

ABSTRACT
Immune check point inhibitors targeting programmed cell death protein-1 (PD-1) and its ligand (PD-L1) have shown clinical success in treatment of human malignancies. Triple negative breast cancer (TNBC), which is primarily characterized by high heterogeneity and presence of tumor infiltrating lymphocytes, remains therapeutic challenge due to unavailability of approved targeted therapy. Therapeutic potential of immune check point inhibitors for TNBC patients is under active clinical investigation. In this study, we show that FDA-approved anti-PD-L1 antibody, atezolizumab (ATE), potentiates T cell-mediated cytotoxicity and apoptosis of TNBC cells that express higher levels of PD-L1, but does not have significant effect on TNBC cells expressing low levels of PD-L1. PD-L1 knockdown further confirmed that ability of ATE to promote T cell-induced cytotoxicity is PD-L1 expression dependent. Combination of ATE with PD-L1 upregulating agents, such as HDAC, proteasomal, and lysosomal inhibitors, further augmented cytotoxic activity of T cells toward TNBC cells. Based on analysis of breast cancer tissue samples deposited in The Cancer Genome Atlas (TCGA), we found a positive correlation between PD-L1 and focal adhesion kinase (FAK) mRNA expression in PD-L1-positive (PD-L1+) TNBC, suggesting a functional association of FAK and immune checkpoints. We further demonstrate that ATE dramatically downregulates phosphorylation status of FAK, an important regulator of cell invasion and migration, and significantly enhances FAK inhibitor mediated inhibition of cell motility and invasion of PD-L1+ TNBC cells independent of T cells. Taken together, our data suggest that ATE shows promising anti-tumor activity in PD-L1+ TNBC via both T cell-dependent and -independent mechanisms.
KEYWORDS: atezolizumab, immune checkpoint inhibitors, triple negative breast cancer (TRBC), PD-L1, focal adhesion kinase (FAK)
Introduction
Triple negative breast cancer (TNBC), which represents about 10–20% of all mammary tumors, is characterized by lack of molecular therapeutic targets like estrogen receptor (ER), progesterone receptor (PR) and HER2.1,2 TNBCs are highly aggressive tumors with dismal prognosis. Because these tumors lack targetable receptors, they do not respond to hormonal or targeted therapies, hence chemotherapy remains standard of care for such patients.3 Despite aggressive treatment with adjuvant and neoadjuvant chemotherapies, the mortality rate for advanced stage TNBC patients remains very high; therefore, there is an urgent need to develop effective targeted therapies for TNBC patient population. TNBC tumor has abundant presence of tumor infiltrating lymphocytes (TILs; T-lymphocytes, B-lymphocytes, and macrophages), which are used for prognostic purpose in clinical settings.4 Additionally, infiltration of CD8+ intra-tumoral lymphocytes was considered as independent favorable prognostic factor for a subpopulation of TNBC patients.5 Moreover, mutational and neoantigen load, which strongly correlates with infiltration of cancer specific immune cells in tumor microenvironment, is relatively higher in TNBC compare to other histological subtypes of breast cancers.6 Considering the strong immunogenic features of TNBC, immune-checkpoint-based cancer immunotherapies are extensively investigated in clinical settings for such tumors.
Immune checkpoints are a set of inhibitory pathways in the immune system that are required for maintaining self-tolerance and preventing host tissue damage by regulating durability of immune response.7 Programmed cell death ligand 1 (PD-L1), a key immune checkpoint molecule and member of B7 transmembrane protein family, interacts with its receptor, programmed cell death (PD-1), and induces negative regulatory signal by neutralizing cytotoxic T cell activity.8 Early preclinical studies have shown that disrupting PD-L1/PD-1 association by using monoclonal antibodies may enhance anti-tumor immunity and promote tumor regression in in vitro and in vivo models.9,10 Data from recent clinical studies have successfully demonstrated that blockade of PD-1/PD-L1 axis can produce overall survival benefit in patients with solid tumors leading to FDA approval of several check point inhibitors for variety of cancers.11 Cell-surface expression of PD-L1 in variety of solid cancers primarily serve as resistance mechanism, which allows tumors to escape from host immune response.12 Although impact of PD-L1 expression on tumor and immune cells remains unclear, both tumor and host immune cells PD-L1 expression could predict the therapeutic response to agents blocking PD-1/PD-L1 axis.13 Analysis of The Cancer Genome Atlas (TCGA) RNA sequencing data and breast tumor tissue microarrays showed significant higher PD-L1 expression in TNBC patient subgroup than that in non-TNBC population.14 Another study, which evaluated PD-L1 expression in breast cancer patient biopsies, reported that PD-L1 expression was observed in 30% of patients with hormone receptor–negative and triple-negative status, and strong correlation was observed in PD-L1 and TILs.15 These immunogenic features of TNBC tumors strongly advocate that immune checkpoint inhibitors could be viable therapeutic agents for these patient population.
Several anti-PD-L1 (atezolizumab (ATE), avelumab, and durvalumab) have been approved by FDA for treatment of solid malignancies. ATE, which selectively targets PD-L1 and inhibit binding of PD-L1 to receptor PD-1, showed improved clinical utility against urothelial and non-small cell lung carcinomas, and later received market approval for such patient populations.16,17 ATE, formerly known as MPDL3280A, was isolated from a single phage clone by screening human phage display library directed against extracellular domain-Fc fusion of human PD-L1.18 Although clinical activity of ATE is explored in variety of cancer types, more recently, a phase 3 clinical trial using ATE with nab-paclitaxel in patients with locally advanced or metastatic TNBC patients showed significantly longer progression-free survival compared with placebo-nab-paclitaxel treated group.19 Earlier, a phase 1b clinical trial evaluating the clinical activity of ATE in metastatic TNBC patients reported that ATE monotherapy can provide durable clinical benefit in those patients.20 Combining immune checkpoint inhibitors with chemotherapeutic agents can expand the clinical benefit of immune therapies to a larger patient population by multiple mechanisms including activation of immune effector cells, depletion of immune suppressive cells, and generation of tumor-associated antigens.21 Currently, numerous clinical trials are ongoing to study the therapeutic efficacy of ATE alone and in combinations in breast cancer subtypes including TNBC.
In this study, we subcategorized TNBC cells based on cell surface expression of PD-L1 and explored the efficacy of ATE in potentiating Tcell-mediated cytotoxicity of TNBC cells. Extending our investigation to novel combination approaches, we discovered that combination of ATE and agents that can increase PD-L1 expression in TNBC cells can further enhance T cell-dependent cytotoxicity. To further explore combination therapy to enhance the therapeutic efficacy of PD-L1 by analyzing TCGA, we found a positive correlation of PD-L1 and FAK mRNA expressions in TNBC patients and demonstrated that ATE inhibited FAK phosphorylation in TNBC cells without involvement of T cells. Our data suggest that ATE has a bimodal function: T cell-mediated cell cytotoxicity and non-T cell-mediated anti-cancer properties via FAK-mediated signaling.
