T (LOXL1–141Arg-153Gly-407Phe) and T-G-T (LOXL1–141Leu-153Gly- 407Phe) haplotypes were cloned. We retested all six haplotype constructs together for differences in cell-cell adhesion (Supplementary Figure 7), and continue to observe that the introduction of the rare protective p.407Phe (rs201011613-T) allele significantly increased physiological cell-cell adhesion regardless of background p.Arg141Leu and p.Gly153Asp alleles. In contrast, the haplotypes carrying the wild-type baseline p.407Tyr (rs201011613-A) allele had significantly lower cell-cell adhesion also regardless of the p.Arg141Leu and p.Gly153Asp alleles (P<1×10−4 for all comparisons; Supplementary Figure 7).
GWAS identifies five new loci associated with XFS
As XFS is a complex disease, we also sought to identify new genetic loci associated with this disorder. For the GWAS discovery stage, we directly genotyped a total of 9,035 XFS cases and 17,008 controls enrolled from 24 countries across six continents using the Illumina OmniExpress Beadarray (Supplementary Table 2). After quality checks, we were able to analyze 683,397 directly genotyped autosomal SNP markers for association with XFS disease status (Supplementary Dataset 2)(see Supplementary Note for full details).
The GWAS discovery meta-analysis revealed consistent and significant association (OR = 1.17, P=2.97 × 10−10, l2 = 0%; Supplementary Figure 8) at a novel locus defined by SNP rs7329408 mapping to FLT1-POMP-SLC46A3 on chromosome 13. We also observed a clear excess of smaller-than-expected P-values at the tail end of the quantile-quantile distribution (P ≤ 1 × 10−4; Supplementary Figure 837,46,47), suggesting that there are additional loci to be identified. We forwarded all markers showing P ≤ 1 × 10−4 in the GWAS discovery stage for further assessment in the replication stage (see Supplementary Table 10 for power calculations) comprising 4,803 XFS cases and 93,267 controls independently ascertained from 18 countries (Supplementary Table 2). SNPs at five distinct new loci showed consistent evidence of replication, and meta-analysis of all 13,838 cases and 110,275 controls from the GWAS and replication series revealed genome-wide significant association at the five loci (1.56 × 10−16 ≤ P ≤ 1.5 × 10−8, Table 4, Figure 2, and Supplementary Figure 9; see Supplementary Table 11 for ethno-geographical stratified analysis of the five new loci). These loci are FLT1-POMP-SLC46A3 rs7329408 (chromosome 13), TMEM136-ARHGEF12 rs11827818 (chromosome 11) AGPAT1 rs3130283 (chromosome 6), RBMS3 rs12490863 (chromosome 3), and SEMA6A rs10072088 (chromosome 5). Regional association maps showing the genomic organization of the five loci within a 1 Mb flanking region of the sentinel SNPs are appended as Supplementary Figure 10. We verified the genotyping at the sentinel SNPs for all five loci to be of good quality (Supplementary Figure 11). We also confirm genome-wide significant association at the previously reported CACNA1A rs4926244 (Supplementary Table 12). Of the five new loci, only rs7329408 showed a significant latitude gradient effect, with the odds ratio of the risk allele highest in polar regions and lowest in equatorial regions (Supplementary Note and Supplementary Table 13).
Table 4.
Summary of genetic associations for the five newly identified loci.
| Association tests | Heterogeneity tests | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome | SNP (effect/reference) |
Position | Gene locus |
Stage | OR | L95 | U95 | P-value | Phet | I2 index |
| 13 | rs7329408 (A/G) | 29166671 | FLT1 -POMP | GWAS discovery | 1.17 | 1.11 | 1.22 | 2.97 × 10−10 | 0.9 | 0.00% |
| Replication summary | 1.18 | 1.11 | 1.25 | 9.63 × 10−8 | 0.17 | 23.20% | ||||
| All data summary | 1.17 | 1.13 | 1.22 | 1.56 × 10−16 | 0.62 | 0.00% | ||||
| European Caucasian summary ∥ | 1.22 | 1.15 | 1.29 | 7.82 × 10−12 | ||||||
| Asian summary | 1.13 | 1.07 | 1.19 | 1.61 × 10−5 | ||||||
| 11 | rs11827818 (G/A) | 120198728 | TMEM136 | GWAS discovery | 1.10 | 1.05 | 1.16 | 0.0001 | 0.10 | 28.10% |
| Replication summary | 1.18 | 1.11 | 1.25 | 1.96 × 10−8 | 0.36 | 8.00% | ||||
| All data summary | 1.14 | 1.09 | 1.18 | 5.86 × 10−11 | 0.09 | 23.10% | ||||
| European Caucasian summary ∥ | 1.14 | 1.08 | 1.20 | 2.09 × 10−6 | ||||||
| Asian summary | 1.15 | 1.08 | 1.22 | 4.35 × 10−6 | ||||||
| 6 | rs3130283 (A/C) | 32138545 | AGPAT1 | GWAS discovery | 1.19 | 1.11 | 1.27 | 1.29 × 10−6 | 0.38 | 5.60% |
| Replication summary | 1.15 | 1.07 | 1.24 | 0.00013 | 0.96 | 0.00% | ||||
| All data summary | 1.17 | 1.11 | 1.23 | 7.62 × 10−10 | 0.81 | 0.00% | ||||
| European Caucasian summary ∥ | 1.13 | 1.06 | 1.22 | 0.00034 | ||||||
| Asian summary | 1.24 | 1.14 | 1.34 | 2.27 10−7 | ||||||
| 3 | rs12490863 (A/G) | 29907310 | RBMS3 | GWAS discovery | 1.15 | 1.09 | 1.22 | 4.9 × 10−7 | 0.69 | 0.00% |
| Replication summary | 1.12 | 1.04 | 1.20 | 0.002 | 0.13 | 26.50% | ||||
| All data summary | 1.14 | 1.09 | 1.19 | 7 × 10−9 | 0.23 | 12.50% | ||||
| European Caucasian summary ∥ | 1.19 | 1.11 | 1.27 | 1.64 × 10−6 | ||||||
| Asian summary | 1.12 | 1.05 | 1.20 | 0.00053 | ||||||
| 5 | rs10072088 (G/A) | 116019417 | SEMA6A | GWAS discovery | 0.89 | 0.85 | 0.94 | 2.3 × 10−5 | 0.85 | 0.00% |
| Replication summary | 0.88 | 0.83 | 0.94 | 0.00017 | 0.10 | 36% | ||||
| All data summary | 0.89 | 0.85 | 0.93 | 1.5 × 10−8 | 0.66 | 0.00% | ||||
| European Caucasian summary ∥ | 0.90 | 0.85 | 0.94 | 2.83 × 10−5 | ||||||
| Asian summary | 0.88 | 0.81 | 0.96 | 0.0024 | ||||||
This summary includes 7,113 cases and 95,863 controls from North America, Northern-, Southern-, Eastern-, and CentralWestern Europe. This summary does not include Latin and South America.
Figure 2.
