

Abstract
Bioengineered dental tissues and whole teeth that exhibit features and properties of natural teeth can functionally surpass currently used artificial dental implants. However, no biologically based alternatives currently exist for clinical applications in dentistry. Here, we describe a newly established bioengineered tooth bud model for eventual applications in clinical dentistry. We also describe methods to fabricate and analyze bioengineered tooth tissues, including cell isolation, in vivo implantation, and post-harvest analyses.
Keywords: Tooth tissue engineering, Hydrogel scaffolds, Primary dental cell culture, Odontogenesis
1. Introduction
Currently, artificial dental implants are commonly used to replace human teeth that have been lost due to trauma, cancer resection, or birth defects. Although this therapy has been proven to be a successful approach to replace lost teeth in many individuals, a variety of common patient complications can occur, such as gingival tissue and bone loss, peri-implantitis, and even implant failure [1–4]. It has been suggested that biological-based living tooth replacements can overcome many of the complications observed in synthetic dental implant therapy [5–7]. One area of current dental research aims to fabricate bioengineered whole teeth by using natural tooth development as a guide [6, 8, 9]. Using this approach, tissue engineering strategies were used to identify suitable cell populations, and scaffold materials and designs, to support dental cell attachment, viability, proliferation, and differentiation [6, 8, 10]. It was shown that gelatin methacrylate (GelMA) hydrogel scaffolds can be tuned to mimic certain features of immature natural dental tissues and to support the differentiation of encapsulated dental cells, including elaboration of mineralized dental tissue formation [9]. Here, we describe in detail the fabrication and analytical methods used to create and validate a novel bioengineered 3D GelMA tooth bud model.
2. Materials
Prepare all solutions using ultrapure water (prepared by purifying deionized water, to attain a sensitivity of 18 MΏ-cm at 25 °C) and analytical grade reagents. Prepare and store all reagents at room temperature (unless indicated otherwise). Diligently follow all waste disposal regulations when disposing waste materials. We do not add sodium azide to reagents. All tissue culture solutions are sterilized by autoclaving and/or filtration through 22 micron filters.
2.1. Primary Dental Cell Isolation, In Vitro Expansion, and Cryopreservation
Collagenase/dispase solution: Prepare 5–10 min prior to use. Dissolve 20 mg collagenase type II and 10 mg dispase in 50 mL pBS.
Dental mesenchymal (DM) culture media (for cells/tissues harvested from human or porcine pulp organ): Advanced DMEM/F12 media supplemented with 10% FBS, 25 μg/mL ascorbic acid, 1 × PSA, and 1 × Glutamax.
Dental epithelial (DE) culture media (for cells/tissues harvested from human or porcine enamel organ): LHC-8 media supplemented with 10% FBS, 0.5 μg/mL epinephrine, and 1 × PSA.
2.2. Preparation and Fabrication of GelMA-Encapsulated Dental Cell Constructs
Bioengineered Tooth Bud (BTB) Culture Media: Combine 250 mL advanced DMEM/F12 media with 250 mL LHC-8 media, supplemented with 10% FBS, 0.5 μg/mL epinephrine, 100 nM dexamethasone, 10 mM beta-glycerophosphate, 50 μg/mL ascorbic acid, and 1× PSA.
Lyophilized Gelatin Methacrylate (GelMA): The GelMA lyo-philizate used to establish these methods was a generous gift from Dr. Ali Khademhosseini. The synthesis of GelMA lyophi- lizate has been thoroughly described [11, 12].
2.3. Live/Dead Analysis of In Vitro Cultured GelMA Tooth Bud Constructs
Live/Dead Staining Kit: Live/Dead Viability/Cytotoxicity Kit (Molecular Probes). Thaw reagents to 37 °C prior to use.
2.4. Subcutaneous Implantation, Harvest, and Fixation of GelMA Tooth Bud Constructs
Rat host: Immunocompromised 5-month-old female Rowett Nude rats supplied by Charles River Laboratories.
2.5. Processing and Analyses of GelMA Tooth Bud Constructs
2.5.1. Decalcification and Processing of GelMA Tooth Bud Constructs
Decalcification Solution (22.5% formic acid + 10% sodium citrate): Prepare 45% formic acid in DI H2O by slowly adding 225 mL of 98% formic acid to 275 mL of DI H2O. Next prepare 20% sodium citrate by slowly adding 100 g of sodium citrate to 400 mL of DI H2O, and then add H2O to bring to 500 mL volume. Carefully combine 500 mL of 20% sodium citrate to 500 mL of 45% formic acid.