Results
PD-L1 is expressed in TNBC cells
PD-L1 expression was analyzed in a broad panel of five different TNBC cells using Western blotting. Among the five TNBC cells tested, MDA-MB-231 cells showed highest expression of PD-L1 while BT-20 cells contain relatively low PD-L1 compared with MDA-MB-231 cells (Figure 1(a)). MDA-MB-468 cells did not show any PD-L1 expression in our Western blotting experiment (Figure 1(a)). Flow cytometry experiment was performed to assess cell surface content of PD-L1 in TNBC cells (Figure 1(b)). Mean fluorescent intensity (MFI) data from flow cytometry reveal that MDA-MB-231 and BT-20 cells possess significantly higher cell surface PD-L1 expression compared to isotype control. Consistent with Western blot analysis, MDA-MB-468 cells appear to have very low PD-L1 expression in flow cytometry analysis. To further extend our findings from Western blotting and flow cytometry, immunofluorescence experiments were performed to monitor subcellular distribution of PD-L1 expression, which revealed that PD-L1 was localized on membrane lamellipodia, as well as intracellular component in both MDA-MB-231 and BT-20 cells (Figure 1(c)), but not in MDA-MB-468 cells (Figure 1(c)). Based on our PD-L1 expression analysis, we conclude that two TNBC cell lines, MDA-MB-231 and BT-20 are PD-L1+ cells, and one TNBC cell line, MDA-MB-468, is PD-L1− cells.
Figure 1.
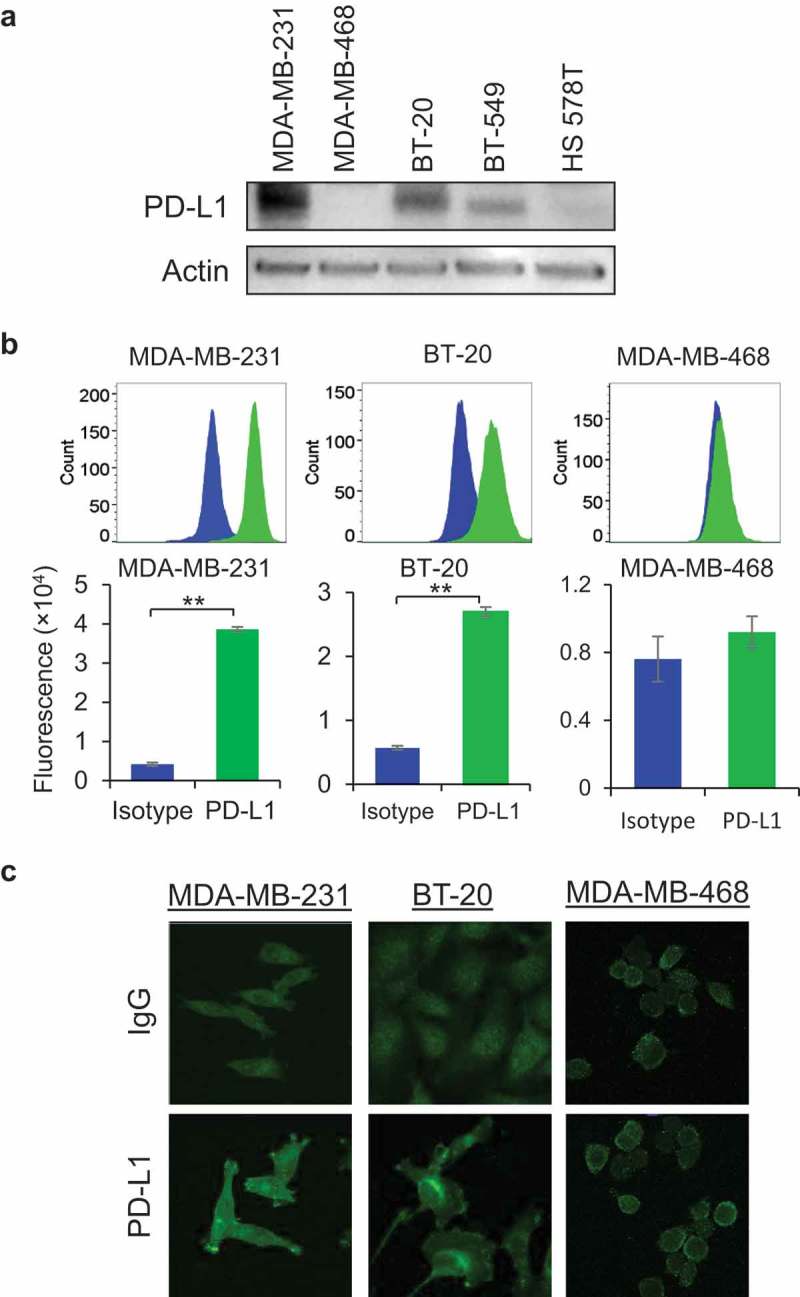
Expression of PD-L1 in TNBC cells. (a) Protein expression of PD-L1 was determined by Western blotting analysis in five different TNBC cells namely MDA-MB-231, MDA-MB-468, BT-20, BT-549, and HS578T cells. Equal loading of samples was confirmed by actin expression. (b) Cell surface expression of PD-L1 in MDA-MB-231, BT-20, and MDA-MB-468 cells was determined by flow cytometry after staining cells with anti-PD-L1 antibody (Green color) and control cells were stained with isotype IgG. Fluorescent intensity data was presented as flow cytometric histograms (upper panel) and quantitative data was represented as bar diagrams (lower panel). (c) Immunofluorescence staining of PD-L1 was performed in MDA-MB-231, BT-20, and MDA-MB-468 cells and fluorescent images were captured by LSM 880 confocal microscope. Rabbit IgG was used as negative control.
ATE significantly potentiates T cell-mediated cytotoxicity of PD-L1+ TNBC cells
ATE elicits its anti-tumor immune activity by disrupting the interaction of PD-1 on cytotoxic T cells with PD-L1 on tumor cells. We then investigated anti-tumor immune activity of ATE in presence of T cells to inhibit TNBC cell growth. For this purpose, MDA-MB-231, BT-20, and MDA-MB-468 cells were co-cultured with pre-anti-CD3/CD28-activated primary human pan T cells in presence or absence of ATE, and in vitro T cell-mediated tumor cell cytotoxicity assay was performed. Counting of viable tumor cells labeled with calcein AM dye was done using Celigo Imaging System. As shown in Figure 2(a), ATE significantly potentiated Tcell-mediated cytotoxicity of MDA-MB-231 cells in dose-dependent manner, in which 100 µg/ml concentration of ATE showed significant efficacy at 4 and 24 h incubation. Similar results were also obtained in BT-20 (Figure 2(b)). In contrast to MDA-MB-231 and BT-20 cells, ATE-induced T cell-mediated cytotoxicity was not observed in PD-L1− MDA-MB-468 cells (Figure 2(c)), suggesting that PD-L1 expression is crucial for ATE to potentiate T cell-mediated cytotoxicity in TNBC cells. Phase contrast microscopic images showed that incubation of T cells + ATE displayed more apoptotic feature such as cell shrinkage in MDA-MB-231 and BT-20 cells, but not MDA-MB-468 cells, when compared with T cell only incubation (Figure 2(d)). Cellular apoptosis was further examined in TNBC cells by co-culturing with T cells in presence or absence of 100 µg/ml ATE for 24 h and then stained with FITC-conjugated anti-annexin V antibody and DAPI for nucleus staining. As shown in Figure 2(e–g), immunofluorescent intensity of annexin V was significantly increased in ATE-treated MDA-MB-231 and BT-20 cells (Figure 2(e–g)). These data suggest that ATE potentiates T cell-mediated anti-tumor activity by enhancing cell apoptosis in PD-L1+ TNBC cells and confirm that PD-L1 enrichment on cancer cells is pre-requisite for ATE to enhance T cell-mediated anti-tumor activity in TNBC cells.
Figure 2.