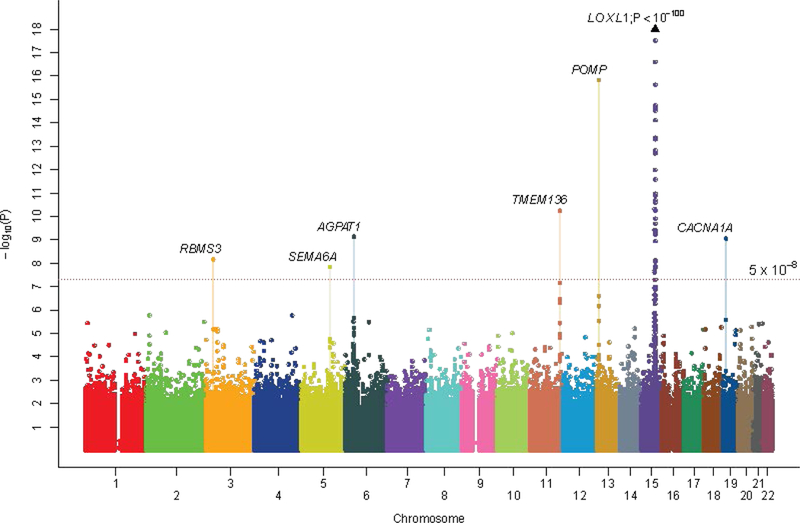
Manhattan plot of the results from the GWAS discovery plus replication meta-analysis comprising 13,620 XFS cases and 109,837 controls. Genetic markers are plotted according to chromosomal location on the horizontal axis and statistical significance on the vertical axis. SNP markers at seven independent loci surpass genome-wide significance (defined as P<5×10−8). They are LOXL1, CACNA1A, POMP, TMEM136, AGPAT1, SEMA6A and RBMS3.
Biological insights from GWAS associated loci.
We annotated 33 genes mapping to or located closest (genomic region within a 150,000bp flanking region both 5’ and 3’ to the sentinel SNPs and SNP markers showing r2>0.5 with the sentinel SNPs (Supplementary Table 14), or as defined by credible set analysis37,48,49, see Methods) to the seven genome-wide significant loci. Except for AGPAT rs3130283 which was located within the broad MHC locus on chromosome 6 which is well known for showing long range complex LD patterns, we observed that defining an ‘associated locus region’ either generically as ±150 Kb from the index variant, or as the region containing proxy SNPs with r2>0.5 with the index variant, all highlight the same genes (Supplementary Figure 10 and Supplementary Table 14). All credible sets for the 7 genome-wide significant loci were located within relatively narrow regions (<100,000 base-pairs), and within the region bound by the proxy SNPs showing r2>0.5 with the index variant (inclusive of the index variant; Supplementary Table 14).
We next assessed the potential biological contribution for each of the 33 genes using the following criteria: 1) genes expressed in anterior segment tissues such as the iris and ciliary body from publicly available databases50–54 . 2) Presence of relevant eye-related phenotypes in knockout mice. 3) cis-QTL genes55,56 . 4) Genes prioritized by text mining in Pubmed. 5) Genes showing pleiotropy with other forms of glaucoma. 6) Highlighted genes from unbiased genome-wide molecular pathway analysis. We summarize these additional information in Supplementary Table 15. A genome-wide search using publicly available databases57 revealed potential molecular interactions between several of the 33 genes located within the 7 genome-wide significant loci (Supplementary Table 15), suggesting that the significantly associated loci could be implicating broader yet undescribed disease biological pathways. A search of the UCSC genome browser revealed that none of the 7 genome-wide significant loci harbor any long non-coding RNA except for LOXL1 (which harbors LOXL1-AS1 in the opposite direction) and the FLT1-POMP-SLC46A3 locus (Supplementary Figure 12). Further interrogation using the INRICH (interval based enrichment analysis tool) software package58, designed for detecting enriched association signals of LD-independent genomic regions within biologically relevant gene sets did not reveal any statistically significant biological pathways highlighted by the 7 genome-wide significant loci (Supplementary Dataset 3).
We next studied the expression of genes associated with the 3 most significantly associated loci. These loci (and genes) were 13q12 (POMP, FLT1, SLC46A3), 11q23.3 (TMEM136, ARHGEF12), and 6p21 (AGPAT1). Expression for these 6 genes was tested in fresh ocular tissues obtained from human donor eyes with appropriate research consent (see Methods). For mRNA expression analyses, 41 normal eyes with no known ocular disease (mean age, 77.1 ± 8.1 years; 20 female, 21 male) and 21 eyes with XFS (mean age, 80.1 ± 7.9 years; 11 female, 10 male) were used. Messenger RNA (mRNA) expression of all 6 genes was detected at moderate levels in the panel of eye tissues analysed (Supplementary Figure 13). The highest expression levels were observed in tissues relevant for the synthesis of exfoliation material (iris, ciliary body) and for glaucoma pathophysiology (retina). Expression levels in these tissues did not significantly correlate with genotypes of the sentinel SNPs underlying the 3 loci (Supplementary Figure 14).
Comparing tissues from XFS and control eyes, mRNA expression levels of POMP and TMEM136 were significantly reduced by up to 41% in anterior segment tissues of XFS eyes, such as iris and ciliary body compared to age-matched control eyes (Supplementary Figure 15). These results in XFS-relevant tissues suggest that POMP (rather than FLT1 and SLC46A3 which are located nearby) is the likely disease gene in the chromosome 13 locus and that TMEM136 (rather than the neighbouring ARHGEF12) is the likely disease gene for the chromosome 11 locus.
POMP and TMEM136 protein expression was further analysed by Western blot and immunofluorescence microscopy. POMP, a proteasome maturation protein, was shown to be expressed in most ocular cell types by immunofluorescence (Figure 3). However, POMP protein expression was significantly reduced in iris (−45%) and ciliary body (−33%) specimens from XFS eyes compared to control eyes when investigated using Western blots (Figure 3G and Supplementary Figure 16) and using immunofluorescence microscopy (Figures 3H, 3J, and 3K, and Figures 3L, 3M, and 3N). These results are consistent with the differential mRNA expression shown in Supplementary Figure 15.
Figure 3.
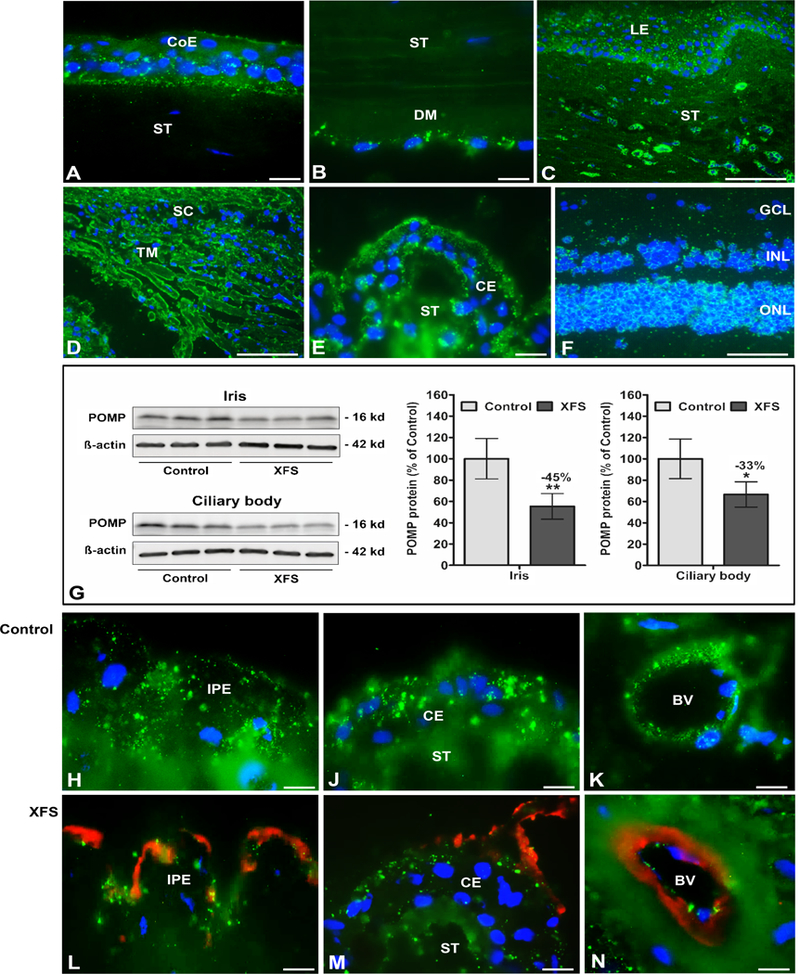
Expression of POMP protein in ocular tissues of normal human donor eyes and donor eyes with XFS, as determined by Western blotting and immunohistochemistry. Immunofluorescence labelling of normal eye tissues shows punctate POMP immune-positivity (green fluorescence) in the cytoplasm of the corneal epithelium (A), the corneal endothelium (B), limbal epithelium and stromal cells (C), trabecular meshwork endothelium (D), ciliary epithelium (E), and retinal cell layers (F).
