

ABSTRACT
Adoptive transfer of chimeric antigen receptor (CAR)-equipped T cells have demonstrated astonishing clinical efficacy in hematological malignancies recently culminating in the approval of two CAR T cell products. Despite this tremendous success, CAR T cell approaches have still achieved only moderate efficacy against solid tumors. As a major obstacle, engineered conventional T cells (Tconvs) face an anti-inflammatory, hostile tumor microenvironment often infiltrated by highly suppressive regulatory T cells (Tregs). Thus, potent CAR T cell treatment of solid tumors requires efficient activation of Tconvs via their engrafted CAR to overcome Treg-mediated immunosuppression. In that regard, selecting an optimal intracellular signaling domain might represent a crucial step to achieve best clinical efficiency. To shed light on this issue and to investigate responsiveness to Treg inhibition, we engrafted Tconvs with switchable universal CARs (UniCARs) harboring intracellularly the CD3ζ domain alone or in combination with costimulatory CD28 or 4-1BB. Our studies reveal that UniCAR ζ-, and UniCAR BB/ζ-engineered Tconvs are strongly impaired by activated Tregs, whereas UniCARs providing CD28 costimulation overcome Treg-mediated suppression both in vitro and in vivo. Compared to UniCAR ζ- and UniCAR BB/ζ-modified cells, UniCAR 28/ζ-armed Tconvs secrete significantly higher amounts of Th1-related cytokines and, furthermore, levels of these cytokines are elevated even upon exposure to Tregs. Thus, in contrast to 4-1BB costimulation, CD28 signaling in UniCAR-transduced Tconvs seems to foster a pro-inflammatory milieu, which contributes to enhanced resistance to Treg suppression. Overall, our results may have significant implications for CAR T cell-based immunotherapies of solid tumors strongly invaded by Tregs.
KEYWORDS: Chimeric antigen receptor, intracellular signaling domain, regulatory T cells, immunosuppression, tumor immunotherapy, solid tumor
Introduction
The clinical application of chimeric antigen receptor (CAR)-armed T cells have been proven to be highly successful for the treatment of various hematological malignancies thereby revolutionizing cancer immunotherapy.1,2 CARs are typically composed of an extracellular binding moiety specifically interacting with an antigen of interest, a flexible hinge region and a transmembrane domain fused to an intracellular signaling unit. Originally, first-generation CARs were designed, in which T cell activation is triggered by an isolated CD3ζ domain. In order to enhance T cell proliferation, effector function and persistence, at least one additional costimulatory region was integrated into the CAR architecture leading to the development of second- or third-generation CARs.3 To date, costimulatory domains primarily utilized in clinical trials derive from CD28 or 4-1BB (CD137). Likewise, both CAR T cell products being recently approved by the Food and Drug Administration for the treatment of CD19+ B cell acute lymphoblastic leukemia and/or diffuse large B cell lymphoma harbor a BB/ζ (tisagenlecleucel) or a 28/ζ (axicabtagene ciloleucel) endodomain.4 Despite tremendous complete remission rates obtained with these therapeutic cell products,5–7 CAR technology still has to surmount several major challenges.
On the one hand, administration of CAR T cells is often accompanied by severe adverse events among which the two most frequent ones are cytokine release syndrome and CAR T cell-related encephalopathy syndrome.8–10 Both toxicities can result in life-threatening or even fatal outcome, as recently reported for several patients who died from cerebral edema.8,11,12 Furthermore, on-target/off-tumor responses in patients with solid tumors have provoked severe, partly lethal toxicity.13–15 For a better control of side effects, it is of greatest importance to accurately adjust activity of CAR-engrafted T cells. To fulfill this urgent need, our group has developed a switchable, universal CAR (UniCAR) system which allows precise tuning of T cell functionality between an “ON” and “OFF” status.16–18 In contrast to conventional CARs, interaction between UniCAR-armed T cells and target cells is mediated by a separate, targeting module (TM), binding on the one hand an antigen of interest and comprising, on the other hand, a small peptide epitope recognized by the UniCAR. As a consequence, UniCAR-modified T cells are activated only in the presence of both the cross-linking TM and the targeted antigen. This innovative and well-controllable technology has already demonstrated remarkable efficacy in vitro as well as in vivo and might, therefore, broaden current treatment modalities for cancer patients.17–24
Another major obstacle hampering a wide-spread application of CAR T cell therapies remains their moderate efficacy in the setting of solid tumors. Although stable disease or partial responses were achieved in some patients, therapeutic success remains far behind clinical outcomes obtained in hematological malignancies.25–28 A substantial hurdle for CAR-modified T cells in solid tumors constitutes the hostile tumor microenvironment containing a plethora of suppressive factors. The establishment of this anti-inflammatory milieu is especially fostered by regulatory T cells (Tregs) which are capable of hampering effector cells by multiple mechanisms such as IL-2 consumption, cell-contact dependent inhibition or secretion of suppressive cytokines.29,30 Moreover, in most tumor infiltrates enrichment of Tregs correlates with a poor survival prognosis for cancer patients underlining the detrimental effect of Tregs on treatment outcome.31–34 As endogenous, tumor-resident Tregs might also negatively affect efficacy of CAR-modified T cells, it is of utmost importance to provide powerful (co)stimulatory signals to trigger optimal CAR T cell activation in the face of Treg-mediated immunosuppression. To shed light on this issue, we aimed to investigate the performance of T cells that were armed with UniCARs delivering either CD28- or 4-1BB-derived costimulation in the presence of highly suppressive Tregs (Figure 1). Within this study, we provide the first experimental evidence that UniCAR 28/ζ-engrafted conventional T cells (Tconvs) overcome Treg inhibition both in vitro and in vivo, whereas UniCAR BB/ζ-armed Tconvs fails to resist Treg-mediated immunosuppression.
Figure 1.
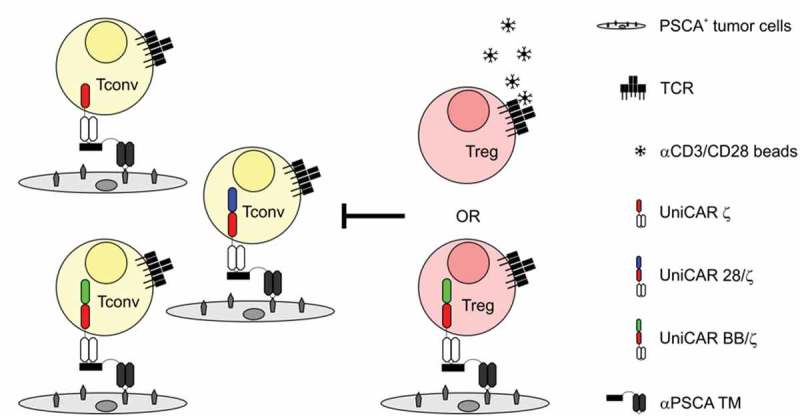
Schematic representation of the experimental setup. Tconvs engrafted with UniCAR ζ, UniCAR BB/ζ or UniCAR 28/ζ were redirected to prostate stem cell antigen (PSCA)-expressing target cells via an αPSCA targeting module (TM). To determine performance of these responder cells in an immunosuppressive milieu, Tregs were added. For stimulation, suppressor cells were either pre-activated with αCD3/CD28 beads via their endogenous T cell receptor (TCR) or redirected via UniCAR BB/ζ engraftment.
Results
Generation of UniCAR-armed Tconvs with different intracellular signaling domains
Human Tconvs were isolated from peripheral blood of healthy donors and transduced with UniCAR constructs harboring in addition to the CD3ζ signaling unit either a costimulatory signaling domain of 4-1BB (UniCAR BB/ζ) or CD28 (UniCAR 28/ζ). As control, the UniCAR ζ construct was employed lacking any costimulation. Aside from these differences, UniCAR ζ, UniCAR 28/ζ, and UniCAR BB/ζ share identical extracellular and transmembrane proportions (Figure 2(a)).17,18,23 All UniCARs were expressed on the surface of genetically modified Tconvs as validated by extracellular detection of the epitope tag E7B6 incorporated into the UniCAR architecture (Figure 2(b)). Importantly transduction efficacy was comparable between the individual UniCAR constructs yielding no statistically significant differences (Figure 2(c); UniCAR ζ: 87.4 ± 6.1%, UniCAR BB/ζ: 85.7 ± 6.2%, UniCAR 28/ζ: 92.3 ± 3.2%, n = 7).
Figure 2.