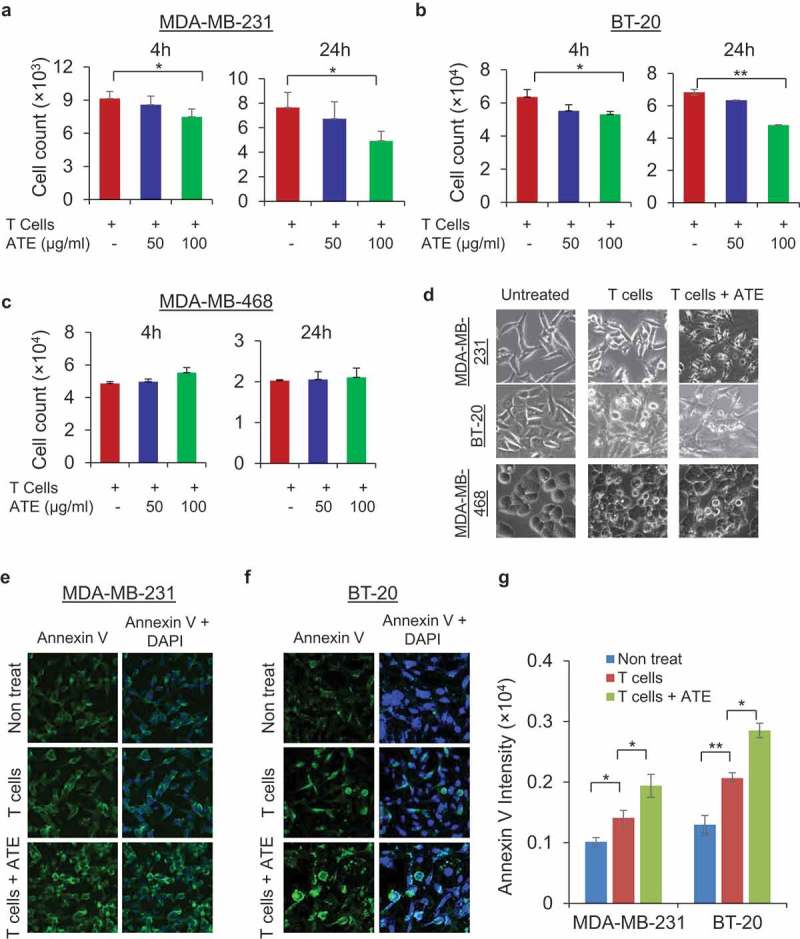
ATE promoted T cell-mediated cytotoxicity and apoptosis in PD-L1+ TNBC cells: (a–c) T cell-mediated cytotoxicity assay was performed in MDA-MB-231, BT-20, and MDA-MB-468 cells, in which cancer cells were co-cultured with activated T cells alone or activated T cells with ATE at 50 µg/ml and 100 µg/ml concentrations for 4 and 24 h time. After incubation, calcein AM-labeled cells were counted by Celigo Imaging system. (d) Bright field images of MDA-MB-231, BT-20, and MDA-MB-468 cells were captured after co-culturing cancer cells with T cells alone or T cells + ATE. (e–f) Annexin V apoptosis assay was performed in MDA-MB-231 and BT-20 cells after co-incubating cancer cells with T cells or T cells + ATE for 24 h and after staining, images were captured using confocal microscope. Annexin V-stained apoptotic cells are shown in green and DAPI was stained with blue color. (g) Quantitative analysis of fluorescence intensity of annexin V-labeled cells was determined by Zeiss software (Carl Zeiss Microscopy) and data was presented in form of bar diagram.
Silencing of PD-L1 reverses ATE-induced T cell-mediated cytotoxicity
Next, we examined whether knockdown of PD-L1 can escape tumor cells from ATE-induced Tcell-mediated cytotoxicity due to loss of target. To this end, PD-L1 expression was silenced by using RNAi technology in MDA-MB-231 and BT-20 cells, and then T cell-mediated cytotoxicity assay was performed. Knockdown of PD-L1 was confirmed in both cell lines at 24 and 48 h with most robust knockdown was observed at 48 h post-transfection of PD-L1 siRNA (Figure 3(a)). As shown in Figure 3(b), PD-L1 knockdown rescued both TNBC cell lines from ATE-induced T cell-mediated cytotoxicity effects, thus further validating the necessity of PD-L1 expression for ATE to induce anti-tumor immune response.
Figure 3.
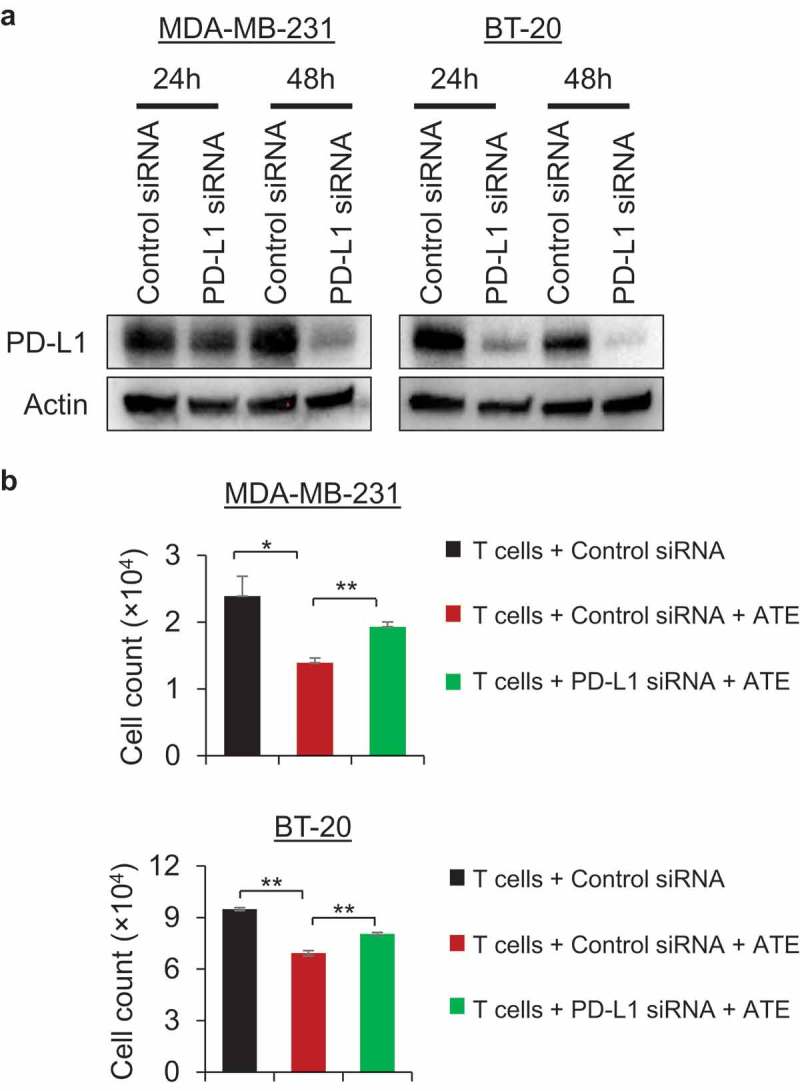
PD-L1 knockdown reverses ATE-induced T cell-mediated cytotoxicity in PD-L1+ TNBC cells. (a) PD-L1 silencing was accomplished by using PD-L1 siRNA transfection and Western blotting was performed post 24 and 48 h transfection to confirm PD-L1 silencing. (b) T cell-mediated cytotoxicity assay was performed in MDA-MB-231 and BT-20 cells after transfecting cancer cells with control siRNA or PD-L1 siRNA. After transfection, cancer cells were cocultured with T cells alone or T cells + ATE and fluorescent labeled cells were counted by using Celigo Imaging system.