Reduced POMP protein expression levels in iris and ciliary body tissues of XFS eyes compared to age matched controls are shown by Western blot analysis (G), and by immunofluorescence labelling of iridal (H,L) and ciliary epithelia (J,M) as well as vascular endothelia in the iris (K,N). Reduced staining intensity in XFS tissues is associated with LOXLI-positive exfoliation material accumulations (red immunofluorescence) on the surface of the iris pigment epithelium (L), ciliary epithelium (M) and iris blood vessel walls (N). Western blot (cropped images) and densitometry analysis shows reduced POMP protein expression in iris and ciliary body tissue lysates of XFS eyes compared to control eyes (G). Data are shown as the POMP/B-actin ratio (n=6 for each group; mean ± standard deviation; *P<0.01; **P<0.005); uncropped versions of all Western blots are shown in Supplementary Figure 16. (BV blood vessel, CE ciliary epithelium, CoE corneal epithelium, DM Descemet membrane, GCL retinal ganglion cell layer, INL inner nuclear layer, IPE iris pigment epithelium, LE limbal epithelium, ONL outer nuclear layer, SC Schlemm’s canal, ST stroma, TM trabecular meshwork; DAPI nuclear counterstain in blue; scale bars = 100 μm in C,D,F and 20 μm in A,B,E,H-N).
TMEM136, a transmembrane protein of unknown function, was primarily immunolocalized to vascular endothelial cells of blood vessels in eye tissues (Figure 4). Analysis of TMEM136 protein expression by Western blot and tissue from eyes with XFS showed significantly reduced expression levels in iris (−26%) and ciliary body (−32%) (Figure 4G and Supplementary Figure 17) compared to that observed in control eyes, also consistent with the differential mRNA expression analysis shown in Supplementary Figure 15. Similar findings showing reduced TMEM136 protein staining in epithelial and endothelial cells in XFS eyes compared to controls eyes were also observed using immunofluorescence microscopy (Figures 4H, 4J, 4K, and Figures 4L, 4M, and 4N).
Figure 4.
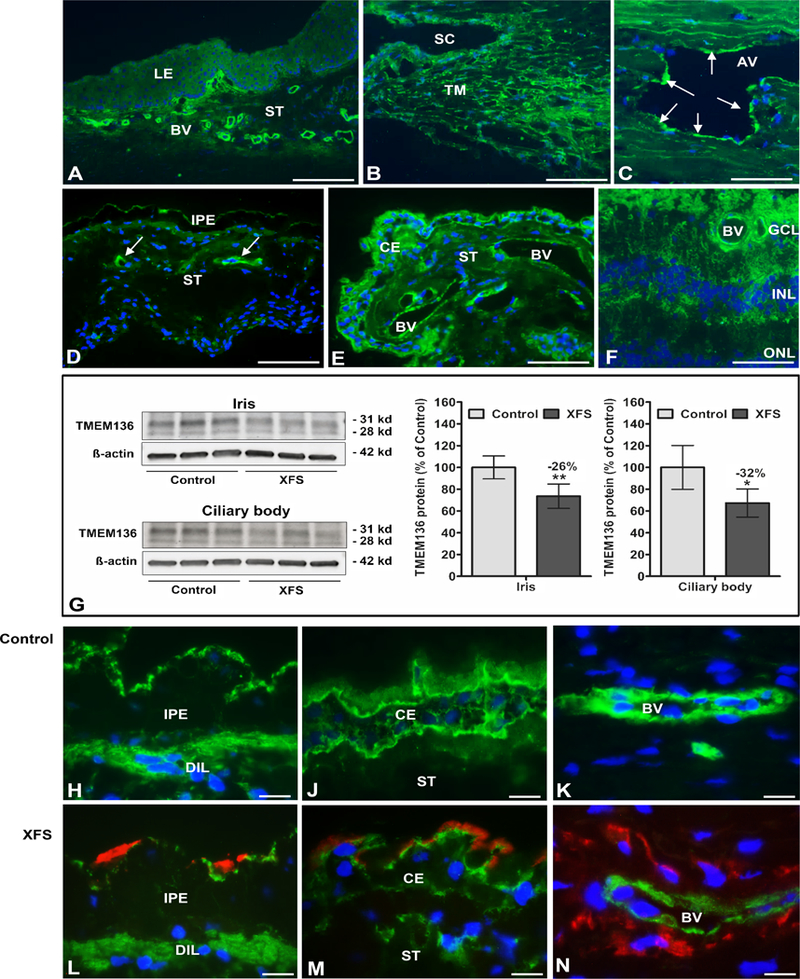
Expression of TMEM136 protein in ocular tissues of normal human donor eyes and donor eyes with XFS, as determined by Western blotting and immunohistochemistry. Immunofluorescence labelling of normal eye tissues shows cytoplasmic TMEM136 immunopositivity (green fluorescence) in limbal blood vessels (A), trabecular meshwork and Schlemm’s canal endothelium (B), walls of aqueous veins (arrows) (C), blood vessels of the iris (arrows) (D), blood vessels and epithelia of the ciliary body (E), and retinal blood vessels and cell layers (F).
Reduced TMEM136 protein expression levels in iris and ciliary body tissues of XFS eyes compared to age matched controls are shown by Western blot analysis (G), and by immunofluorescence labelling of iridal (H,L) and ciliary epithelia (J,M) as well as vascular endothelia in the iris (K,N). Reduced staining intensity in XFS tissues is associated with LOXL1- positive exfoliation material accumulations (red immunofluorescence) on the surface of the iris pigment epithelium (L), ciliary epithelium (M) and iris blood vessel walls (N). Western blot (cropped images) and densitometry analysis shows reduced TMEM136 protein (isoform 1 at 28 KD and isoform 3 at 31 KD) expression in iris and ciliary body tissue lysates of XFS eyes compared to control eyes (G). Data are shown as the TMEM136/B-actin ratio (mean ± standard deviation; n=6 for each group; *P<0.01; **P<0.005); uncropped versions of all Western blots are shown in Supplementary Figure 17. (AV aqueous vein, BV blood vessel, CE ciliary epithelium, DIL dilator muscle, GCL retinal ganglion cell layer, INL inner nuclear layer, IPE iris pigment epithelium, LE limbal epithelium, ONL outer nuclear layer, SC Schlemm’s canal, ST stroma, TM trabecular meshwork; DAPI nuclear counterstain in blue; scale bars = 200 μm in A, 100 μm in B-F and 20 μm in H-N).