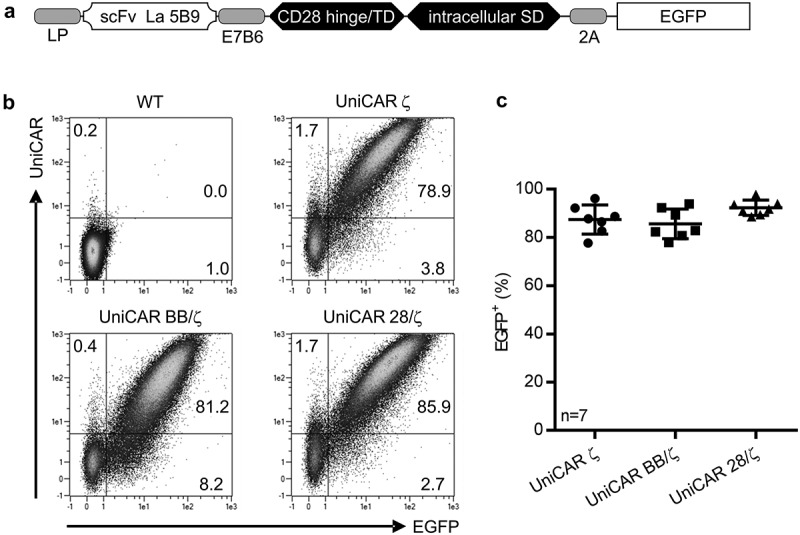
Genetic modification of Tconvs with UniCARs. After isolation, Tconvs were bead-stimulated in the presence of exogenous cytokines and transduced by using a lentiviral gene transfer system to stably express the UniCAR constructs. (a) Schematic representation of UniCAR construct design. All constructs possess an N-terminal leader peptide (LP), followed by a single-chain fragment variable (scFv) originating from the αLa 5B9 mAb, an epitope tag (E7B6) as well as a CD28-derived hinge/transmembrane domain (TD). The intracellular signaling domain (SD) differs between individual UniCARs and contains either CD3ζ SD alone (UniCAR ζ) or in addition a costimulatory signal of 4-1BB (UniCAR BB/ζ) or CD28 (UniCAR 28/ζ). Moreover, EGFP was fused C-terminally to the UniCAR construct separated by a 2A ‘cleavage’ site. (b) Surface staining of UniCARs using a monoclonal antibody against the E7B6 epitope tag located in the extracellular hinge region. Signal is blotted against the co-expressed intracellular EGFP marker. Blots of one representative donor are shown. (c) To assess transduction efficacy, EGFP signal of seven individual donors is shown as mean ± SD. No statistical difference was determined between the individual UniCAR constructs (1-way ANOVA with Bonferroni post hoc test).
Inhibition of UniCAR-engrafted Tconvs by autologous Tregs in vitro
Having confirmed a uniform surface expression of all UniCAR constructs, we aimed to explore the influence of different costimulatory signaling domains on Tconv responsiveness to Treg-mediated suppression (see also Figure 1 for experimental setup). To that end, autologous CD4+CD25+CD127dimCD45RA+ Tregs were isolated to high purity and subsequently expanded in the presence of Proleukin® S and αCD3/CD28 beads. Lineage stability of ex vivo cultured Tregs was confirmed by a high FOXP3+ expression (96.4 ± 3.1% CD4+FOXP3+, n = 7, Supplementary Fig. 1C).
In order to examine responsiveness to Treg repression, UniCAR-endowed Tconvs were retargeted to PC3 cells expressing the prostate stem cell antigen (PSCA) by using a cross-linking αPSCA TM in the absence or presence of T cell receptor (TCR)-stimulated autologous Tregs. Tregs that were not pre-activated with conventional αCD3/CD28 beads served as control. As anticipated, addition of resting Tregs did not markedly influence expansion of either of the investigated UniCAR Tconv populations (Figure 3(a)). However, Tregs that were activated via their endogenous TCR prior to the assay significantly repressed UniCAR BB/ζ-engrafted Tconvs at all tested ratios (76 ± 20% and 31 ± 5% for the highest and lowest ratio, respectively, n = 3). In contrast, UniCAR 28/ζ-armed Tconv expansion was only impaired at the highest Treg to Tconv ratio (63 ± 19%, n = 3). Thus, UniCAR 28/ζ-endowed Tconvs are more resistant to Treg-mediated suppression than Tconvs with a BB/ζ signaling domain, which was most pronounced when low Treg numbers were added (Figure 3(b)). In line with previously published results,35 Tconvs engrafted with a control UniCAR ζ construct were highly prone to inhibition by TCR-stimulated Tregs at all Treg to Tconv ratios tested, underlining a significant difference to both second-generation UniCARs (Figure 3(b)).
Figure 3.
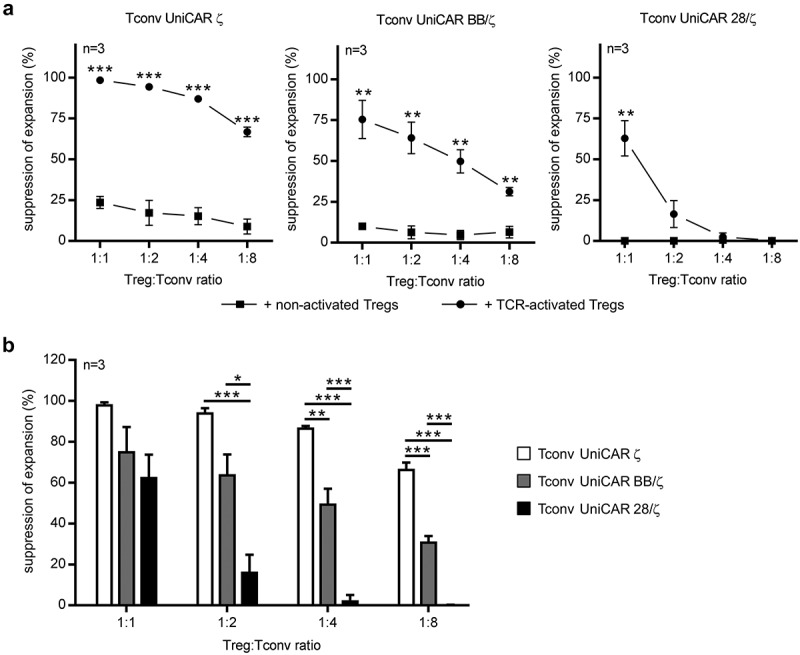
Suppression of UniCAR-equipped Tconvs by autologous TCR-activated Tregs. 0.5 × 104 eFluor670-labeled, UniCAR-endowed Tconvs were cocultured with PC3-PSCA cells (effector to target cell ratio of 5:1) in the absence or presence of 6 pmol αPSCA TM. For immunosuppression, autologous, eFluor450-stained Tregs, which were either non-activated or pre-stimulated with αCD3/CD28 for 24 h prior to the assay, were added at indicated Treg:Tconv ratios. After four days, absolute cell number of eFluor670-labeled Tconvs was determined by flow cytometry and percent of suppression was calculated. Summarized data of three independent donors are depicted as mean ± SEM in the presence of (a) non- or (a,b) TCR-activated Tregs. Statistical significance was calculated by using (a) unpaired, two-tailed Student’s t-test or (b) 1-way ANOVA with Bonferroni post hoc test (*p < .05, **p < .01, ***p < .001).
Next, in order to mimic an antigen-specific immunosuppression bringing both responder and suppressor cells into immediate proximity, Tregs were genetically modified to express a UniCAR BB/ζ construct. Previous investigations have revealed that a BB/ζ signaling domain is most favorable for Treg modification, whereas signaling via a UniCAR 28/ζ heightened their pro-inflammatory potential and triggered a certain level of antigen-independent Treg activation.23 Thus, UniCAR BB/ζ-armed Tregs (80.5 ± 6.4% CD4+EGFP+ cells, n = 6, Supplementary Fig. 1A and 1B) were applied in all experiments and redirected to PC3-PSCA cells using the same αPSCA TM as for stimulation of UniCAR Tconvs. Therefore, Tregs engrafted with a UniCAR stop construct lacking any intracellular signaling domain (83.9 ± 11.7% CD4+EGFP+ cells, n = 6, Supplementary Fig. 1A and 1B) were used as additional control in order to exclude that effects observed in this experimental setup were caused by competitive TM usage. As depicted in Figure 4(a), neither UniCAR stop- nor non-modified Tregs were capable of markedly influencing expansion of cocultured second-generation UniCAR Tconvs (Figure 4(a)). Of note, control Tconvs expressing a first-generation UniCAR were suppressed by UniCAR stop-armed Tregs with statistical significance at the highest ratio tested, although cross-linkage of these Tregs to tumor cells does not trigger an activation signal23 (Figure 4(a), left panel). As shown in Figure 4(a) (middle panel), UniCAR-stimulated Tregs considerably impaired expansion of Tconvs harboring a costimulatory 4-1BB signaling domain, with suppression rates ranging from 89 ± 8% (1:1, n = 6) to 42 ± 15% (1:8, n = 3). Further, in contrast to the aforementioned assay using TCR-activated Tregs, the responsiveness of UniCAR BB/ζ-armed Tconvs to Treg-induced immunosuppression was comparable to that of control Tconvs expressing a UniCAR ζ construct (99 ± 2% (1:1, n = 6) to 36 ± 30% (1:8, n = 3)). On the other hand, UniCAR 28/ζ-engrafted Tconvs were only affected by UniCAR-redirected activated Tregs at a 1:1 ratio (37 ± 24%, n = 6), but not influenced upon coculture with less amounts of Tregs (Figure 4(a), right panel). Overall, in comparison to UniCAR ζ- and UniCAR BB/ζ-mediated stimulation, Tconv activation via a combined 28/ζ signaling domain conferred a significant resistance to immunosuppressive Tregs (Figure 4(b)). To further support the aforementioned findings, suppression of Tconv proliferation by TCR- or UniCAR BB/ζ-activated Tregs was analyzed. As shown in Supplementary Fig. 2 and Supplementary Fig. 3, proliferation of UniCAR ζ- and UniCAR BB/ζ-modified Tconvs was more effectively impaired by stimulated Tregs as compared to UniCAR 28/ζ-armed cells, clearly supporting superiority of the latter.