Enhancing PD-L1 expression with histone deacetylases (HDAC), proteasomal, and lysosomal inhibitors further promotes ATE-induced T cell-mediated tumor cell cytotoxicity
Given that only 10% of patients with metastatic disease respond to single-agent activity of immune checkpoint inhibitors,7 current efforts are being invested in developing immunotherapy/chemotherapy combinations that can further enhance responses and overcome resistance to checkpoint inhibitors. For this purpose, we extended our investigation to screen novel combination approaches to amplify the anti-tumor efficacy of ATE in TNBC models. Recent studies have shown histone deacetylase (HDAC) inhibitors can improve the anti-tumor activity of immunotherapies by increasing PD-L1 expression levels.22,23 First, we confirmed that mocetinostat (moce), a HDAC inhibitor, in combination with ATE increased PD-L1 expression in MDA-MB-231 and BT-20 cells (Figure 4(a)). However, the expression of PD-L1 was not increased in PD-L1− MDA-MB-468 cells when treated with ATE and moce (Figure 4(a)). As shown in Figure 4(b), combination of ATE + moce significantly inhibited cell growth in T cell co-incubated MDA-MB-231 and BT-20 cells, but not in MDA-MB-468 cells, compared with ATE or moce treatment alone. These data suggest that moce-mediated enrichment of PD-L1 can promote tumor recognition through T cell receptor thus allowing ATE to elicit its anti-tumor activity against TNBC cells.
Figure 4.
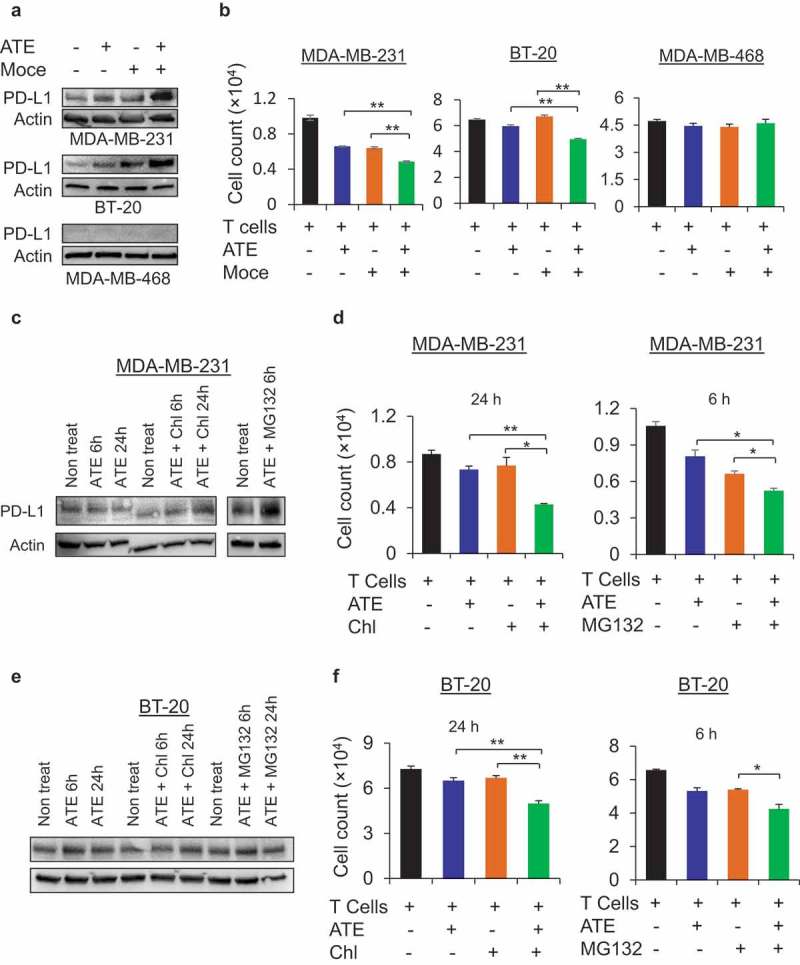
Combination of ATE and PD-L1-upregulating agents further increase Tcell-mediated cytotoxicity in PD-L1+ TNBC cells. (a) Expression levels of PD-L1 was determined by Western blotting after treating MDA-MB-231, BT-20, and MDA-MB-468 cells with ATE, Moce, ATE + Moce or left untreated for 24h. (b) Tcell-mediated cytotoxicity was assessed in MDA-MB-231, BT-20 and MDA-MB-468 cells after co-culturing with T cells and treated with ATE, Moce, ATE + Moce or left untreated. (c) Western blotting was performed to assess PD-L1 expression in MDA-MB-231 cells after treatments with ATE, ATE+ Chl, and ATE + MG132 at indicated time points. (d) T cell-mediated cytotoxicity assay was performed in MDA-MB-231 cells for 24 h with ATE, Chl, and ATE + Chl treatments, and for 6 h with ATE, MG132, and ATE + MG132 treatments. (e) PD-L1 expression was determined in BT-20 cells after treatment with ATE, ATE+ Chl, and ATE + MG132 at indicated time points. (f) Tcell-mediated cytotoxicity was determined in BT-20 cells with treatment conditions described in (c).
Previous study has shown that proteasomal inhibitor MG132 can increase PD-L1 expression in prostate cancer cells by blocking its degradation,24 hence we hypothesized that combining ATE with proteasomal and lysosomal inhibitors in immune-dependent cytotoxicity assays can further increase efficacy of ATE against TNBC cells by preventing proteasomal or lysosomal-mediated degradation of PD-L1. Western blotting experiments showed that MG132, a proteasomal inhibitor and Chloroquine (Chl), a lysosomal inhibitor, increased expression of PD-L1 when combined with ATE (Figure 4(c)). T cell cytotoxicity data in Figure 4(d) showed that either MG132 or Chl in combination with ATE significantly promoted T cell-mediated cytotoxicity in MDA-MB-231 compared with single agent treatments. Likewise, enhanced PD-L1 expression and subsequently increased T cell-mediated cytotoxicity induced by combination approach was also observed in BT-20 cells (Figure 4(e and f)). Furthermore, we also assessed the effect of ATE on cellular growth independent of T cell activity in PD-L1+ cells and PD-L1− cells and observed that growth rate remains unchanged when cells were exposed to ATE alone or in combination with anti-EGFR monoclonal antibody cetuximab under T cell-free environment (Figures S1A, S1B, and S1C). These data suggest that combining ATE with chemotherapeutic compounds that can upregulate PD-L1 expression would maximize immune-mediated therapeutic efficacy of ATE in PD-L1+ TNBC models, thus providing valuable insights into designing of combination clinical approaches for immune checkpoint inhibitors.
ATE-inhibited focal adhesion kinase (FAK) phosphorylation
ATE was recently approved for treatment of TNBC patients with metastatic disease.19 It is possible that ATE could potentially affect cellular invasive and motility properties in PD-L1+ TNBC cells, independent of T cell involvement. Focal adhesion kinase (FAK) signaling pathways play crucial role in regulating metastasis of cancers to distant organs; therefore, we investigated the effect of ATE on FAK phosphorylation levels after exposing PD-L1+ TNBC cells with ATE in time-dependent manner. Western blotting data show that FAK phosphorylation at Tyr397 was notably decreased after 24 h treatment of ATE in both MDA-MB-231 and BT-20 cells (Figure 5(a), upper panel). Phosphorylation levels of extracellular-regulated signal kinase (ERK) were remain unchanged after incubation with ATE in both MDA-MB-231 and BT-20 cells (Figure 5(a), lower panel). Next, we investigated the association of endogenous PD-L1 and FAK using coimmunoprecipitation assays. Data in Figure 5(b) showed that FAK was detected in ATE-immunoprecipitate, but not in IgG-immunoprecipitate, suggesting that endogenous PD-L1 associates with FAK either directly or indirectly. Immunoprecipitated PD-L1 by ATE was confirmed by Western blotting under non-reducing conditions to avoid overlap with IgG heavy chain (Figure 5(b)). Taken together, these results suggest that PD-L1 has functional connection with FAK activity in TNBC cells. Down-regulation of FAK by ATE may inhibit FAK-regulated cellular behaviors such as cell invasion and motility activity in TNBC models but didn’t affect ERK-mediated proliferative pathways.