We then replicated the immunofluorescence microscopy findings in tissues from a further 3 donor eyes with XFS and 3 matched controls using an independent batch of antibodies, co-staining for LOXL1, POMP, and TMEM136. We confirmed reduced expression of POMP and TMEM136 in important ocular anterior segment structures in XFS eyes compared to controls, on both low magnification (Supplementary Figure 18) and higher magnification (Supplementary Figures 19 and 20).
Discussion
XFS is a disorder of aging involving the ECM, with LOXL1 hypothesized to play a central role in disease pathogenesis. As all common LOXL1 haplotypes detected by the resequencing effort showed reversal of genetic effect, their functional consequences remain in doubt. Even a recent study describing a region in intron 1 of LOXL1 and 5’ upstream of LOXL1-AS1 containing a promoter that influences LOXL1-AS1 expression showed that all strongly associated variants influencing the promoter activity of LOXL1-AS1 also have genome-wide significant reversal of genetic effect21. The common allele reversals at LOXL1 are unlikely to be due to sampling differences within the same population in light of multiple replications confirming the reversal. It is also unlikely to be due to different LD structures across different populations due to the reversal occurring across all phased haplotypes along the broad LOXL1 locus (Supplementary Dataset 1) in the absence of gross differences in LD architecture (Supplementary Figure 21).
Deep resequencing of LOXL1 enabled us to observe that the rare p.Tyr407Phe variant had a protective effect strong enough (ORfor resistance=25) to surpass genome-wide significance on its own. Although strong, the protection is not absolute, as the variant was observed in 2 XFS patients. Both patients also carry a copy of the TMEM136 rs11827818-G risk allele, in keeping with XFS being a complex disease. Our experiments confirm a clear functional and physiological role for the rare p.407Phe allele, but less so for the common p.153Gly>Asp and p.141Leu>Arg polymorphisms.
The protective p.Tyr407Phe substitution is located in the evolutionarily conserved catalytic domain of LOXL159. Follow up biological experiments suggests that the protective effect may be a consequence of stabilization of the ECM due to increased elastin and fibrillin-1 deposition. This notion is supported by histopathological observations demonstrating decreased elastic fiber formation and tissue stiffness as well as impaired cell adhesion in ocular tissues of patients with XFS60,61. We speculate that carrying the rare protective p.407Tyr variant could maintain cellular integrity and render cells more resistant to environmental stressors which destabilize or disrupt the ECM. One limitation of this approach is that the biological mechanisms for p.Tyr407Phe were not tested at the RNA level, as there is a possibility that p.Tyr407Phe could affect RNA stability62–64.
The seven loci emerging from the GWAS study do not implicate a single pathogenesis pathway, supporting the hypothesis that XFS is a complex systemic disease of aging that can arise from genetic lesions in multiple pathways and different tissue types. For example, the marked downregulation of POMP (Figure 3), a ubiquitously expressed proteasome maturation protein in XFS tissues suggests a reduction of ubiquitin conjugating enzymes in XFS tissues65. Abnormalities in the closely related autophagy pathway have also been implicated in XFS66, a process that also involves ubiquitin-proteasome signaling67,68. TMEM136 expression was predominantly localized to vascular endothelia. Interestingly, a pronounced and early vasculopathy, partly involving XFS material deposits around ocular blood vessels (Figure 4) appears to play a significant role in XFS pathophysiology69,70.
In addition to POMP and TMEM136, in this study we also identified 3 other XFS susceptibility loci that map to: AGPAT1 in the class III MHC region (6p21), to RBMS3 (3p24), and near SEMA6A (5q23). AGPAT1 has been identified as a susceptibility locus for Omega6 (n6) polyunsaturated fatty acids (PUFAs) levels in the CHARGE consortium, which may be related to cardiovascular risk in aging populations71. The MHC locus (where AGPAT1 resides) has also been reported to be involved in conditions such as Alzheimer’s and Parkinson’s disease72,73, which, like XFS, are age-related conditions. Although the biological roles of AGPAT1, RBMS3 and SEMA6A are not well understood, the consistent evidence of association at these loci thus opens up further avenues for research into XFS disease biology.
In summary we now show that a rare LOXL1 variant, p.Tyr407Phe, strongly protects against XFS, raising the possibility of potential pharmacological targeting of LOXL1 for therapeutic purposes29,74–76. In addition, we have identified 5 new XFS loci that implicate new biological pathways which could be important for disease pathogenesis.
Online methods
Patient collections
DNA and tissue samples from all patients with XFS and exfoliation glaucoma together with normal controls without XFS were obtained after informed written consent from each participant. All human samples were obtained in strict adherence to the tenets of the Declaration of Helsinki. Details for each XFS case control collection are appended in Supplementary Note.
Genotyping of samples
For the GWAS discovery stage performed in 24 countries (considered across 25 strata due to Russia contributing two distinct collections, one from St Petersburg and one from the Republic of Bashkortostan; Supplementary Table 2), genome-wide genotyping was undertaken using the Illumina OmniExpress Beadchip, as previously described14. To minimize bias between different genotyping arrays and platforms, all 9,035 XFS cases and 17,008 controls for the GWAS discovery stage were genotyped using the Illumina OmniExpress array, ensuring that the primary discovery analysis used only directly genotyped SNP markers uniformly genotyped in cases and controls. The absence of imputation on primary discovery analysis means that issues due to imputation uncertainty and insufficient information content for imputed SNPs with varying minor allele frequencies across the different ethnic groups is minimized77.
The replication stage included XFS collections from 18 countries (Supplementary Table 2). Details on the genotyping and analysis for the replication stage are appended in Supplementary Note.
Statistical analysis for the GWAS discovery stage
Stringent quality control checks were performed for each SNP marker and each individual sample. Our statistical analysis protocol for the GWAS discovery stage only included directly genotyped SNP markers genotyped by the Illumina OmniExpress bead array. We removed poorly performing SNP markers showing genotyping completion rates of <95%, as well as SNPs showing significant deviation from Hardy-Weinberg equilibrium (P < 1 × 10−6 for deviation). SNPs with minor allele frequency of <1% were also removed from further GWAS discovery analysis. Each sample was similarly checked, and those with poor genotyping success rate (defined as genotyping completeness < 95%), showing excess heterozygosity defined as > 3 standard deviations from the mean78,79, and have outlying genetic ancestry (defined by more than six standard deviations from the mean on principal component analysis80) were excluded from further analysis.
We verified the biological relationships of all samples remaining after further exclusion of samples using the principle of variability in allele sharing. We used the PLINK software (See URLs) to derive information for identify-by-state status for each sample pair comparison. For each sample pair showing evidence of cryptic relatedness (IBD > 0.1875)78, the sample with the lower genotyping completeness rate was removed from further analysis.
We performed principal component analysis to assess the degree of genetic stratification and population substructure for all samples which underwent genome-wide genotyping, as previously described80. Principal component analysis was performed for each country / site separately to remove samples with outlying ancestry from further analysis. Principal component scores were than calculated from a pruned set of unlinked markers (defined as pair-wise r2 < 0.1) for each country / site separately. These scores were used as covariates to adjust for residual population stratification. Principal component plots were executed using the R statistical program package (See URLs).