Figure 4.
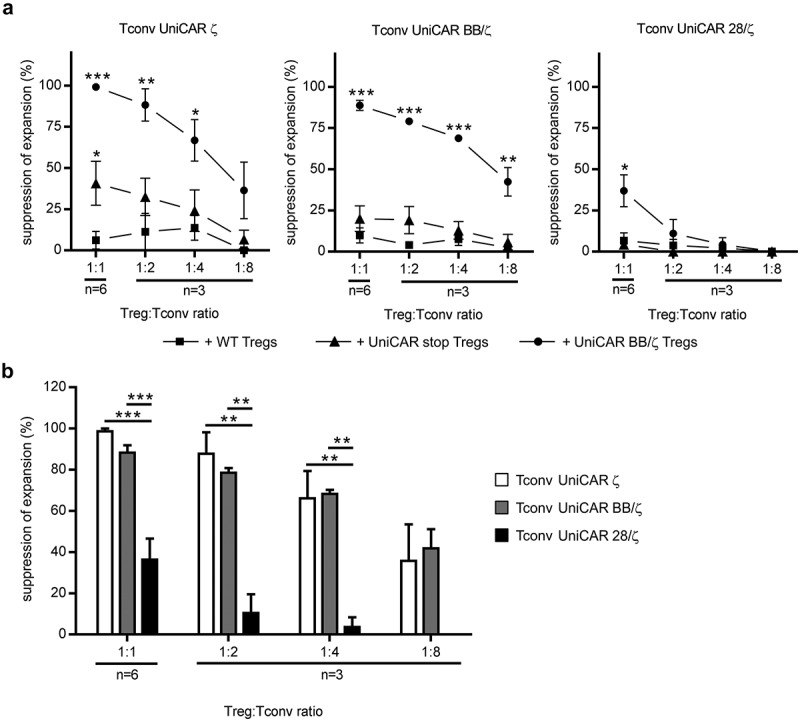
Responsiveness of UniCAR-armed Tconvs to immunosuppression by autologous UniCAR-activated Tregs. 0.5 × 104 eFluor670-stained, UniCAR-engrafted Tconvs were cocultured with autologous, eFluor450-labeled Tregs expressing the UniCAR BB/ζ at indicated Treg:Tconv ratios. As control, either non-transduced (wild type, WT) or UniCAR stop-endowed Tregs were used. Cocultivation was performed in the presence of PC3-PSCA cells at a 5:1 T cell to target cell ratio with or without 6 pmol αPSCA TM for four days. Absolute eFluor670-labeled Tconv numbers were assessed at the endpoint of the assay and percent of suppression was calculated. Data are summarized for three or six individual donors and shown as mean ± SEM in the presence of (a) WT, UniCAR stop- or (a,b) UniCAR BB/ζ-armed Tregs. Statistical significance was determined by 1-way ANOVA with Bonferroni post hoc test (*p < .05, **p < .01, ***p < .001, (a) with respect to control with WT Tregs).
Responsiveness of UniCAR-armed Tconvs to Treg suppression in vivo
So far, the in vitro assays clearly underline that UniCAR 28/ζ-engrafted Tconvs are less efficiently suppressed by Tregs than Tconvs armed with a UniCAR BB/ζ construct. Yet, it remains to be explored whether UniCAR 28/ζ-endowed Tconvs show a better performance in the presence of Tregs also in vivo. For that reason, we investigated the influence of immunosuppressive Tregs on the anti-tumor activity of UniCAR-modified Tconvs in athymic NMRInu/nu mice (Figure 5). To that end, animals were injected with luciferase-expressing PC3-PSCA cells either alone or together with PSCA-redirected Tconvs harboring a UniCAR with costimulatory 4-1BB or CD28 signaling domain. In line with the experimental concept of others,36 we aimed to mimic a hostile, immunosuppressive tumor microenvironment in two groups of mice by co-administration of UniCAR-expressing Tregs at a 1:1 Treg to Tconv ratio. Subsequently, survival of tumor cells was monitored over three weeks by measuring bioluminescence signal. As expected, tumor burden continuously increased in the control group receiving only PC3-PSCA cells (Figure 5). In mice transplanted with UniCAR-endowed Tconvs and the αPSCA TM, luciferase activity was massively reduced, proving that gene-modified UniCAR Tconvs lyse PC3-PSCA cells and completely abolish tumor outgrowth independently of the intracellular domain used for costimulation. However, anti-tumor response of UniCAR BB/ζ-engineered Tconvs was strongly impaired upon administration of immunosuppressive Tregs. By contrast, UniCAR-redirected Tregs were not capable of impeding the tumor eradication activity of UniCAR 28/ζ-engrafted Tconvs verifying that the latter are more resistant to Treg-mediated suppression also in vivo.
Figure 5.
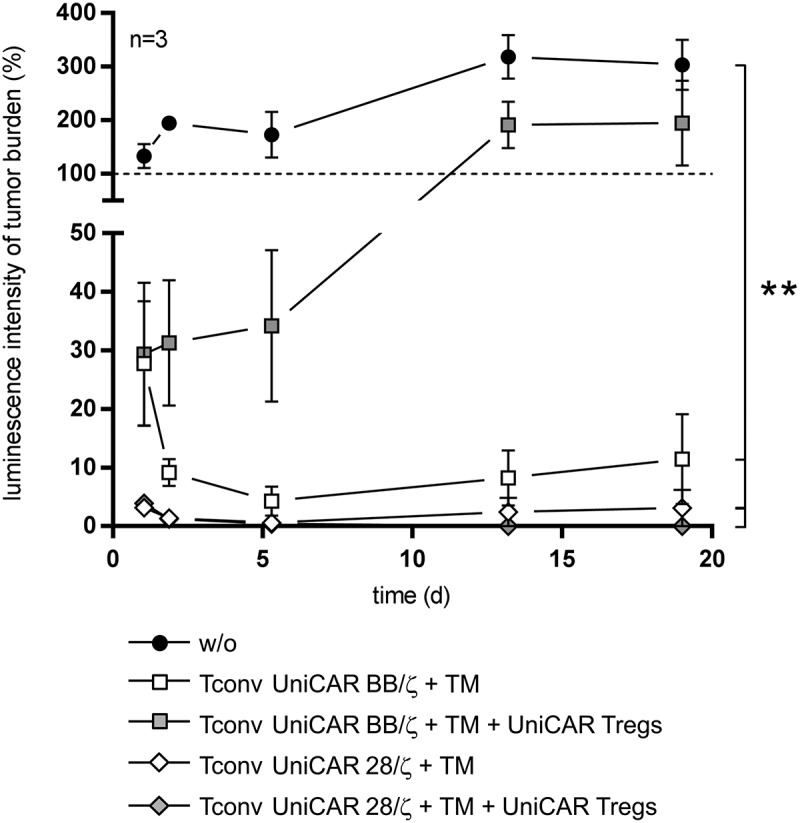
In vivo inhibition of UniCAR-equipped Tconvs by autologous UniCAR-activated Tregs. Athymic NMRInu/nu mice were injected s.c. with luciferase-expressing PC3-PSCA cells either alone or in the presence of αPSCA TM and UniCAR-armed Tconvs containing the costimulatory signaling domain of 4-1BB or CD28. For immunosuppression, autologous UniCAR-engrafted Tregs were added in addition at a 1:1 Treg to Tconv ratio in two groups of mice. To monitor tumor outgrowth, bioluminescence signal of anesthetized mice (n = 3 per group) was assessed 10–15 min after i.p. inoculation of D-luciferin potassium salt for the following 19 days. Values (photons/s/mm2) were correlated to that of day 0. The in vivo experiment was performed in parallel with previously published results. Data are shown as mean ± SEM. Statistical significance was calculated by 1-way ANOVA with Bonferroni post hoc test (**p < .01). The presented data for UniCAR 28/ζ-engrafted Tconvs were confirmed in an independent set of experimental mice (n = 3).