Figure 5.
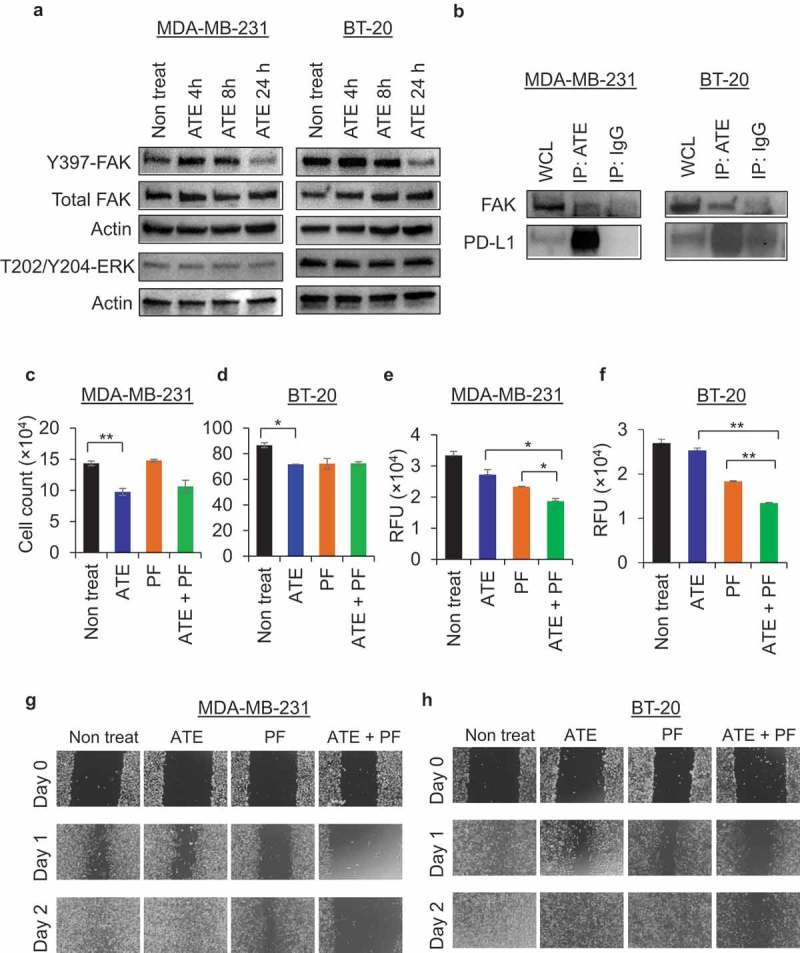
ATE downregulates FAK activity and coordinate with FAK inhibitor to suppress cell invasion and motility (a) Phosphorylation of FAK at Tyr-397 site and ERK at Thr-202/Tyr-204 sites were determined by western blotting after treatment with ATE for 4, 8, 24 h in MDA-MB-231 and BT-20 cells. (b) Whole cell lysate from MDA-MB-231 and BT-20 cells were subjected to co-immunoprecipitation assay to determine the association of FAK with PD-L1. (c–d) T cell-mediated cytotoxicity assay was performed in MDA-MB-231 and BT-20 cells co-cultured with T cells and treated with ATE, PF, ATE + PF or left untreated. (e–f) Cell invasion assay was performed in MDA-MB-231 and BT-20 cells treated with ATE, PF, ATE + PF or left untreated as per manufacturer’s instructions. Quantitative analysis of data was performed after reading the plate with fluorescence plate reader using 480/520 filter set and presented in the form of bar diagram. (g–h) Cellular motility was determined by scratch wound healing assay. MDA-MB-231 and BT-20 cells were exposed to ATE, PF, ATE + PF or left untreated, and then wound healing scratch was performed. Representative brightfield images from wound healing assay were presented post 24 and 48 h treatment.
ATE is unable to inhibit the growth of TNBC cells, but capable of reducing cell invasion and motility activities in combination with FAK-inhibitor in PD-L1+ TNBC
Next, we sought to determine if ATE treatment alone or in combination with FAK inhibitor can affect T cell-mediated cytotoxicity and T cell independent cellular properties, such as cell invasion and migration. For this purpose, we utilized PF573,228 (PF), an ATP-competitive inhibitor of FAK signaling, which can potently block phosphorylation of FAK at Tyr397 and inhibit both chemotactic and haptotactic cellular migration.25 We observed that 1 µM concentration of PF didn’t significantly affect T cell-mediated cytotoxicity in MDA-MB-231 or BT-20 cells (Figure 5(c and d)). The combination of ATE + PF didn’t produce additive or synergistic effect on T cell-mediated cytotoxicity in both MDA-MB-231 and BT-20 cells. Moreover, data from cell invasion assay showed that ATE alone inhibited invasive activity in both MDA-MB-231 and BT-20 cells although statistical significance was not observed (Figure 5(e and f)). PF alone had the ability to inhibit cell invasion, and ATE significantly enhanced the PF-mediated inhibition of invasive activity of MDA-MB-231 and BT-20 cells after 48 h incubation (Figure 5(e and f)). Furthermore, we examined the motility of TNBC cells by performing wound healing scratch assay after treating cells with ATE, PF, or ATE + PF. Data from wound healing assay showed that cellular migration was dramatically halted in TNBC cells treated with combination regimen compared to individual treatments in both MDA-MB-231 and BT-20 cells (Figure 5(g and h)). Taken together, these data suggest that ATE, when combined with FAK inhibitor, suppress metastatic potential of TNBC independent of immune-mediated responses.
Correlation of mRNA expression between PD-L1 and focal adhesion kinase (FAK) is observed in TNBC
The RNA-seq data from breast cancer patients’ samples available in the TCGA database were used to analyze the correlation between the expression of PD-L1 and FAK. The total 1105 samples were sub-grouped into TNBC and non-TNBC, and we searched for molecule(s) whose expression is correlated with that of PD-L1 in TNBC samples. Firstly, we observed significant increase in the mRNA expression levels of PD-L1 in the TNBC subgroup compared with non-TNBC subgroup (Figure 6(a)), whereas FAK mRNA expression levels didn’t change significantly between TNBC and non-TNBC populations (Figure 6(b)). The mean PD-L1 mRNA value (in log2) for the TNBC was 1.93, significantly higher than the non-TNBC subgroup of 0.72. Furthermore, we analyzed the correlation coefficient of PD-L1 and FAK in TNBC patient population and non-TNBC patient population and observed that there was a positive correlation between FAK and PD-L1 mRNA expression in TNBC samples (Figure 6(c)), but not in non-TNBC samples (Figure 6(d)). Based on TCGA analysis data, it appears that there is a strong correlation between FAK and PD-L1 in TNBC population, suggesting that the therapeutic efficacy of ATE could be potentially potentiated by using pharmacological inhibitors of FAK for TNBC treatments.
Figure 6.