Association between SNP genotypes and XFS disease status was measured using logistic regression for each separate country strata before meta-analysis was conducted, as described elsewhere15,81,82. For the GWAS discovery stage, association analysis was additionally adjusted for the first three principal components of genetic stratification for sample collections to minimize residual population stratification. The genome-wide association summary statistics of all SNP markers from the GWAS are appended as Supplementary Dataset 2, available online. The genomic inflation estimate (λgc) was calculated using directly genotyped SNPs only, using the median regression test statistic, which is distributed in a chi- square manner83. λgc is listed for each individual GWAS discovery strata (and also pictured in Supplementary Figure 22) as well as for the GWAS meta-analysis (Supplementary Table 2). In line with well-described methodologies, results for the seven genome-wide significant loci have underwent double gc correction which corrects for genomic inflation first at the individual population strata and then once again during meta-analysis84,85.
Meta-analysis was performed via the inverse-variance, fixed effects model using genomic control corrected summary results (adjusted odds ratios and standard errors) from each separate country /site. At no point in the analysis were samples pooled within or across continental groups for association analysis, as this measure does not appropriately account for population stratification. The meta-analysis method validates strong reversal at LOXL1 (Supplementary Figure 23).
Statistical analysis for the replication stage.
SNPs showing association with XFS surpassing P≤1 × 10−4 in the GWAS discovery stage were brought forward to the replication stage, and analyzed in a manner similar to that performed for the discovery stage (Supplementary Note).
Genotype imputation
For the five newly identified genome-wide significant loci, we sought to improve on genetic resolution provided by the directly genotyped SNPs currently included on standard content GWAS arrays via imputation fine-mapping using samples and SNP markers passing strict quality control checks. This would also allow for better delineation of the credible sets underlying each of the five newly identified loci48. The imputation and phasing of genotypes were carried out using the IMPUTE2 software (See URLs) with reference panel constructed from cosmopolitan population haplotypes based on data obtained from 2535 individuals from 26 distinct populations around the world. This data is part of the 1000 Genomes project Phase 3 (Jun 2014) release, as described elsewhere. To minimize the effect of imputation uncertainty, we only included imputed genotypes with an information score of ≥0.95. Allele dosages were used for the imputed data association analyses with the software SNPTEST in order to average across imputation uncertainty.
Credible set analysis
Credible sets of SNPs were defined, as previously described, as the minimum number of genetic variants which account for >95% probability of driving each locus-specific association signal48,49. For the construction of credible sets, we included all genotyped variants with genotyping success rates of > 95 percent and minor allele frequency >1 percent. Fine-mapping imputation variants were included using similar thresholds of minor allele frequency > 1 percent and imputation information content >0.95 to reduce the impact of imputation quality on the credible set analysis. For the fine-mapping imputation step, allele dosages were used for the imputed data association analyses with the software SNPTEST in order to average across imputation uncertainty.
Statistical test for interaction with geographical latitude
Statistical tests for interaction between genetic markers and geographical latitude were undertaken for the five newly identified loci showing genome-wide significant association with XFS. The odds ratios and standard error for the odds ratio estimate for each SNP to be tested are assigned a latitude band for the country (or zone, whichever is more precise) where XFS cases and controls were drawn from (see Supplementary Table 2).
The odds ratios and standard error used in this test have already undergone PCA adjustment and genomic control correction during the GWAS analysis. We then conduct a trend test to assess whether the odds ratios for disease increases with increasing 10-degree increments of geographical latitude.
Mouse model phenotypes
For the 33 genes implicated by the 7 genome-wide significant SNPs on 7 distinct loci (Supplementary Table 15), we looked up the Mouse Genome Informatics publicly available database86. The output is manually checked and curated, with the relevant references detailing the mouse models appended as footnotes in Supplementary Table 15.
Deep sequencing of LOXL1 and CACNA1A
Deep sequencing was performed on a total of 5,570 XFS cases and 6,279 controls from 9 countries (Supplementary Table 1). Both the LOXL1 and CACNA1A genetic loci (exons, introns, 5’ and 3’ flanking regions) spanning coordinates chr15:74,200,000 to 74,260,000 and Chr19:13,307,000 to 13,745,000 were captured using the Roche Nimblegen SeqCap Easy probe kit. Enrichment and amplification of the libraries were then created using well-described, routine laboratory techniques36. Sequencing was performed using 2×101 paired end reads using the Illumina Hi-Seq 2500 platform. We required that >95% of the samples to be covered at least 10X87. Mean coverage for sequencing across the samples was observed to be 60X.
For the analysis of LOXL1 p.Y407F, the initial 2,827 cases and 3,013 controls from Japan which underwent re-sequencing were enrolled from December 2007 to January 2015. A further 1,082 exfoliation syndrome cases and 2,325 controls from Japan were enrolled for the replication stage. These samples were collected between February 2015 and December 2016 and did not undergo deep sequencing of the entire LOXL1 locus.
Read-mapping, variant detection and annotation.
All sequence reads in each individual were aligned to the human reference genome (hg19) using the Burrows-Wheeler Aligner software, which is well described36. Consensus genotypes were called using the GATK best practices guidelines. Only high quality variants assigned ‘PASS’ scores by variant quality score recalibration and individuals with variant genotype calling completeness >95% were brought forward for further statistical analysis.
Power calculations for genetic association study
Power calculations for the GWAS discovery and replication stages were performed on an additive genetic model using well described methods (See URLs), and is shown in Supplementary Table 10. These power calculations take into account the asymmetric number of cases and controls, as is well reported in genetic association studies82,88.
Haplotype phasing for the LOXL1 locus
To ensure accurate phasing in order to reliably capture both common and rare haplotypes, we only included individuals with a genotype call completeness rate of 100% from both the sequencing and GWAS experiments. LOXL1 haplotypes were phased using the BEAGLE89 and PLINK90 software packages, as previously described35.
Tissue specimens for analysis of the GWAS loci.
Human donor eyes used for corneal transplantation with appropriate research consent were obtained and processed within 15 hours after death. For RNA and DNA extractions, 21 donor eyes with XFS (mean age, 80.1 ± 7.9 years; 11 female, 10 male) and 41 normal, healthy, age-matched control eyes (mean age, 77.1 ± 8.1 years; 20 female, 21 male) without any known ocular disease were used. Ocular tissues were prepared under a dissecting microscope and were snap frozen in liquid nitrogen. The presence of characteristic exfoliation material deposits was assessed by macroscopic inspection of anterior segment structures and confirmed by electron microscopic analysis of small tissue sectors.
For immunostaining experiments, ocular tissue samples obtained from 10 donor eyes with XFS (mean age, 78.7 ± 9.7 years; 6 female, 4 male) and 10 normal human donor eyes (mean age, 72.3 ± 11.6 years; 5 female, 5 male) were embedded in optimal cutting temperature compound and frozen in liquid nitrogen. DNA samples obtained from ocular tissues and cells were genotyped by Sanger sequencing (Supplementary Note).