Cytokine production profile of UniCAR-expressing Tconvs in the absence or presence of Tregs
The obtained data unambiguously demonstrate a superior functionality of UniCAR 28/ζ-endowed Tconvs over UniCAR BB/ζ-transduced cells in the face of Treg-mediated suppression. As it has already been published by several groups that pro-inflammatory challenge negatively affects Treg stability and function,37–40 we wanted to clarify whether UniCAR signaling via the 28/ζ domain enhances pro-inflammatory cytokine secretion which in turn might account for the observed resistance to Tregs. In order to shed light on this issue, cytokine amounts were analyzed in supernatants of Tconvs that were stimulated via their UniCAR (ζ, BB/ζ or 28/ζ) using a cross-linking αPSCA TM. Alternatively, conventional αCD3/CD28-coated beads were used for Tconv activation via the endogenous TCR (Figure 6(a)). Expectedly, both stimulation types caused a substantial release of Th1-related cytokines IFN-γ, TNF, GM-CSF, and IL-2. For cells transduced with the UniCAR 28/ζ construct, amounts of pro-inflammatory cytokines (IFN-γ, TNF and GM-CSF) triggered via TCR or UniCAR stimulation were comparable, whereas the latter resulted in significantly elevated IL-2 levels. By contrast, UniCAR BB/ζ-armed Tconvs produced similar IL-2 amounts upon TCR or CAR activation, whilst TNF levels were markedly lower upon TM-compared to bead-mediated redirection. Thereby, the cytokine secretion profile of UniCAR BB/ζ-engrafted Tconvs rather resembles the one of control Tconvs expressing a first generation CAR without an additional costimulatory signal. When directly comparing cytokine concentrations triggered by the individual second-generation UniCARs, considerable differences could be detected. As depicted in Figure 6(b), analysis of seven independent T cell donors revealed that UniCAR 28/ζ-engrafted Tconvs produce significantly higher amounts of all tested cytokines (IFN-γ, TNF, GM-CSF, and IL-2) than UniCAR BB/ζ-expressing cells.
Figure 6.
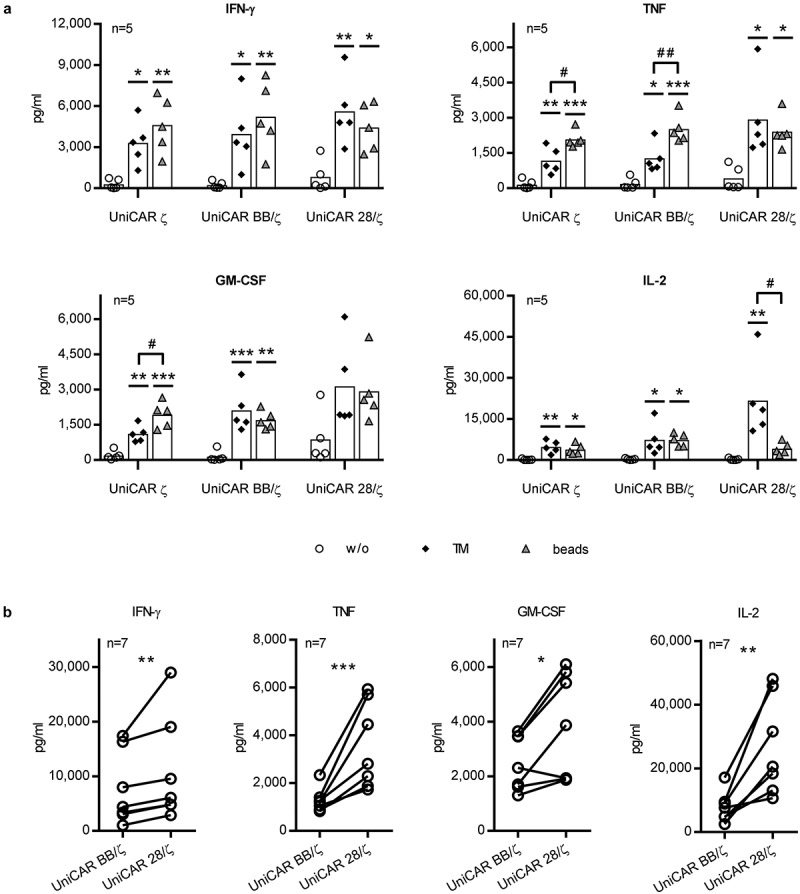
Cytokine secretion profile of UniCAR-equipped Tconvs. UniCAR-transduced Tconvs were incubated with PC3-PSCA cells (effector to target cell ratio of 5:1) in the absence or presence of 6 pmol αPSCA TM. For T cell stimulation via the endogenous TCR, αCD3/CD28 beads were added. After 24 h, cell-free supernatants were harvested and analyzed using ELISA or bead-based flow cytometry analysis. Concentration of predominantly secreted cytokines (IFN-γ, TNF, GM-CSF, and IL-2) are shown. Statistical significance was assessed by (a) 1-way ANOVA with Bonferroni post hoc test or (b) ratio paired, two-tailed Student’s t-test for five and seven individual donors, respectively (*p < .05, **p < .01, ***p < .001, (a) with respect to control without any stimulus; #p < .05, ##p < .01).
In light of these results, it can be reasoned that UniCAR 28/ζ-armed Tconvs create a strong pro-inflammatory milieu, which in turn contributes to a reduced responsiveness to Treg immunosuppression. Yet, since Tregs are known to interfere with cytokine secretion of responder cells,41,42 cytokine levels in cocultures of Tregs and Tconvs remained to be analyzed in order to corroborate our hypothesis. As illustrated in Figure 7, neither TCR- nor UniCAR BB/ζ-activated Tregs were capable of completely inhibiting cytokine release of cocultured Tconvs. In comparison to cocultures containing UniCAR BB/ζ- or UniCAR ζ-engrafted Tconvs, elevated levels of IFN-γ, TNF, GM-CSF, and IL-2 were measured in supernatants containing UniCAR 28/ζ-armed Tconvs, which is especially pronounced at lower Treg to Tconv ratios (Figure 7(a)). Similar results were obtained when using UniCAR BB/ζ-redirected instead of pre-activated, bead-stimulated Tregs as suppressor cells. As summarized for three different donors in Figure 7(b), concentrations of all four cytokines were higher in the presence of UniCAR 28/ζ-expressing Tconvs than in conditions containing UniCAR BB/ζ-engrafted cells, thus, supporting our assumption that costimulatory CD28 signaling creates an enhanced pro-inflammatory milieu obstructing Treg suppression.
Figure 7.
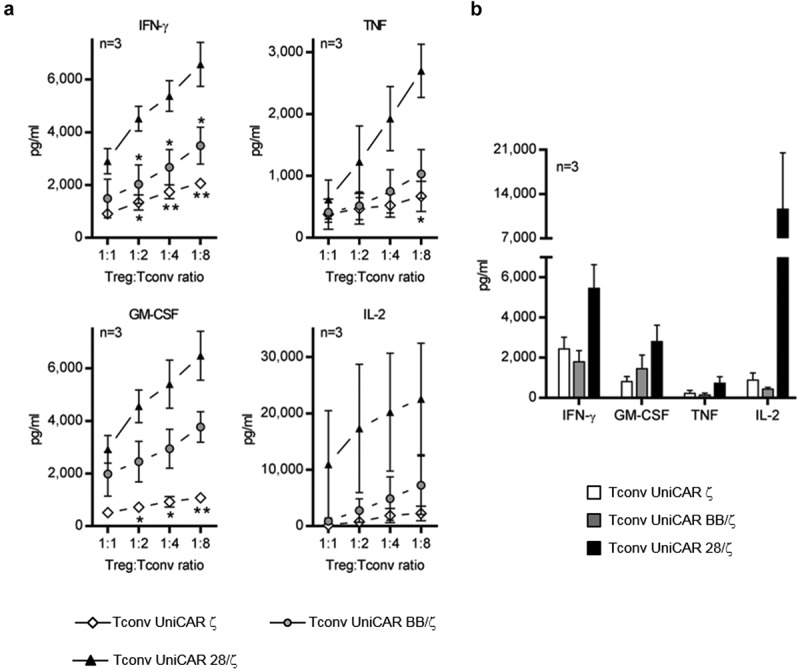
Analysis of cytokines in cocultures of UniCAR-armed Tconvs and autologous Tregs. Cocultivation of UniCAR-endowed Tconvs and (a) TCR- or (b) UniCAR-activated Tregs was performed in the presence of 6 pmol αPSCA TM (a) at indicated Treg:Tconv ratios or (b) at a 1:1 ratio of both T cell populations. PC3-PSCA cells were added at a 5:1 UniCAR T cell to target cell ratio. After 48 h, cell-free supernatants were collected and analyzed for cytokine amounts using a MACSPlex Cytokine Kit. Concentration of indicated cytokines are shown as mean ± SEM for three individual donors. Statistical significance was calculated between individual UniCAR constructs using 1-way ANOVA with Bonferroni post hoc test (*p < .05, **p < .01, (a) with respect to UniCAR 28/ζ-engrafted Tconvs).