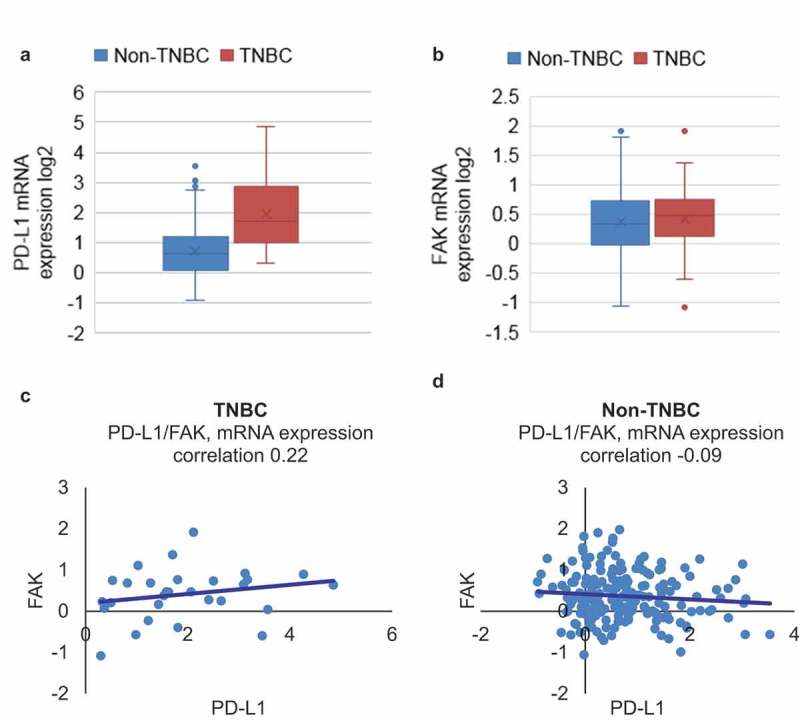
PD-L1 associates with FAK in TNBC patient sample database. (a–b) RNAseq data from the two queried genes, PD-L1 and FAK, were downloaded from TCGA database and expression of PD-L1 mRNA and FAK mRNA was analyzed in TNBC and non-TNBC patient population. (c–d) Correlation between PD-L1 and FAK in TNBC and non-TNBC samples obtained from TCGA analysis was performed.
Discussion
Usage of immune checkpoint inhibitors for treatment of TNBC patient population is under active clinical investigation. In this pre-clinical investigation, we observed that ATE can significantly potentiate Tcell-mediated cytotoxicity in PD-L1+ TNBC cells. PD-L1 knockdown experiments showed that ATE-induced anti-cancer immune activity is PD-L1 expression dependent. To extend anti-tumor effects of ATE, combination approaches are warranted. Therefore, when combining ATE with PD-L1 enriching reagents such as moce, MG132, and Chl, ATE further enhanced T cell-mediated cancer cell death. Our study, for the first time, discovered the positive correlation between PD-L1 and FAK in TNBC patient samples from TGCA analysis. Furthermore, ATE remarkably downregulated FAK phosphorylation independent of T cell-mediated cytotoxicity, suggesting that functional correlation between PD-L1 and FAK existed. Metastatic cellular attributes such as motility and invasion were suppressed by ATE in association with FAK inhibitor thus underscoring the utility of ATE for T cell-independent anti-cancer properties.
Our preclinical study for targeting TNBC tumors using ATE supports clinical trials that have shown encouraging therapeutic response in metastatic TNBC patient population.19,20 Other preclinical studies have also investigated the blockage of PD-1/PD-L1 interaction for therapeutic efficacy in TNBC and other solid tumor models. Li et al. demonstrated that an antibody-drug conjugate targeting glycosylated PD-L1 elicited remarkable anti-tumor activity in TNBC cellular and syngeneic mouse models.26 Our study is in agreement with previously published report where HDAC inhibitor significantly potentiated anti-cancer immune response of PD-1/CTLA-4 blocking therapy in triple negative 4T1 mouse model, which was associated with increased T cell infiltration in tumor microenvironment, reduction in tumor growth and increased survival.22
The status of PD-L1 as predictive biomarker in breast cancer has not yet been validated. Based on data from PD-L1 knockdown experiments and PD-L1 upregulating pharmacological inhibitors, we observed that ATE’s ability to enhance T cell-mediated cytotoxicity of TNBC was PD-L1-expression dependent and low-PD-L1 expressing cells such as MDA-MB-468 escaped ATE-induced cytotoxicity. In clinical settings, patients overexpressing PD-L1 on immune cells as determined by IHC have shown improved clinical outcomes with immune check point therapy. Recent clinical trial studying the effect of ATE in TNBC patients reported that patient population with PD-L1-positive tumors showed improved clinical outcomes emphasizing the need for adding checkpoint inhibitor to standard chemotherapy for the first-line treatment of metastatic TNBC.19 Majority of clinical data suggest that patients whose tumor overexpress PD-L1 exhibit better clinical response to immune checkpoint therapy, however, some patients with low PD-L1 expression also show anti-tumor immune responses thus complicating the issue of PD-L1 as an exclusive predictive biomarker.27 Rather than being an oncogenic driver of tumor progression, PD-L1 overexpression appears to be immune protection mechanism that shields cancer cells from immunogenic tumor microenvironment.27 Activation of PI3K/AKT/mTOR pathway drives PD-L1 expression and promotes immune escape in lung cancer model.28 Therefore, immunotherapy can provide clinical benefit to patients whose tumors have both high TILs and PD-L1 expression thus overcoming adaptive immune resistance to endogenous anti-tumor activity.29
Cancer metastasis, one of most life-threatening event in cancer patients, occurs due to deleterious invasion of tumors cells into surrounding tissues and distant organs.30,31 Although metastatic potential of TNBC tumors are similar to other breast cancer subtypes; these patients have much shorter median time from relapse to death.32,33 Previously published report suggests that high PD-L1 expression significantly correlate with lymph node metastasis and poor prognosis in cervical carcinoma.34 Tissue microarray analysis from colorectal cancers reveal that PD-L1 is predominantly expressed in metastatic tumors compared with primary tumor suggesting that PD-L1 expression rises during metastatic spread of tumor.35 Likewise, analysis of PD-L1 expression in lymph-node positive TNBC patients suggest that PD-L1 is expressed significantly higher in tumor cells and lymphocytes of metastasizing lymph nodes.36 FAK is key driver of cancer cell invasion and metastasis and pharmacological inhibition of FAK has been reported to induce tumor suppressive effects on tumor microenvironment in pancreatic cancer mice model.37 In this study, we discovered a novel functional correlation between PD-L1 and FAK and showed that ATE can remarkably downregulate FAK activity, and combination of ATE with FAK inhibitor can dramatically inhibit tumor cell invasion and migration. Our cell invasion and migration data support the findings from recent clinical study in which ATE + chemotherapy has shown better clinical response in TNBC patients with metastatic disease,19 therefore adding FAK antagonists in combination with immunotherapy in clinical settings could be attractive strategy to arrest the metastatic spread of tumors and extend patient’s life.
In conclusion, our findings provide novel mechanistic insights into anti-tumor immune response for ATE in immunogenic TNBC cellular models. Combining ATE with chemotherapeutic compounds that can upregulate PD-L1 cell surface expression or minimize PD-L1 degradation can synergistically enhance T cell-mediated cytotoxicity of TNBC tumor cells. In addition, this study for the first time suggest a possible role of anti-PD-L1 antibody in suppressing cell invasion and migration property by regulating FAK-dependent signaling pathway. Overall, our investigation adds to growing body of evidence demonstrating that immune checkpoint inhibitors alone or in combination with PD-L1 modulating drugs provide novel therapeutic strategy for TNBC tumors.