Real-time PCR of human eye tissues
Ocular tissues were extracted using the Precellys 24 homogenizer and lysing kit together with the AllPrep DNA/RNA kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. This includes an on-column DNase I digestion step. First-strand cDNA synthesis from 0.5 μg of total RNA was performed with Superscript II reverse transcriptase (Invitrogen, Karlsruhe, Germany) in a 20 μl reaction volume. Quantitative real-time PCR was performed using the CFX Connect thermal cycler and software (Bio-Rad Laboratories, München, Germany). PCR reactions (25 μl) were run in duplicate and contained 2 μl of first-strand cDNA, 0.4 μM each of upstream- and downstream-primer, and SsoFast EvaGreen Supermix (Bio-Rad). Exon-spanning primers (Eurofins Genomics, Ebersberg, Germany), designed using Primer 3 software (See URLs), and PCR conditions are appended in Supplementary Table 16. For normalization of gene expression levels, mRNA ratios relative to the house-keeping gene GAPDH were calculated by the comparative CT method (2 −ΔCT). Amplification specificity was checked using melt curve and sequence analyses using the Prism 3100 DNA-sequencer (Applied Biosystems, Foster City, CA).
Western blot analysis of human eye tissues
Total protein was extracted from iris and ciliary body tissues of 6 eyes with XFS and 6 normal eyes using RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% DOC, 0.1% SDS). Protein concentrations were determined by the Micro-BCA protein assay kit (Thermo Scientific). Proteins (10 μg per lane) were separated by 4–15% SDS-polyacrylamide gel electrophoresis under reducing conditions (6% DTT) and transferred onto nitrocellulose membranes with the Trans-Blot Turbo transfer system (Bio-Rad). Membranes were blocked with SuperBlock T20 (Thermo Scientific) for 30 minutes and incubated for 1h at room temperature or overnight at 4°C with antibodies against POMP (Abcam) and TMEM136 (Abcam) diluted in PBST/10% SuperBlock T20. Equal loading was verified with mouse anti-human p- actin antibody (clone AC-15; Sigma-Aldrich) in PBST/10% SuperBlock T20. In negative control experiments, the primary antibody was replaced by PBST. Immunodetection was performed with a horseradish peroxidase-conjugated secondary antibody in PBST/10% SuperBlock T20 and the Super Signal West Femto ECL kit (Thermo Scientific), and band intensity was analysed by computerized densitometry.
Immunohistochemistry of human eye tissues follow routine laboratory procedures, and are appended in Supplementary Note.
LOXL1 constructs
The full-length cDNA encoding LOXL1 was amplified using the primers shown in Supplementary Table 16. The restriction enzymes sites, EcoRI and Sa/I were added in a second amplification using a second set of primers (Supplementary Table 17). The LOXL1 fragment (~1.7kb) was then subcloned into a HA- tagged pcipuro vector. Four haplotypes LOXL1-Arg141-Gly153-Tyr407 (G-G-A), LOXL1-Leu141-Gly153- Tyr407 (T-G-A), LOXL1-Arg141-Asp153-Tyr407 (G-A-A), and LOXL1-Arg141-Asp153-Phe407 (G-A-T) were generated and contained genetic variants in the following order; rs1048661 (G>T)-rs3825942 (G>A)-rs201011613 (A>T). The first haplotype generated was LOXL1-Arg141-Asp153-Tyr407 (G-A-A) and it served as the template plasmid for subsequent site-directed mutagenesis.
Targeted base-substitution was generated by site-directed mutagenesis using a PCR-based strategy with Transformer™ site-directed mutagenesis kit (Clontech) with respective oligonucleotide primer pairs (Supplementary Table 17) onto the LOXL1-Arg141-Asp153-Tyr407 (G-A-A) haplotype. All accuracy of haplotypes constructed were confirmed by Sanger sequencing of the full length clone. All oligonucleotide primer pairs used to create the LOXL1 constructs are appended in Supplementary Table 17.
Nano luciferase secretion assay for LOXL1
The secretion assay for LOXL1 with respect to the four tested haplotypes LOXL1-Arg141-Gly153-Tyr407 (G-G-A), LOXL1-Leu141-Gly153-Tyr407 (T-G-A), LOXL1-Arg141-Asp153-Tyr407 (G-A-A), and LOXL1- Arg141-Asp153-Phe407 (G-A-T) were measured using the NanoLuc luciferase assay. More details for this assay are appended in Supplementary Note.
Details for Western blot analysis of the different hemagglutinin-tagged LOXL1 haplotypes on elastin, fibronectin, and collagen IV follow routine laboratory procedures, and are appended in Supplementary Note.
Spheroid cultures
The human lens epithelial cell line (HLEC (B-3)) was obtained from the American Tissue Culture Collection (ATCC). The cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 20% fetal bovine serum (Sigma-Aldrich) and 2mM Glutamax (Invitrogen) at 37oC with 5% CO2, and passaged every 2–3 days in a 1:4 ratio. The cell line was tested for mycoplasma and was found to be negative.
Nucleofected HLECs were trypsinized 48 hours post-nucleofection and seeded in their growth media in low attachment 6-well plates with hydrophobic surfaces (Greiner Bio-One GmbH) at 300,000 cells per well. Cells were left to form spheroids and collected at 72 hours. The spheroids were fixed with 4% paraformaldehyde for 10min at room temperature and stored at 4oC. Fixed spheroids were washed with PBST (0.1% Tween-20 in 1× PBS) and blocked in blocking buffer (3% BSA in PBS) for 1 hour at room temperature. They were subsequently incubated overnight at 4oC with the following primary antibodies: mouse or rabbit antibody to HA (1:100 dilution; sc-7392; sc-805; Santa Cruz), goat antibody to elastin (1:100 dilution; sc-17581; Santa Cruz), mouse antibody to fibrillin-1 (1:100 dilution; ab6328; Abcam), and rabbit antibody to collagen IV (1:100 dilution; ab6586; Abcam). All antibodies were diluted with the blocking buffer. After incubation with the primary antibody, the spheroids were washed three times with PBST and labeled with their respective secondary antibodies for 1 hour at room temperature. Secondary antibodies used were with FITC, Cy3 or AlexaFluor 647-conjugated anti-mouse, anti-rabbit, or anti-goat secondary antibody (1:300 dilution; Jackson Laboratories). The spheroids were stained with DAPI (1ug/mL) and mounted on microscope glass slides using a cytocentrifuge (Thermo Fisher Scientific Inc.) and FluorSave Reagent (Merck Millipore). Immunolabelled spheroids images were acquired at the Advanced Bioimaging Core at the Academia, Singapore Health Services with a Leica TCS SP8 confocal laser scanning platform, Z-planes were imaged in 1μm steps. Analysis of spheroids was done on a maximal projection image of 5 Z-planes onto a single image. Heat map of respective immunofluorescent signals were generated relative to the minimum and maximum fluorescence intensity values of the same scale defined by the color range as indicated within the figures. Each experiment was repeated independently three times, with images also acquired independently three times.
Cell-cell adhesion assay
HLECs nucleofected with LOXL1-Arg141-Gly153-Tyr407 (G-G-A), LOXL1-Leu141-Gly153-Tyr407 (T-G- A), LOXL1-Arg141-Asp153-Tyr407 (G-A-A), and LOXL1-Arg141-Asp153-Phe407 (G-A-T) haplotype constructs were plated into 96-well plates designed for the xCELLigence RTCA SP instrument (ACEA Biosciences Inc) at a density of 120,000 cells per well. The 96-well plates were incubated at 37°C with 5% CO2 and monitored on the xCELLigence RTCA SP system at 15-min intervals for the first 24 h and at 30-min intervals for the subsequent 48 h. The impedance data was extracted from the RTCA software and analyzed for differences in cell-cell adhesion between the different LOXL1 haplotypes. Triplicates were performed for HLECs nucleofected with each haplotype to allow for robust statistical evaluation of the results obtained. The readings for each variant were normalized against their respective initial readings at the first time point, and the normalized readings were subsequently compared against the LOXL1-Arg141-Asp153-Phe407 (G-A-T) haplotype. The assay shown in Figure 1e was repeated four independent times, with the follow up independent experiment (shown in Supplementary Figure 7) repeated seven independent times.