Discussion
In B cell-derived malignancies, 28/ζ- and BB/ζ-based αCD19 CAR therapies reached extraordinary clinical results culminating in the recent approval of two CAR T cell products for cancer patients.1,4 Both second-generation CAR formats were extensively examined in several comparative, pre-clinical in vitro and in vivo studies demonstrating for instance that 4-1BB signaling facilitates mitochondrial biogenesis, reprogramming towards a central memory T cell phenotype and increased in vivo persistence, whereas incorporation of CD28 into the CAR architecture promotes glycolysis, effector memory maturation, rapid tumor eradication but also exhaustion.43–45 However, to our best knowledge there is so far no data available directly comparing the responsiveness of CAR 28/ζ- and CAR BB/ζ-armed human Tconvs to Treg suppression. Especially in light of a CAR-based T cell therapy for solid tumors, identification of an intracellular signal fostering highly effective Tconvs that are able to escape strong Treg-mediated inhibition would be of greatest importance. In that regard, Loskog et al. demonstrated in vitro superiority of αCD19 CAR 28/ζ-armed Tconvs over cells engrafted with a first-generation CAR in the presence of Tregs or the recombinant cytokines IL-10 and TGF-β both known to be naturally secreted by these suppressor cells.35 These data, at least in part, were confirmed by Koehler and colleagues. They show that TGF-β substantially hampers CAR ζ-modified Tconvs with specificity for carcinoembryonic antigen (CEA), whereas αCEA CAR 28/ζ-engrafted cells are less affected.46 More recently, an in vitro comparison between Tconvs transduced with different αCEA CARs revealed that TGF-β-mediated inhibition of proliferation can be overcome by CD28/ζ but not by BB/ζ or ζ signaling alone.47
In the present study, we are now demonstrating that UniCAR 28/ζ-engrafted Tconvs clearly outcompete UniCAR BB/ζ-modified cells upon exposure to both TCR- and UniCAR-activated Tregs in vitro. We were further able to corroborate these findings in experimental mice, in which UniCAR-modified Tregs were co-transferred with Tconvs in order to mimic a hostile tumor microenvironment of solid cancers being heavily infiltrated with these suppressor cells. In that regard, outgrowth of tumor cells mirrors strong immunosuppression of UniCAR BB/ζ-armed Tconvs by Tregs, whereas Tconvs overcome inhibition and vigorously eradicate tumor cells when integrating CD28 instead of 4-1BB into the UniCAR architecture. In accordance with the concept of others,36 we injected all cellular components s.c., as this experimental setup allows for a good control over all parameters like Tconv to tumor cell ratio or responder to suppressor cell ratio, thereby minimizing confounding effects and ensuring little variability between individual animals and groups. Nonetheless, s.c. inoculation exhibits some limitations as for instance migratory behavior and infiltration of genetically modified Tconvs into the tumor site cannot be studied. Moreover, co-injected Tregs and Tconvs are stimulated simultaneously, whereas an established tumor microenvironment harbors highly pre-activated Tregs. Nevertheless, the applied mouse model provides a first insight how UniCAR-endowed Tconvs might perform in vivo upon immunosuppressive exposure.
In line with previously published data,35 control Tconvs engrafted with a UniCAR ζ lacking any costimulatory signal were most compromised by Treg-mediated suppression. This is reflected by profound reduction of their expansion potential in the presence of TCR-activated Tregs, which significantly differs from that of both second-generation UniCAR-modified Tconvs. In addition, impairment of first-generation UniCAR-armed Tconvs was even detectable in cocultures with UniCAR stop-engrafted Tregs. As previously published by our group, Tregs equipped with a UniCAR stop construct are not activated upon TM-mediated cross-linkage to target cells.23 Consequently, close proximity of responder and suppressor cells was already sufficient to provoke significant inhibition of UniCAR ζ-modified Tconvs underlining their high responsiveness to Treg suppression. Conflicting data, however, have been provided by Kofler et al. as they show that engraftment with an αCEA CAR ζ results in elevated in vivo anti-tumor activity of transduced Tconvs in the presence of Tregs in contrast to CARs incorporated with an additional CD28 endodomain.36 We reason that a number of substantial experimental differences including targeted antigen, CAR architecture, Treg population used for suppression and activation status of these cells might be responsible for the observed discrepancies to our findings.
Within our experiments, UniCAR 28/ζ-endowed Tconvs produce significantly greater amounts of IL-2 as compared to cells engrafted with a UniCAR BB/ζ. Consumption of IL-2 as well as transcriptional repression of IL-2 gene expression represents an important regulatory mechanism by which Tregs can efficiently interfere with proliferation of activated effector cells.41,42 Thus, vast amounts of IL-2 secreted by UniCAR 28/ζ-modified Tconvs might serve as an efficient countermeasure to this Treg-mediated suppression mechanism. In line with this assumption, several groups demonstrate that addition of exogenous IL-2 to suppression assays abrogates Treg inhibition.38,41,48 Moreover, UniCAR 28/ζ-engrafted Tconvs secrete significantly higher levels of pro-inflammatory cytokines (IFN-γ, GM-CSF, and TNF) than UniCAR BB/ζ-transduced cells. These data indicate that costimulation via a CD28 endodomain results in stronger activation of effector functions, thereby fostering an inflammatory milieu and helping to evade Treg-mediated control. This hypothesis is supported by previously published data demonstrating that hyperactivation of the phosphatidylinositol-3-kinase/Akt pathway, which is potently induced by CD28 signaling, decreases responsiveness of Tconvs to Treg inhibition.49–51 In accordance with these data, it was proven that addition of an agonistic anti-CD28 antibody to in vitro suppression assays abrogates Treg influence on cocultured Tconvs.40,41
Nevertheless, we cannot rule out the possibility that ligand-independent tonic signaling contributes to the observed disparities between UniCAR BB/ζ- and UniCAR 28/ζ-armed Tconvs. As various studies have revealed that pre-activated or memory T cells can less efficiently be suppressed by Tregs,52–54 sustained signaling via the engrafted 28/ζ endodomain might facilitate an enhanced resistance to Treg inhibition. Given that tonic signaling is influenced by multiple factors55 and conflicting data exist in terms of the effects of CD28 vs. 4-1BB costimulation,45,56–58 we cannot draw any conclusion at present regarding the UniCAR constructs utilized in this study. Hence, the influence of tonic signaling on Tconv responsiveness to Tregs remains to be addressed in future experiments.
Taken together, our data clearly underline that arming Tconvs with a UniCAR 28/ζ conveys enhanced capability to escape Treg control and, therefore, this construct proves to be preferentially suited for treatment of solid cancers. On the other hand, escape from Treg regulation might be accompanied by a higher risk for unwanted side effects leading to severe or even life-threatening toxicities. This potential safety hazard could, however, easily be overcome by our switchable UniCAR platform technology which allows for a precisely controlled activation of highly potent 28/ζ-engrafted T cells. Consequently, UniCAR 28/ζ-armed Tconvs combine both increased safety and vigorous anti-tumor efficiency despite the presence of strongly activated Tregs.
Materials and methods
Cell lines
All cell lines were obtained from the American Type Culture Collection (ATCC). The prostate cancer cell line PC3 (ATCC CRL-1435), transduced with the tumor-specific antigen PSCA as previously published,59 was cultured in RPMI 1640 supplemented with 10% FCS, 1% non-essential amino acids, 100 µg/ml penicillin/streptomycin, 2 mM N-acetyl-L-alanyl-L-glutamine and 1 mM sodium pyruvate (complete RPMI 1640 medium, all obtained from Biochrom, Berlin, Germany). Human embryonic kidney cells HEK293T (ATCC CRL-11268) used for virus particle production and murine fibroblast cells 3T3 (ATCC CRL-1658) genetically modified to express the soluble αPSCA TM were kept in DMEM (Invitrogen, ThermoFisher Scientific, Schwerte, Germany) completed with 10% FCS, 1% non-essential amino acids and 100 µg/ml penicillin/streptomycin. Cell culture was performed at 37°C in a humidified atmosphere with 5% CO2.
Production and purification of the αPSCA TM
Design, production, and purification of the αPSCA TM were already reported elsewhere.18,23,60 Briefly, 2 × 106 TM-expressing 3T3 cells were seeded in T175 cell culture flasks and supernatant was harvested after 96 h. His-tagged recombinant proteins were purified by Ni-NTA affinity chromatography (Qiagen, Hilden, Germany). After two consecutive washing steps, column-bound αPSCA TM was eluted with ice-cold PBS containing 350 mM imidazole and 150 mM NaCl. Dialysis was performed overnight against PBS at 4°C. Purity and concentration of the αPSCA TM were determined after SDS-PAGE and Coomassie brilliant blue G250 staining61 using Image Lab™ software (Bio-Rad Laboratories GmbH, Munich, Germany).