Saturated Ammonium Oxalate: Fully dissolve 10 g of ammonium oxalate in 100 mL of H2O. Slowly add additional ammonium oxalate until saturated, and then filter.
2.5.2. Paraffin Embedding and Sectioning
Molten paraffin: Heat paraffin to 65 °C manually or via automatic Thermo Shandon Citadel 2000 Tissue Processor (Thermo Fisher Scientific) and/or Microm EC 350–1 Paraffin Embedding Center (Microm International GmbH).
65 °C oven or hot plate to hold molten paraffin and samples.
Sectioning microtome: Microm HM 355S (Thermo Fisher Scientific).
2.5.3. Hematoxylin and Eosin Staining
Mayor’s hematoxylin working solution: Dissolve 5 g of hematoxylin in 800 mL H2O. Bring to a boil; then cool to room temperature and cover with aluminum foil to keep in dark. Stir overnight with a magnetic stirrer over a stir plate at room temperature. Add 1 g of sodium iodate and 50 g aluminum ammonium sulfate. Once dissolved, use a pipette to add 200 mL glycerol. Next, add 8 mL glacial acetic acid and mix for 2 h. Filter and adjust pH to 2.4–2.8.
Diluted hydrochloric acid: Carefully add 2 mL of 37% hydrochloric acid to 250 mL H2O.
Ammonium hydroxide working solution: Add 4 mL (28–30%) ammonium hydroxide to 250 mL H2O.
Eosin stock solution: Dissolve 1 g of eosin Y in DI H2O to total volume 100 mL.
Phloxine B stock: Dissolve 1 g phloxine B in 100 mL H2O total volume.
Eosin working solution: Carefully combine 100 mL eosin stock solution, 10 mL phloxine B, 780 mL 95% ethanol, and 8 mL glacial acetic acid. Mix well and adjust pH to 4.5.
2.5.4. Immunohistochemical Staining Using Vectastain Kit and DAB Detection
3% H2O2: Add 25 mL of 30% H2O2 to 225 mL 100% methanol, keeping solutions in the dark.
TE buffer (10 mM Tris +1 mM EDTA): Dissolve 1.210 g Trizma and 0.372 g EDTA into 900 mL H2O. Bring to total volume with H2O.
Blocking solution (10% donkey serum): Add 0.5 mL of 100% normal donkey serum to 4.5 mL PBS, final volume 5.0 mL.
2% donkey serum: Mix 2 mL of 10% donkey serum with 8 mL of PBS, total volume 10 mL.
ABC reagent (Vectastain ABC Kit): In the dark, add 2.5 mL of PBS to a 15 mL conical tube covered in aluminum foil. Add 1 drop of Reagent A and mix well. Then add 1 drop of Reagent B and mix well. Allow mixing for 45 min prior to use.
DAB substrate (Sigma Fast DAB Tablets): Prepare within 5–10 min of use. In the dark, add 5 mL of distilled water in a 15 mL conical tube covered in aluminum foil. Add one urea tablet and mix until dissolved. Add one DAB tablet and mix well.
0.2% Fast Green: Dissolve 0.2 g Fast Green into 500 mL H2O at 37 °C for 1 h and then filter.
2.5.5. Immunofluorescence Staining
10 mM Citric Acid Solution: Dissolve 1.07 g of citric acid monobasic in 500 mL H2O and adjust to pH 6.0. Store at 4 °C up to 1 week.
Blocking Solution (5% Bovine Serum Albumin, BSA): Dissolve 0.5 g in 10 mL H2O.
1% BSA: Mix 2 mL of 5% BSA with 8 mL of H2O.
3. Methods
3.1. Primary Dental Cell Isolation, In Vitro Expansion, and Cryopreservation of Primary Dental Cells
Isolate unerupted molar tooth buds from harvested mandibular jaws of 3–5-month-old pigs, and place in PBS. Briefly identify depression in the jaw indicating location of unerupted molar tooth bud. Use a hammer and chisel to create a window in the mandibular bone, and remove bone flap. Remove unerupted tooth bud using sterile forceps. Wash isolated tooth bud in 1× PBS 3×, 5 min each. Under aseptic conditions, place washed tooth bud in a petri dish containing PBS. Use a dissecting microscope to dissect out the enamel organ, any mineralized tooth cusps if present, and the dental pulp organ, by cutting at the cervical margin using a #10 scalpel and forceps. Place the dissected enamel organ in a separate petri dish containing PBS. Do not allow tissues to become dry. Place the harvested pulp organ in a clean petri dish containing PBS; separate from the enamel organ. At all times, keep the isolated enamel organ and pulp organ tissues and cells separate and hydrated in PBS.