Statistical procedures for biological analysis.
Statistical evaluation of expression differences between patients and controls was performed using SPSS v.20 software (IBM, Ehningen, Germany) with an unpaired two-tailed t test. P < 0.05 was considered statistically significant. Data for adhesion and secretion assays were analyzed by an unpaired homoscedastic t test, and two-tailed P-values were calculated. We considered P < 0.05 as statistically significant.
Supplementary Material
Acknowledgments
This research is supported by the Biomedical Research Council, Agency for Science, Technology, and Research, Singapore (to C.C.K.), by the Glaucoma Foundation of New York (to C.C.K.), by grants from the National Medical Research Council, Singapore (NMRC/TCR/002-SERI/2008 to T.A. and NMRC/CBRG/0032/2013 to E.N.V.), by the Interdisziplinäres Zentrum für Klinische Forschung (IZKF-E23) from Germany to F.P. and to U.S-S, and by grants from the National Institutes of Health, USA: UM1 CA186107, R01 CA49449, R01 EY015473 to J.L.W. For XFS cases in the USA GWAS dataset, genotyping services were provided through a grant to J.L.W. (HG008597) by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number HHSN268201200008I. We dedicate this article to the ophthalmologist Eva Forsman from Finland, who passed away after diagnosing all of the exfoliation syndrome cases from Finland.
Footnotes
URLs
PLINK software, http://pngu.mgh.harvard.edu/~purcell/plink/; R statistical program package, https://www.r-project.org/; IMPUTE2 software, https://mathgen.stats.ox.ac.uk/impute/impute_v2.html; Power calculator for genetic association studies, http://pngu.mgh.harvard.edu/~purcell/gpc/; Primer3 software, http://primer3.ut.ee.
Data Availability Statement
The primary dataset comprising genome-wide association summary statistics of all SNP markers reflected in the Figure 2 Manhattan plot are appended as Supplementary Dataset 2, available online. Secondary analysis datasets are appended as Supplementary Dataset 1 (LOXL1 phased haplotype analysis) and Supplementary Dataset 3 (INRICH analysis).
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Vesti E & Kivela T Exfoliation syndrome and exfoliation glaucoma. Prog Retin Eye Res 19, 345–68 (2000). [DOI] [PubMed] [Google Scholar]
- 2.Ritch R & Schlotzer-Schrehardt U Exfoliation syndrome. Surv Ophthalmol 45, 265–315 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Leske MC et al. Factors for glaucoma progression and the effect of treatment: the early manifest glaucoma trial. Arch Ophthalmol 121, 48–56 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Ritch R, Schlotzer-Schrehardt U & Konstas AG Why is glaucoma associated with exfoliation syndrome? Prog Retin Eye Res 22, 253–75 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Olawoye OO et al. Exfoliation syndrome in Nigeria. Middle East Afr J Ophthalmol 19, 402–5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kivela T Ocular Pseudoexfoliation Syndrome and Life Span: Act 2. EBioMedicine 2, 640–1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ritch R Ocular and systemic manifestations of exfoliation syndrome. J Glaucoma 23, S1–8 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Ritch R Exfoliation syndrome-the most common identifiable cause of open-angle glaucoma. J Glaucoma 3, 176–7 (1994). [PubMed] [Google Scholar]
- 9.Allingham RR et al. Pseudoexfoliation syndrome in Icelandic families. Br J Ophthalmol 85, 702–7 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orr AC et al. Exfoliation syndrome: clinical and genetic features. Ophthalmic Genet 22, 171–85 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Thorleifsson G et al. Common sequence variants in the LOXL1 gene confer susceptibility to exfoliation glaucoma. Science 317, 1397–400 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Williams SE et al. Major LOXL1 risk allele is reversed in exfoliation glaucoma in a black South African population. Mol Vis 16, 705–12 (2010). [PMC free article] [PubMed] [Google Scholar]
- 13.Wiggs JL & Pasquale LR Expression and regulation of LOXL1 and elastin-related genes in eyes with exfoliation syndrome. J Glaucoma 23, S62–3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aung T et al. A common variant mapping to CACNA1A is associated with susceptibility to exfoliation syndrome. Nat Genet 47, 387–92 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Al Olama AA et al. A meta-analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat Genet 46, 1103–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu JZ et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 47, 979–86 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paternoster L et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat Genet 47, 1449–56 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ehret GB et al. The genetics of blood pressure regulation and its target organs from association studies in 342,415 individuals. Nat Genet (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu C et al. Meta-analysis identifies common and rare variants influencing blood pressure and overlapping with metabolic trait loci. Nat Genet (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Surendran P et al. Trans-ancestry meta-analyses identify rare and common variants associated with blood pressure and hypertension. Nat Genet (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hauser MA et al. Genetic variants and cellular stressors associated with exfoliation syndrome modulate promoter activity of a lncRNA within the LOXL1 locus. Hum Mol Genet 24, 6552–63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H et al. Ethnicity-based subgroup meta-analysis of the association of LOXL1 polymorphisms with glaucoma. Mol Vis 16, 167–77 (2010). [PMC free article] [PubMed] [Google Scholar]
- 23.Fan BJ et al. LOXL1 promoter haplotypes are associated with exfoliation syndrome in a U.S. Caucasian population. Invest Ophthalmol Vis Sci 52, 2372–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ozaki M et al. Association of LOXL1 gene polymorphisms with pseudoexfoliation in the Japanese. Invest Ophthalmol Vis Sci 49, 3976–80 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Tanito M et al. LOXL1 variants in elderly Japanese patients with exfoliation syndrome/glaucoma, primary open-angle glaucoma, normal tension glaucoma, and cataract. Mol Vis 14, 1898–905 (2008). [PMC free article] [PubMed] [Google Scholar]
- 26.Pasutto F et al. Association of LOXL1 common sequence variants in German and Italian patients with pseudoexfoliation syndrome and pseudoexfoliation glaucoma. Invest Ophthalmol Vis Sci 49, 1459–63 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Mori K et al. LOXL1 genetic polymorphisms are associated with exfoliation glaucoma in the Japanese population. Mol Vis 14, 1037–40 (2008). [PMC free article] [PubMed] [Google Scholar]
- 28.Nakano M et al. Novel common variants and susceptible haplotype for exfoliation glaucoma specific to Asian population. Sci Rep 4, 5340 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flannick J et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet 46, 357–63 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonnefond A et al. Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat Genet 44, 297–301 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Majithia AR et al. Rare variants in PPARG with decreased activity in adipocyte differentiation are associated with increased risk of type 2 diabetes. Proc Natl Acad Sci U S A 111, 13127–32 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Majithia AR et al. Prospective functional classification of all possible missense variants in PPARG. Nat Genet (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Do R et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature 518, 102–6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo Y et al. Exploring the genetic architecture of inflammatory bowel disease by whole-genome sequencing identifies association at ADCY7. Nat Genet (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raychaudhuri S et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat Genet 43, 1232–6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seddon JM et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age- related macular degeneration. Nat Genet 45, 1366–70 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fritsche LG et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet 48, 134–43 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schlotzer-Schrehardt U, von der Mark K, Sakai LY & Naumann GO Increased extracellular deposition of fibrillin-containing fibrils in pseudoexfoliation syndrome. Invest Ophthalmol Vis Sci 38, 970–84 (1997). [PubMed] [Google Scholar]