Structural design of UniCAR constructs
Generation and detailed structure of UniCAR 28/ζ, UniCAR ζ, UniCAR BB/ζ, and UniCAR stop were previously described.17,23 In brief, all constructs contain extracellularly an αLa 5B9 single-chain fragment variable62 followed by a short peptide epitope tag (E7B6) for UniCAR identification on the surface of transduced cells.17 Hinge and transmembrane region are derived from human CD28. Intracellularly, UniCARs possess either no signaling chain (UniCAR stop), a human CD3ζ signaling domain in isolation (UniCAR ζ) or, in addition, a costimulatory signaling unit of human CD28 (UniCAR 28/ζ) or human 4-1BB (UniCAR BB/ζ). As marker gene EGFP is fused to the C-terminal UniCAR sequence separated by a 2A protease ‘cleavage’ site, allowing a separate translation of the two reading frames according to a ribosomal skip mechanism.63
T cell isolation and expansion
The study was approved by the local ethics committee of the university hospital of the medical faculty of “Carl Gustav Carus” TU Dresden (EK27022006). Virus particle production was performed as published elsewhere.64,65 CD4+CD25− Tconvs and CD4+CD25+CD127dimCD45RA+ Tregs were isolated, cultured and transduced with the respective UniCAR construct as previously outlined in detail.18,19,23,66 In brief, peripheral blood mononuclear cells were obtained from buffy coats (German Red Cross, Dresden, Germany) by Ficoll gradient centrifugation with informed consent of healthy donors. Enrichment of CD4+ T cells was performed using a human CD4+ T cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) yielding a purity of 96.5% ± 1.9% CD3+CD4+ cells (n = 7). After magnetic labeling with human CD25+ Microbeads II (Miltenyi Biotec), CD4+CD25− Tconvs were obtained as untouched cells in the flow through. Eluted CD4+CD25+ T cells were subsequently sorted on a FACSAria™ Fusion (BD Biosciences Pharmingen, Heidelberg, Germany) to gain highly pure CD4+CD25+CD127dimCD45RA+ Tregs. After isolation, both Tconvs and Tregs were activated with DynaBeads® Human T-Activator CD3/CD28 (Invitrogen, ThermoFisher Scientific) and transduced with lentiviral particles on day 1–4 by adding concentrated virus supernatant. Beads were removed from Tconvs after 72 h. One day prior to experiments, Tconvs were maintained in complete RPMI 1640 medium without any exogenously added cytokines. Regarding Treg cultures, cells were restimulated with αCD3/CD28 DynaBeads® at a 1:2 bead to T cell ratio on day four or five for 48 h to 72 h. After bead removal, Tregs were rested for 2 to 3 days prior to assays and characterized regarding FOXP3, EGFP and UniCAR expression (Supplementary Fig. 1) as well as IL-10 secretion and latency-associated peptide (LAP) upregulation (Supplementary Fig. 4).
Flow cytometry
Cell surface staining for molecules of interest was performed with monoclonal antibodies (mAbs) directed against CD3 (BW264/56), CD4 (VIT4), CD25 (4E3), CD45RA (T6D11), CD127 (MB15-18C9) and IgG1 (isotype control, IS5-21F5) all purchased from Miltenyi Biotec as well as LAP (TW4-6H10) obtained from BioLegend (London, UK). To verify UniCAR expression on the T cell surface, an αLa 7B6 mAb67 targeting the epitope tag E7B6 was applied which was detected with a fluorochrom-conjugated goat-α-mouse IgG F(ab´)2 antibody (Immunotech Beckman Coulter, Krefeld, Germany). For identification of intracellular FOXP3, cells were incubated with a Zombie Red™ dye (Zombie Red™ Fixable Viability Kit, BioLegend) allowing live-dead cell discrimination, further processed with a FoxP3 Staining Buffer Set (Miltenyi Biotec) and finally stained with a mAb against FOXP3 (259D) or the respective isotype control IgG1 (MOPC-21) (both from BioLegend).adapted from68 Flow cytometry data were acquired on a MACSQuant® Analyzer and evaluated using MACSQuantify® software (Miltenyi Biotec).
In vitro suppression assay
To discriminate the individual T cell populations within suppression assays, CD4+CD25+CD127lowCD45RA+ Tregs were labeled with 10 µM cell proliferation dye eFluor™450 (eBioscience, ThermoFisher Scientific), whereas CD4+CD25− Tconvs were stained with 5 µM cell proliferation dye eFluor™670 (eBioscience, ThermoFisher Scientific) according to the manufacturers protocol. One day later, 5 × 104 Tconvs were cocultured with PC3-PSCA cells at a 5:1 T cell to target cell ratio either in the absence (unstim.) or in the presence (stim.) of 6 pmol αPSCA TM. To investigate responsiveness of UniCAR-armed Tconvs to Treg suppression, expanded autologous TCR- or UniCAR-activated CD4+CD25+CD127lowCD45RA+ Tregs were added. For TCR stimulation, wild type (WT) Treg cells were incubated with αCD3/CD28 DynaBeads® at a 1:1 ratio for 24 h. Subsequently, beads were removed and TCR-activated WT Tregs were added at indicated Treg to Tconv ratios. Non-activated WT Tregs served as control. To mimic antigen-specific Treg stimulation, UniCAR BB/ζ-engrafted Tregs were cultured together with Tconvs at indicated ratios. As control, WT Tregs and UniCAR stop-endowed Tregs were applied. To maintain a 5:1 T cell to target cell ratio for cross-linkage, the respective amount of PC3-PSCA cells was accordingly supplemented into each well. All samples were analyzed as triplicates. After 96 h, absolute Tconv numbers were determined using a MACSQuant® Analyzer as previously explained.23 In brief, cultures were resuspended carefully and an aliquot of 20 µl was transferred to 80 µl propidium iodide (1 µg/ml, Invitrogen) immediately before MACSQuant® measurement. To determine number of living cells/ml, dead cells and cell debris were excluded based on size, granularity, and propidium iodide staining.
Proliferation of eFluor670-labeled, UniCAR-armed Tconvs was determined on the basis of eFluor670 dilution over time. Division index of Tconvs was calculated using FlowJo 9.9.6 software (TreeStar, Ashland, USA). Percentage of Tconv suppression by Tregs was calculated as follows: 100-[(sample-unstim.)/(stim.-unstim.)] x 100. Values lower than 0 or higher than 100 were set to 0% or 100%, respectively.
Responsiveness to Treg suppression in vivo
The animal experiment was performed with 8-week-old male NMRI-Foxn1nu/Foxn1nu mice (Janvier Labs, St. Berthevin, France) at the Helmholtz-Zentrum Dresden-Rossendorf according to the guidelines of German Regulations of Animal Welfare and was approved by the local authorities (Landesdirektion Dresden, 24–9165.40–4, 24.9168.21–4/2004–1). Luciferase-expressing PC3-PSCA cells (1 x 106) were injected s.c. into the right flank of experimental mice either alone or in the presence of 10 µg αPSCA TM and 1 × 106 UniCAR BB/ζ- or UniCAR 28/ζ-armed Tconvs. To investigate responsiveness to Treg suppression, 1 × 106 UniCAR BB/ζ-endowed Tregs were additionally added in two groups of mice. As reference value bioluminescence signal was determined 2 h after cell injection. For that purpose, mice were narcotized as described elsewhere.19 Next, 200 µl D-luciferin potassium salt (15 mg/ml) (ThermoFisher Scientific) was inoculated i.p. and the bioluminescence signal was measured by using an In-Vivo-Xtreme imaging system 10–15 min later (exposure time of 60 s, Bruker, Germany). Tumor burden was assessed over 19 days. Resulting data were quantified as previously published23 by using Bruker MI SE software (Bruker, Germany) and correlated to that of day 0.
Detection of cytokine levels
For analysis of cytokine production profiles of Tconvs or Tregs, cell-free supernatants were collected after 24 h or 48 h of cocultures of 2*104 PC3-PSCA cells and 1*105 UniCAR-endowed Tcells (ratio 5:1) being either non-stimulated, stimulated via 6 pmol αPSCA TM or via αCD3/CD28 DynaBeads® (1:5 bead to cell ratio). For assessment of cytokine secretion in the presence of Tregs, cell-free supernatants were harvested from suppression assays after 48 h. After storage at −80°C, analysis of human cytokines was performed using OptEIA™ Human ELISA Kits (BD Biosciences Pharmingen) or bead-based flow cytometry (MACSPlex Cytokine 12 Kit, Miltenyi Biotec) according to the manufacturer’s instructions.
Statistical analysis
Statistical tests were performed as indicated in figure legends by using GraphPad Prism 7 software (GraphPad software Inc., La Jolla, CA, USA). P values of <0.05 were considered as statistically significant.
Funding Statement
This work was supported by grants provided to MBP including those by the German government Bundesministerium für Bildung und Forschung (BMBF) [01EK1513 (TurbiCAR)].
Acknowledgments
The authors thank Julia Lagler for excellent technical assistance.
Author contributions
AK and SK conceived and conducted all experiments, analyzed the data and wrote the manuscript. RB and NB performed the animal imaging. CA and AF cloned and established the TM. AH, MB and MS provided valuable input and materials. MPB secured funding, contributed to data interpretation, critically reviewed the manuscript and supervised the research.