Materials and methods
Cell lines and culture conditions
TNBC cells MDA-MB-231, BT-20, MDA-MB-468, BT-549, HS-578T cells were obtained from ATCC. MDA-MB-231, MDA-MB-468, BT-549, HS-578T cells were cultured in RPMI media (Lonza) while BT-20 cells were maintained in EMEM media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin (P/S). All the cell lines were propagated at 37°C under 5% CO2. Human Peripheral Blood Pan-T Cells (T cells) were purchased from Stem Cell Technologies which were isolated from peripheral blood mononuclear cells using negative immunomagnetic separation techniques. T cells were cultured in RPMI media containing 10% FBS and 1% P/S and activated by adding 25 µl of ImmunoCult™ Human CD3/CD28 T Cell Activator (Stem Cell Technologies) per 1 ml of cell suspension.
T cell-mediated cytotoxicity assay
Target cancer cells were seeded in 24-well plate for T cell-mediated cytotoxicity assay. Next day, cancer cells were labeled with calcein AMcell permeant dye, which is converted to a green-fluorescent calcein after acetoxymethyl ester hydrolysis by intracellular esterases in live cells. After observing the green fluorescence, cells were washed with PBS to remove residual dye. Further, cancer cells were co-incubated with T cells alone or T cells with ATE at desired concentrations for indicated time. The ratio of cancer cells and T cells was optimized to 1:3 for MDA-MB-231 and MDA-MB-468 cells, and 1:2 for BT-20 cells. After incubation, culture plate was washed with PBS to remove residual T cells and calcein-labeled cancer cells were counted using Celigo cell imaging system.
Annexin V-FITC apoptosis assay
Annexin V-FITC apoptosis assay kit was obtained from Cell Signaling Technologies and assay was performed per manufacturer’s instructions. During early stages of apoptosis, phosphatidylserine translocates from inner side of plasma membrane to outer surface, where fluorochrome-conjugated Annexin V binds to it with strong affinity. Briefly, MDA-MB-231, BT20 and MDA-MB-468 cells were cocultured with activated T cells in chamber slides and treated with ATE at 100 µg/ml or left untreated for 24 h in humidified incubator. Following incubation, cells were washed twice with PBS and stained with FITC-conjugated annexin V antibody. After staining, cells were fixed in 4% paraformaldehyde for 10 min. Fixation was performed after annexin V staining to avoid non-specific binding of annexin to PS after cell membrane disruption. Chamber slides were mounted with coverslips using Prolong Gold Anti-Fade Reagent with DAPI. Images were captured on a Zeiss LSM-880 confocal microscope using a 40 ×/1.4 NA plan Apo objective.
PD-L1 expression by flow cytometry
To assess cell surface expression of PD-L1, MDA-MB-231, BT-20, MDA-MB-468 cells were harvested by trypsinization and stained with FITC conjugated anti-PD-L1 antibody for 1 h at 4°C. Cells were washed twice with PBS and analyzed with BD LSR Flow cytometer using 488 filter. Flowcytometry histograms were further analyzed by FlowJo software and fluorescence intensity was quantified from three replicates.
Cell invasion assay
Cell invasion assay kit was obtained from Millipore (cat#ECM554) and assay was executed according to manufacturer’s instructions as described previously.38 The assay kit contains invasion chamber or inserts with 8 µm pore size polycarbonate membrane and coated with thin layer of ECMatrix. Briefly, MDA-MB-231 and BT-20 cells were trypsinized and cell suspension was pre-incubated with ATE, FAK inhibitor (PF), ATE + PF or left untreated for 30 min at 37°C. Cell invasion chamber or inserts were rehydrated by adding 300 µl prewarmed media for 20 min at room temperature. After pre-incubation, 250 µl of cell suspension containing 2.5 × 105 cells were added to each insert and 500 µl of FBS containing media was added to the lower chamber. Cell plate was incubated for 48h in CO2 incubator at 37°C. Then, inserts were placed in 24-well plate containing 225 µl of cell detachment and cells were dislodged by gently tilting inserts back and forth. Dye solution was prepared in lysis buffer at 1:75 ratio and 75 µl of this dye solution was added to 225 µl of cell detachment buffer containing invaded cells. After 15 min incubation, 200 µl of cell mixture was transferred to a new 96-well plate and read with fluorescence plate reader using 480/520 filter set.
PD-L1 siRNA transfection
PD-L1 siRNA (cat# L-015836–01-0005) and control siRNA were purchased from GE Dharmacon and transfection experiments were performed using lipofectamine 3000 (Invitrogen) per manufacturer’s protocol and described previously.39 Briefly, cells were seeded in 6-well plate at 1 × 105 per well seeding density. Lipofectamine 3000 reagent and siRNA were diluted separately in Opti-MEM media and mixed together. Lipid-siRNA complex was further incubated for 5 min and then added to respective wells. Post 24 and 48 h of transfection, cells were harvested by trypsinization and western blotting was performed to assess PD-L1 knockdown.
Scratch wound healing assay
Cancer cells were grown in 6-well plate containing media supplemented with 10% FBS and allowed to reach 70–80% confluency. A monolayer of cells was scratched using a 1 ml pipette tip across the center of well and another straight line was scratched perpendicular to the first line to create a cross in each well. After scratching, each well was washed gently with PBS and replenished with fresh media. Next day, cells were treated with ATE, FAK inhibitor, and ATE + FAK inhibitor or left untreated. Following 24 and 48 h post treatment, bright field images were captured using microscope.
TCGA data retrieval and analysis
RNAseq data from the two queried genes, CD274 (PD-L1) and PTK2 (FAK), were downloaded from the cbioportal (http://www. cbioportal.org/public-portal/). Correlation between CD274 and PTK2 mRNA expression in TNBC and non-TNBC patients was analyzed with Spearman correlation coefficients calculated using Microsoft Excel software. The expression level of CD274 and PTK2 was also evaluated.
Statistical analysis
For statistical analysis, GraphPad Prism and Microsoft Excel software were used. Statistical significance was determined by Student’s t-test (*, P < .05; **, P < .01). Data are expressed as mean ± standard deviation (STDEV) or mean ± standard error of mean (SEM).
Abbreviations
- PD-1
Programmed Cell Death Protein-1
- PD-L1
Programmed Cell Death Ligand 1
- TNBC
Triple Negative Breast Cancer
- ATE
Atezolizumab
- TCGA
The Cancer Genome Atlas
- FAK
Focal Adhesion Kinase
- ER
Estrogen Receptor
- PR
Progesterone Receptor
- TILs
Tumor Infiltrating Lymphocytes
- MFI
Mean Fluorescent Intensity
- HDAC
Histone Deacetylase
- Chl
Chloroquine
- ERK
Extracellular-regulated Signal Kinase
- FBS
Fetal Bovine Serum
- P/S
Penicillin And Streptomycin
- STDEV
Standard Deviation
- SEM
Standard Error of Mean
Author contributions
N.M. and W.J.W. developed, designed, and conceived the project. N.M., S.H., J.Z., Y.S., X.L., J.J., and Y.E. performed experiments. N.M., S.H., J.Z., and W.J.W. analyzed and interpreted data. N.M., Y.E., and W.J.W. wrote the manuscript with input from S.H., J.Z., X.L., J.J., and W.J.W. supervised the study.
Competing interests/Disclaimer
All authors declared no competing financial interests with industry. This article reflects the views of the authors and should not be construed to represent FDA’s views or policies.