- 39.Schlotzer-Schrehardt U, Zenkel M, Kuchle M, Sakai LY & Naumann GO Role of transforming growth factor-beta1 and its latent form binding protein in pseudoexfoliation syndrome. Exp Eye Res 73, 765–80 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Schlotzer-Schrehardt U Molecular pathology of pseudoexfoliation syndrome/glaucoma--new insights from LOXL1 gene associations. Exp Eye Res 88, 776–85 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Lucero HA & Kagan HM Lysyl oxidase: an oxidative enzyme and effector of cell function. Cell Mol Life Sci 63, 2304–16 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu X et al. Elastic fiber homeostasis requires lysyl oxidase-like 1 protein. Nat Genet 36, 178–82 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Atienza JM et al. Dynamic and label-free cell-based assays using the real-time cell electronic sensing system. Assay Drug Dev Technol 4, 597–607 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Solly K, Wang X, Xu X, Strulovici B & Zheng W Application of real-time cell electronic sensing (RT-CES) technology to cell-based assays. Assay Drug Dev Technol 2, 363–72 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Urcan E et al. Real-time xCELLigence impedance analysis of the cytotoxicity of dental composite components on human gingival fibroblasts. Dent Mater 26, 51–8 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Okbay A et al. Genome-wide association study identifies 74 loci associated with educational attainment. Nature 533, 539–42 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gaulton KJ et al. Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat Genet 47, 1415–25 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wellcome Trust Case Control, C. et al. Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat Genet 44, 1294–301 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y et al. Serial analysis of gene expression (SAGE) in normal human trabecular meshwork.Mol Vis 17, 885–93 (2011). [PMC free article] [PubMed] [Google Scholar]
- 51.Wagner AH et al. Exon-level expression profiling of ocular tissues. Exp Eye Res 111, 105–11(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wistow G et al. NEIBank: genomics and bioinformatics resources for vision research. Mol Vis 14, 1327–37 (2008). [PMC free article] [PubMed] [Google Scholar]
- 53.Bowes Rickman C et al. Defining the human macula transcriptome and candidate retinal disease genes using EyeSAGE. Invest Ophthalmol Vis Sci 47, 2305–16 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Springelkamp H et al. ARHGEF12 influences the risk of glaucoma by increasing intraocular pressure. Hum Mol Genet 24, 2689–99 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Ward LD & Kellis M HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res 40, D930–4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Westra HJ et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet 45, 1238–43 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Orchard S et al. The MIntAct project--IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res 42, D358–63 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee PH, O’Dushlaine C, Thomas B & Purcell SM INRICH: interval-based enrichment analysis for genome-wide association studies. Bioinformatics 28, 1797–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barker HE, Cox TR & Erler JT The rationale for targeting the LOX family in cancer. Nat Rev Cancer 12, 540–52 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Braunsmann C et al. Evaluation of lamina cribrosa and peripapillary sclera stiffness in pseudoexfoliation and normal eyes by atomic force microscopy. Invest Ophthalmol Vis Sci 53, 2960–7 (2012). [DOI] [PubMed] [Google Scholar]
- 61.Schlotzer-Schrehardt UM, Dorfler S & Naumann GO Corneal endothelial involvement in pseudoexfoliation syndrome. Arch Ophthalmol 111, 666–74 (1993). [DOI] [PubMed] [Google Scholar]
- 62.Sauna ZE & Kimchi-Sarfaty C Understanding the contribution of synonymous mutations to human disease. Nat Rev Genet 12, 683–91 (2011). [DOI] [PubMed] [Google Scholar]
- 63.Shabalina SA, Spiridonov NA & Kashina A Sounds of silence: synonymous nucleotides as a key to biological regulation and complexity. Nucleic Acids Res 41, 2073–94 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hunt RC, Simhadri VL, Iandoli M, Sauna ZE & Kimchi-Sarfaty C Exposing synonymous mutations. Trends Genet 30, 308–21 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Zenkel M, Kruse FE, Naumann GO & Schlotzer-Schrehardt U Impaired cytoprotective mechanisms in eyes with pseudoexfoliation syndrome/glaucoma. Invest Ophthalmol Vis Sci 48, 5558–66 (2007). [DOI] [PubMed] [Google Scholar]
- 66.Want A et al. Autophagy and Mitochondrial Dysfunction in Tenon Fibroblasts from Exfoliation Glaucoma Patients. PLoS One 11, e0157404 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Korolchuk VI, Menzies FM & Rubinsztein DC Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 584, 1393–8 (2010). [DOI] [PubMed] [Google Scholar]
- 68.Kraft C, Peter M & Hofmann K Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 12, 836–41 (2010). [DOI] [PubMed] [Google Scholar]
- 69.Schlotzer-Schrehardt U & Naumann GO Ocular and systemic pseudoexfoliation syndrome.Am J Ophthalmol 141, 921–937 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Kivela T, Hietanen J & Uusitalo M Autopsy analysis of clinically unilateral exfoliation syndrome. Invest Ophthalmol Vis Sci 38, 2008–15 (1997). [PubMed] [Google Scholar]
- 71.Guan W et al. Genome-wide association study of plasma N6 polyunsaturated fatty acids within the cohorts for heart and aging research in genomic epidemiology consortium. Circ Cardiovasc Genet 7, 321–31 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lambert JC et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45, 1452–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nalls MA et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 46, 989–93 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cohen JC, Boerwinkle E, Mosley TH Jr. & Hobbs HH Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 354, 1264–72 (2006). [DOI] [PubMed] [Google Scholar]
- 75.Jonsson T et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488, 96–9 (2012). [DOI] [PubMed] [Google Scholar]
- 76.Nioi P et al. Variant ASGR1 Associated with a Reduced Risk of Coronary Artery Disease. N Engl J Med 374, 2131–41 (2016). [DOI] [PubMed] [Google Scholar]
- 77.Consortium UIG et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet 41, 1330–4 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Anderson CA et al. Data quality control in genetic case-control association studies. Nat Protoc 5, 1564–73 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mells GF et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet 43, 329–32 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Price AL et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38, 904–9 (2006). [DOI] [PubMed] [Google Scholar]
- 81.Verhoeven VJ et al. Genome-wide meta-analyses of multiancestry cohorts identify multiple new susceptibility loci for refractive error and myopia. Nat Genet 45, 314–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kiryluk K et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet 46, 1187–96 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bronson PG et al. Common variants at PVT1, ATG13-AMBRA1, AHI1 and CLEC16A are associated with selective IgA deficiency. Nat Genet 48, 1425–1429 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Okada Y et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–81 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kooner JS et al. Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat Genet 43, 984–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Blake JA et al. The Mouse Genome Database: integration of and access to knowledge about the laboratory mouse. Nucleic Acids Res 42, D810–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Foo JN et al. Analysis of non-synonymous-coding variants of Parkinson’s disease-related pathogenic and susceptibility genes in East Asian populations. Hum Mol Genet 23, 3891–7(2014). [DOI] [PubMed] [Google Scholar]
- 88.Cheng TH et al. Five endometrial cancer risk loci identified through genome-wide association analysis. Nat Genet 48, 667–74 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Browning BL & Browning SR A unified approach to genotype imputation and haplotype- phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet 84, 210–23 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Purcell S et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–75 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.