Disclosure and potential conflicts of interest
MPB is the inventor of the UniCAR platform and is a shareholder of the company GEMoaB which holds licenses, patents and has filed patent applications related to the UniCAR platform. The remaining authors declare that they have no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Brudno JN, Kochenderfer JN.. Chimeric antigen receptor T-cell therapies for lymphoma. Nat Rev Clin Oncol. 2018;15(1):31–46. doi: 10.1038/nrclinonc.2017.128. [DOI] [PubMed] [Google Scholar]
- 2.Sadelain M, Rivière I, Riddell S.. Therapeutic T cell engineering. Nature. 2017;545(7655):423–431. doi: 10.1038/nature22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cartellieri M, Bachmann M, Feldmann A, Bippes C, Stamova S, Wehner R, Temme A, Schmitz M. Chimeric antigen receptor-engineered T cells for immunotherapy of cancer. J Biomed Biotechnol. 2010;2010:956304. doi: 10.1155/2010/956304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pang Y, Hou X, Yang C, Liu Y, Jiang G. Advances on chimeric antigen receptor-modified T-cell therapy for oncotherapy. Mol Cancer. 2018;17(1):91. doi: 10.1186/s12943-018-0840-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Ö A, Brogdon JL, Pruteanu-Malinici I, Bhoj V, Landsburg D, et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N Engl J Med. 2017;377(26):2545–2554. doi: 10.1056/NEJMoa1708566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, Yeung C, Liles WC, Wurfel M, Lopez JA, et al Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. 2017;7(12):1404–1419. doi: 10.1158/2159-8290.CD-17-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, López JA, Chen J, Chung D, Harju-Baker S, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood. 2017;130(21):2295–2306. doi: 10.1182/blood-2017-06-793141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, Halton E, Wang X, Senechal B, Purdon T, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. 2018. August;8(8):958–971. doi: 10.1158/2159-8290.CD-17-1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reuters Juno ends development of high-profile leukemia drug after deaths. Reuters; 2017. March 01 [accessed 2018 October19]. https://www.reuters.com/article/us-juno-leukemia/juno-ends-development-of-high-profile-leukemia-drug-after-deaths-idUSKBN1685QQ.
- 12.OncLive Kite reports cerebral Edema Death in ZUMA-1 CAR T-cell trial. OncLive; 2017. May 08 [accessed 2018 October19]. https://www.onclive.com/web-exclusives/kite-reports-cerebral-edema-death-in-zuma1-car-tcell-trial.
- 13.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19(3):620–626. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24(13):e20–22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 16.Koristka S, Cartellieri M, Feldmann A, Arndt C, Loff S, Michalk I, Aliperta R, von Bonin M, Bornhäuser M, Ehninger A, et al. Flexible antigen-specific redirection of human regulatory T cells via a novel universal chimeric antigen receptor system. Blood. 2014;124(21):3494. [Google Scholar]
- 17.Cartellieri M, Feldmann A, Koristka S, Arndt C, Loff S, Ehninger A, von Bonin M, Bejestani EP, Ehninger G, Bachmann MP. Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016;6(8):e458. doi: 10.1038/bcj.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feldmann A, Arndt C, Bergmann R, Loff S, Cartellieri M, Bachmann D, Aliperta R, Hetzenecker M, Ludwig F, Albert S, et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology “UniCAR”. Oncotarget. 2017;8(19):31368–31385. doi: 10.18632/oncotarget.15572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Albert S, Arndt C, Feldmann A, Bergmann R, Bachmann D, Koristka S, Ludwig F, Ziller-Walter P, Kegler A, Gärtner S, et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. Oncoimmunology. 2017;6(4):e1287246. doi: 10.1080/2162402X.2017.1287246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitwasi N, Feldmann A, Bergmann R, Berndt N, Arndt C, Koristka S, Kegler A, Jureczek J, Hoffmann A, Ehninger A, et al. Development of novel target modules for retargeting of UniCAR T cells to GD2 positive tumor cells. Oncotarget. 2017;8(65):108584–108603. doi: 10.18632/oncotarget.21017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bachmann D, Aliperta R, Bergmann R, Feldmann A, Koristka S, Arndt C, Loff S, Welzel P, Albert S, Kegler A, et al. Retargeting of UniCAR T cells with an in vivo synthesized target module directed against CD19 positive tumor cells. Oncotarget. 2017;9(7):7487–7500. doi: 10.18632/oncotarget.23556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albert S, Arndt C, Koristka S, Berndt N, Bergmann R, Feldmann A, Schmitz M, Pietzsch J, Steinbach J, Bachmann M. From mono- to bivalent: improving theranostic properties of target modules for redirection of UniCAR T cells against EGFR-expressing tumor cells in vitro and in vivo. Oncotarget. 2018;9(39):25597–25616. doi: 10.18632/oncotarget.25390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koristka S, Kegler A, Bergmann R, Arndt C, Feldmann A, Albert S, Cartellieri M, Ehninger A, Ehninger G, Middeke JM, et al. Engrafting human regulatory T cells with a flexible modular chimeric antigen receptor technology. J Autoimmun. 2018;90:116–131. doi: 10.1016/j.jaut.2018.02.006. [DOI] [PubMed] [Google Scholar]
- 24.Loureiro LR, Feldmann A, Bergmann R, Koristka S, Berndt N, Arndt C, Pietzsch J, Novo C, Videira P, Bachmann M. Development of a novel target module redirecting UniCAR T cells to Sialyl Tn-expressing tumor cells. Blood Cancer J. 2018;8(9):81. doi: 10.1038/s41408-018-0113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lamers CH, Klaver Y, Gratama JW, Sleijfer S, Debets R. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells-a completed study overview. Biochem Soc Trans. 2016;44(3):951–959. doi: 10.1042/BST20160037. [DOI] [PubMed] [Google Scholar]
- 26.Feng K, Guo Y, Dai H, Wang Y, Li X, Jia H, Han W. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci. 2016;59(5):468–479. doi: 10.1007/s11427-016-5023-8. [DOI] [PubMed] [Google Scholar]
- 27.Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, Meechoovet HB, Bautista C, Chang WC, Ostberg JR, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15(4):825–833. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 28.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2(2):112–120. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang BL, Qin DY, Mo ZM, Li Y, Wei W, Wang YS, Wang W, Wei YQ. Hurdles of CAR-T cell-based cancer immunotherapy directed against solid tumors. Sci China Life Sci. 2016;59(4):340–348. doi: 10.1007/s11427-016-5027-4. [DOI] [PubMed] [Google Scholar]
- 30.Shitara K, Nishikawa H. Regulatory T cells: a potential target in cancer immunotherapy. Ann N Y Acad Sci. 2018;1417(1):104–115. doi: 10.1111/nyas.13625 Epub 2018 Mar 22 [DOI] [PubMed] [Google Scholar]
- 31.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 32.Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, Banham AH. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol. 2006;24(34):5373–5380. doi: 10.1200/JCO.2006.05.9584. [DOI] [PubMed] [Google Scholar]
- 33.Sayour EJ, McLendon P, McLendon R, De Leon G, Reynolds R, Kresak J, Sampson JH, Mitchell DA. Increased proportion of FoxP3+ regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer Immunol Immunother. 2015;64(4):419–427. doi: 10.1007/s00262-014-1651-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flammiger A, Weisbach L, Huland H, Tennstedt P, Simon R, Minner S, Bokemeyer C, Sauter G, Schlomm T, Trepel M. High tissue density of FOXP3+ T cells is associated with clinical outcome in prostate cancer. Eur J Cancer. 2013;49(6):1273–1279. doi: 10.1016/j.ejca.2012.11.035. [DOI] [PubMed] [Google Scholar]
- 35.Loskog A, Giandomenico V, Rossig C, Pule M, Dotti G, Brenner MK. Addition of the CD28 signaling domain to chimeric T-cell receptors enhances chimeric T-cell resistance to T regulatory cells. Leukemia. 2006;20(10):1819–1828. doi: 10.1038/sj.leu.2404366. [DOI] [PubMed] [Google Scholar]
- 36.Kofler DM, Chmielewski M, Rappl G, Hombach A, Riet T, Schmidt A, Hombach AA, Wendtner CM, Abken H. CD28 costimulation Impairs the efficacy of a redirected t-cell antitumor attack in the presence of regulatory t cells which can be overcome by preventing Lck activation. Mol Ther. 2011;19(4):760–767. doi: 10.1038/mt.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, EM Shevach, PE Lipsky. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood. 2006;108(1):253–261. doi: 10.1182/blood-2005-11-4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruprecht CR, Gattorno M, Ferlito F, Gregorio A, Martini A, Lanzavecchia A, Sallusto F. Coexpression of CD25 and CD27 identifies FoxP3+ regulatory T cells in inflamed synovia. J Exp Med. 2005;201(11):1793–1803. doi: 10.1084/jem.20050085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nie H, Zheng Y, Li R, Guo TB, He D, Fang L, Liu X, Xiao L, Chen X, Wan B, et al. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-α in rheumatoid arthritis. Nat Med. 2013;19(3):322–328. doi: 10.1038/nm.3085. [DOI] [PubMed] [Google Scholar]