Acknowledgments
The authors thank Drs. Mamatha Garige and Kula Jha for critical internal review of this article.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary data
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA.. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13(11):674–690. doi: 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010. November 11;363(20):1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 4.Adams S, Gray RJ, Demaria S, Goldstein L, Perez EA, Shulman LN, Martino S, Wang M, Jones VE, Saphner TJ, et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol. 2014;32(27):2959–2966. doi: 10.1200/JCO.2013.55.0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu S, Lachapelle J, Leung S, Gao D, Foulkes WD, Nielsen TO. CD8+ lymphocyte infiltration is an independent favorable prognostic indicator in basal-like breast cancer. Breast Cancer Res. 2012;14:R48. doi: 10.1186/bcr3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kato T, Park J, Kiyotani K, Ikeda Y, Miyoshi Y, Nakamura Y. Integrated analysis of somatic mutations and immune microenvironment of multiple regions in breast cancers. Oncotarget. 2017;8(37):62029–62038. doi: 10.18632/oncotarget.v8i37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 9.Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, Rietz C, Flies DB, Lau JS, Zhu G, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089–1096. [PubMed] [Google Scholar]
- 10.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99(19):12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abril-Rodriguez G, Ribas A. SnapShot: immune checkpoint inhibitors. Cancer Cell. 2017. June 12;31(6):848–848.e1. doi: 10.1016/j.ccell.2017.05.010. [DOI] [PubMed] [Google Scholar]
- 12.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lau J, Cheung J, Navarro A, Lianoglou S, Haley B, Totpal K, Sanders L, Koeppen H, Caplazi P, McBride J, et al. Tumour and host cell PD-L1 is required to mediate suppression of anti-tumour immunity in mice. Nat Commun. 2017;8:14572. doi: 10.1038/ncomms14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S, Su X, Wang Y, Gonzalez-Angulo AM, Akcakanat A, et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res. 2014;2(4):361–370. doi: 10.1158/2326-6066.CIR-13-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wimberly H, Brown JR, Schalper K, Haack H, Silver MR, Nixon C, Bossuyt V, Pusztai L, Lannin DR, Rimm DL. PD-L1 expression correlates with tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy in breast cancer. Cancer Immunol Res. 2015;3(4):326–332. doi: 10.1158/2326-6066.CIR-14-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O’Donnell PH, Balmanoukian A, Loriot Y, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909–1920. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, Gadgeel SM, Hida T, Kowalski DM, Dols MC; OAK Study Group et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017. January 21;389(10066):255–265. Epub 2016 Dec 13. doi: 10.1016/S0140-6736(16)32517-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herbst RS, Soria J-C, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Diéras V, Hegg R, Im S-A, Shaw Wright G, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018. October 20;379:2108–2121. doi: 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]
- 20.Emens LA, et al. Long-term clinical outcomes and biomarker analyses of atezolizumab therapy for patients with metastatic triple-negative breast cancer. JAMA Oncol:2019;5:74–82. doi: 10.1001/jamaoncol.2018.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antonia SJ, Larkin J, Ascierto PA. Immuno-oncology combinations: a review of clinical experience and future prospects. Clin Cancer Res. 2014. December 15;20(24):6258–6268. Epub 2014 Oct 23. doi: 10.1158/1078-0432.CCR-14-1457. [DOI] [PubMed] [Google Scholar]
- 22.Terranova-Barberio M, Thomas S, Ali N, Pawlowska N, Park J, Krings G, Rosenblum MD, Budillon A, Munster PN. HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget. 2017;8(69):114156–114172. doi: 10.18632/oncotarget.23169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Briere D, Sudhakar N, Woods DM, Hallin J, Engstrom LD, Aranda R, Chiang H, Sodré AL, Olson P, Weber JS, et al. The class I/IV HDAC inhibitor mocetinostat increases tumor antigen presentation, decreases immune suppressive cell types and augments checkpoint inhibitor therapy. Cancer Immunol Immunother. 2018;67(3):381–392. doi: 10.1007/s00262-017-2091-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, Tan Y, Ci Y, Wu F, Dai X, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553(7686):91–95. doi: 10.1038/nature25015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slack-Davis JK, Martin KH, Tilghman RW, Iwanicki M, Ung EJ, Autry C, Luzzio MJ, Cooper B, Kath JC, Roberts WG, et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem. 2007;282(20):14845–14852. doi: 10.1074/jbc.M606695200. [DOI] [PubMed] [Google Scholar]
- 26.Li CW, Lim SO, Chung EM, Kim YS, Park AH, Yao J, Cha J-H, Xia W, Chan L-C, Kim T, et al. Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell. 2018;33(2):187–201.e10. doi: 10.1016/j.ccell.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14(4):847–856. doi: 10.1158/1535-7163.MCT-14-0983. [DOI] [PubMed] [Google Scholar]
- 28.Lastwika KJ, Wilson W 3rd, Li QK, Norris J, Xu H, Ghazarian SR, Kitagawa H, Kawabata S, Taube JM, Yao S, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. 2016;76(2):227–238. doi: 10.1158/0008-5472.CAN-14-3362 Epub 2015 Dec 4 [DOI] [PubMed] [Google Scholar]
- 29.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127):127ra37. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steeg PS. Targeting metastasis. Nat Rev Cancer. 2016;16(4):201–218. doi: 10.1038/nrc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yilmaz M, Christofori G, Lehembre F. Distinct mechanisms of tumor invasion and metastasis. Trends Mol Med. 2007;13(12):535–541. doi: 10.1016/j.molmed.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 32.Anders CK, Carey LA. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin Breast Cancer. 2009;9(Suppl2):S73–81. doi: 10.3816/CBC.2009.s.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hudis CA, Gianni L. Triple-negative breast cancer: an unmet medical need. Oncologist. 2011;16(Suppl 1):1–11. doi: 10.1634/theoncologist.2011-S1-01. [DOI] [PubMed] [Google Scholar]
- 34.Wang S, Li J, Xie J, Liu F, Duan Y, Wu Y, Huang S, He X, Wang Z, Wu X. Programmed death ligand 1 promotes lymph node metastasis and glucose metabolism in cervical cancer by activating integrin β4/SNAI1/SIRT3 signaling pathway. Oncogene. 2018;37(30):4164–4180. doi: 10.1038/s41388-018-0252-x. [DOI] [PubMed] [Google Scholar]
- 35.Wang HB, Yao H, Li CS, Liang LX, Zhang Y, Chen YX, Fang JY, Xu J. Rise of PD-L1 expression during metastasis of colorectal cancer: implications for immunotherapy. J Dig Dis. 2017;18(10):574–581. doi: 10.1111/1751-2980.12538. [DOI] [PubMed] [Google Scholar]
- 36.Li M, Li A, Zhou S, Xu Y, Xiao Y, Bi R, Yang W. Heterogeneity of PD-L1 expression in primary tumors and paired lymph node metastases of triple negative breast cancer. BMC Cancer. 2018;18(1):4. doi: 10.1186/s12885-017-3916-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stokes JB, Adair SJ, Slack-Davis JK, Walters DM, Tilghman RW, Hershey ED, Lowrey B, Thomas KS, Bouton AH, Hwang RF, et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther. 2011;10(11):2135–2145. doi: 10.1158/1535-7163.MCT-11-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Endo Y, Shen Y, Youssef LA, Mohan N, Wu WJ. T-DM1-resistant cells gain high invasive activity via EGFR and integrin cooperated pathways. MAbs. 2018;10(7):1003–1017. doi: 10.1080/19420862.2018.1503904 Epub 2018 Sep 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mohan N, Shen Y, Dokmanovic M, Endo Y, Hirsch DS, Wu WJ. VPS34 regulates TSC1/TSC2 heterodimer to mediate RheB and mTORC1/S6K1 activation and cellular transformation. Oncotarget. 2016;7(32):52239–52254. doi: 10.18632/oncotarget.10469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.