- 40.van Amelsfort JM, van Roon JA, Noordegraaf M, Jacobs KM, Bijlsma JW, Lafeber FP, Taams LS. Proinflammatory mediator-induced reversal of CD4+,CD25+ regulatory T cell-mediated suppression in rheumatoid arthritis. Arthritis Rheum. 2007;56(3):732–742. doi: 10.1002/art.22414. [DOI] [PubMed] [Google Scholar]
- 41.Takahashi T, Kuniyasu Y, Toda M, Sakaguchi N, Itoh M, Iwata M, Shimizu J, Sakaguchi S. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol. 1998;10(12):1969–1980. doi: 10.1093/intimm/10.12.1969. [DOI] [PubMed] [Google Scholar]
- 42.Oberle N, Eberhardt N, Falk CS, Krammer PH, Suri-Payer E. Rapid suppression of cytokine transcription in human CD4+CD25 T cells by CD4+Foxp3+ regulatory T cells: independence of IL-2 consumption, TGF-beta, and various inhibitors of TCR signaling. J Immunol. 2007;179(6):3578–3587. doi: 10.4049/jimmunol.179.6.3578. [DOI] [PubMed] [Google Scholar]
- 43.Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, Patel PR, Guedan S, Scholler J, Keith B, et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity. 2016;44(2):380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 44.Zhao Z, Condomines M, Sjc VDS, Perna F, Kloss CC, Gunset G, Plotkin J, Sadelain M. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of car T cells. Cancer Cell. 2015;28(4):415–428. doi: 10.1016/j.ccell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koehler H, Kofler D, Hombach A, Abken H. CD28 costimulation overcomes transforming growth factor-beta-mediated repression of proliferation of redirected human CD4+ and CD8+ T cells in an antitumor cell attack. Cancer Res. 2007;67(5):2265–2273. doi: 10.1158/0008-5472.CAN-06-2098. [DOI] [PubMed] [Google Scholar]
- 47.Golumba-Nagy V, Kuehle J, Hombach AA, Abken H. CD28-ζ CAR T cells resist TGF-β repression through IL-2 signaling, which can be mimicked by an engineered IL-7 autocrine loop. Mol Ther. 2018;26(9):2218–2230. doi: 10.1016/j.ymthe.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thornton AM, Donovan EE, Piccirillo CA, Shevach EM. Cutting edge: IL-2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J Immunol. 2004;172(11):6519–6523. doi: 10.4049/jimmunol.172.11.6519. [DOI] [PubMed] [Google Scholar]
- 49.CG King, Kobayashi T, PJ Cejas, Kim T, Yoon K, GK Kim, Chiffoleau E, SP Hickman, PT Walsh, LA Turka, et al. TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nat Med. 2006;12(9):1088–1092. doi: 10.1038/nm1449. [DOI] [PubMed] [Google Scholar]
- 50.Wehrens EJ, Mijnheer G, Duurland CL, Klein M, Meerding J, van Loosdregt J, de Jager W, Sawitzki B, Coffer PJ, Vastert B, et al. Functional human regulatory T cells fail to control autoimmune inflammation due to PKB/c-akt hyperactivation in effector cells. Blood. 2011;118(13):3538–3548. doi: 10.1182/blood-2010-12-328187. [DOI] [PubMed] [Google Scholar]
- 51.Ben Ahmed M, Belhadj Hmida N, Moes N, Buyse S, Abdeladhim M, Louzir H, Cerf-Bensussan N. IL-15 renders conventional lymphocytes resistant to suppressive functions of regulatory T cells through activation of the phosphatidylinositol 3-kinase pathway. J Immunol. 2009;182(11):6763–6770. doi: 10.4049/jimmunol.0801792. [DOI] [PubMed] [Google Scholar]
- 52.Wang XN, Haniffa MA, Holtick U, Collin MP, Jackson G, Hilkens CM, Holler E, Edinger M, Hoffmann P, Dickinson AM. Regulatory T-cell suppression of CD8+ T-cell-mediated graft-versus-host reaction requires their presence during priming. Transplantation. 2009;88(2):188–197. doi: 10.1097/TP.0b013e3181ac14ce. [DOI] [PubMed] [Google Scholar]
- 53.Afzali B, Mitchell PJ, Scottà C, Canavan J, Edozie FC, Fazekasova H, Lord GM, John S, Barber LD, Hernandez-Fuentes MP, et al. Relative resistance of human CD4(+) memory T cells to suppression by CD4(+) CD25(+) regulatory T cells. Am J Transplant. 2011;11(8):1734–1742. doi: 10.1111/j.1600-6143.2011.03635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hagness M, Henjum K, Landskron J, Brudvik KW, Bjørnbeth BA, Foss A, Taskén K, Aandahl EM. Kinetics and activation requirements of contact-dependent immune suppression by human regulatory T cells. J Immunol. 2012;188(11):5459–5466. doi: 10.4049/jimmunol.1101367. [DOI] [PubMed] [Google Scholar]
- 55.Ajina A, Maher J. Strategies to address chimeric antigen receptor tonic signaling. Mol Cancer Ther. 2018;17(9):1795–1815. doi: 10.1158/1535-7163.MCT-17-1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, Kawalekar OU, Guedan S, McGettigan SE, Posey AD, et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res. 2015;3(4):356–367. doi: 10.1158/2326-6066.CIR-14-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gomes Da Silva D, Mukherjee M, Srinivasan M, Dakhova O, Liu H, Grilley B, Ap G, Ss N, Cm R, He H, et al. Direct comparison of in vivo fate of second and third-generation CD19-specific chimeric antigen receptor (CAR)-T cells in patients with B-cell lymphoma: reversal of toxicity from tonic signaling. Blood. 2016;128(22):1851. doi: 10.1182/blood-2016-06-724161. [DOI] [Google Scholar]
- 58.Mamonkin M. Gomes da Silva D, Mukherjee M, Sharma S, Srinivasan M, JS Orange, MK Brenner. Tonic 4-1BB Signaling from Chimeric Antigen Receptors (Cars) Impairs Expansion of T Cells Due to Fas-Mediated Apoptosis. J Immunol. 2016;196:143.7. [Google Scholar]
- 59.Feldmann A, Stamova S, Bippes CC, Bartsch H, Wehner R, Schmitz M, Temme A, Cartellieri M, Bachmann M. Retargeting of T cells to prostate stem cell antigen expressing tumor cells: comparison of different antibody formats. Prostate. 2011;71(9):998–1011. doi: 10.1002/pros.21315. [DOI] [PubMed] [Google Scholar]
- 60.Arndt C, Feldmann A, Töpfer K, Koristka S, Cartellieri M, Temme A, Ehninger A, Ehninger G, Bachmann M. Redirection of CD4+ and CD8+ T lymphocytes via a novel antibody-based modular targeting system triggers efficient killing of PSCA+ prostate tumor cells. Prostate. 2014;74(13):1347–1358. doi: 10.1002/pros.22851. [DOI] [PubMed] [Google Scholar]
- 61.Arndt C, Koristka S, Feldmann A, Bergmann R, Bachmann M. Coomassie brilliant blue staining of polyacrylamide gels In: Kurien B, Scofield REditors. Protein Gel Detection and Imaging. Methods in Molecular Biology. Vol. 1853 New York (NY): Humana Press; 2018. p. 27–30. [DOI] [PubMed] [Google Scholar]
- 62.Koristka S, Cartellieri M, Arndt C, Bippes CC, Feldmann A, Michalk I, Wiefel K, Stamova S, Schmitz M, Ehninger G, et al. Retargeting of regulatory T cells to surface-inducible autoantigen La/SS-B. J Autoimmun. 2013;42:105–116. doi: 10.1016/j.jaut.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 63.Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol. 2004;22(5):589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- 64.Cartellieri M, Koristka S, Arndt C, Feldmann A, Stamova S, von Bonin M, Töpfer K, Krüger T, Geib M, Michalk I, et al. A novel ex vivo isolation and expansion procedure for chimeric antigen receptor engrafted human T cells. PLoS One. 2014;9(4):e93745. doi: 10.1371/journal.pone.0093745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morgenroth A, Cartellieri M, Schmitz M, Günes S, Weigle B, Bachmann M, Abken H, Rieber EP, Temme A. Targeting of tumor cells expressing the prostate stem cell antigen (PSCA) using genetically engineered T-cells. Prostate. 2007;67(10):1121–1131. doi: 10.1002/pros.20608. [DOI] [PubMed] [Google Scholar]
- 66.Koristka S, Cartellieri M, Arndt C, Feldmann A, Töpfer K, Michalk I, Temme A, Ehninger G, Bachmann M. Cytotoxic response of human regulatory T cells upon T-cell receptor-mediated activation: a matter of purity. Blood Cancer J. 2014;4:e199. doi: 10.1038/bcj.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bippes CC, Feldmann A, Stamova S, Cartellieri M, Schwarzer A, Wehner R, Schmitz M, Rieber EP, Zhao S, Schäkel K, et al. A novel modular antigen delivery system for immuno targeting of human 6-sulfo LacNAc-positive blood dendritic cells (SlanDCs). PLoS One. 2011;6(1):e16315. doi: 10.1371/journal.pone.0016315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Koristka S, Cartellieri M, Theil A, Feldmann A, Arndt C, Stamova S, Michalk I, Töpfer K, Temme A, Kretschmer K, et al. Retargeting of human regulatory T cells by single-chain bispecific antibodies. J Immunol. 2012;188(3):1551–1558. doi: 10.4049/jimmunol.1101760. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.