Separately mince enamel organ, and pulp organ tissues using 2 #10 blades to obtain approximately 1–2 mm3-sized pieces. Place minced tissues in a 50 mL conical tube with PBS, and invert tubes several times to wash. Allow the minced tissues to settle to the bottom of the tube, and then gently remove PBS. Repeat wash 2 ×.
After last wash, remove PBS and add 25 mL of collagenase/ dispase solution per tissue group. Incubate minced tissue tubes for 30 min rotating or rocking slowly at 37 °C.
Gently titurate the tissue suspension first with a 25 mL pipette for 5 min and then with a 10 mL pipette for an additional 5 min.
Filter the cell/tissue suspension through a 40 μm cell strainer by gravity, into a new sterile 50 mL conical tube.
Collect tissue from the strainer by rinsing, and place in a T175 flask with 25 mL of corresponding cell/tissue growth media (DE media for enamel organ and DM media for pulp organ tissues) (see Subheading 2).
Centrifuge the filtered cell suspension for 5 min at 1144 × g at room temperature. Remove supernatant and add 30 mL of the appropriate growth media. Centrifuge again for 5 min at 1500 rpm at RT. Remove supernatant, and wash again with 30 mL of growth media. Centrifuge for 5 min at 1500 rpm at RT. Finally, resuspend cells in 40 mL of appropriate growth media.
Use trypan blue stain to count cells. Add 25 μL of trypan blue to 25 μL of cell suspension. Use a hemocytometer or a cell counter (e.g., Countess Automated Cell Counter, Invitrogen) to count cells.
To culture the pulp organ cells, add two to three million cells to each T75 culture flask or five million cells to each T175 culture flask. Next add 15 mL of dental mesenchymal growth media to T75 flasks and 25 mL to T175 flasks.
To culture the enamel organ cells, add 10 million cells per T75 flask or 20 million cells per T175 flask. Add 15 mL of dental epithelial growth media to T75 flasks and 25 mL to T175 flasks.
Culture tissues and cells separately in a humidified incubator with 5% CO2 at 37 °C. Monitor cell growth daily. Change medium when at least 10% of cell attachment can be seen. Then change medium every 2–3 days.
Passage or cryopreserve the cells when the cells reach ~80–95% confluency.
3.2. Preparation and Fabrication GelMA-Encapsulated Dental Cell Constructs
Prepare 20% photoinitiator (PI) (Irgacure2959). Fully dissolve 0.2 g PI in 100% methanol in an amber microcentrifuge tube. Adjust volume to 1 mL with methanol. Keep in the dark and at room temperature prior to use.
Prepare 3–5% GelMA. Measure out appropriate amount of lyophilized GelMA, and place in a 50 mL conical tube. In a biosafety hood, dissolve GelMA with warmed DMEM/F12 media (see Note 1). Maintain in the dark at 60 °C until fully dissolved. Once dissolved, maintain in the dark at room temperature. Right before use, warmthe dissolved GelMA solution at 37 °C, add PI (final concentration 0.1%), and filter with a 0.22 μm vacuum filter system (see Note 2).
Trypsinize flasks of cultured dental epithelial (DE) and dental mesenchymal (DM) cells (see Note 3). Collect, wash, and count the cells, and resuspend DE and DM cell pellets each in the appropriate filtered GelMA/PI solution (3% GelMA for DE cells and 5% GelMA for DM cells) at 30–60 × 106 cells/mL. Mix well (Fig. 1a1).
In the tissue culture hood, place individual 6 mm-inner-diameter polydimethylsiloxane (PDMS) ring-shaped molds in the center of each well of a 24-well tissue culture-treated plate (Fig. 1a2) (see Note 4). Pipette 40–50 μL of GelMA cell suspension into each PDMS mold. Place sample directly under the UV spot curer at a distance of 6 cm, and photocrosslink by UV exposure at 9.16 W/cm2 for 30–35 s (see Note 5) (Fig. 1a3).
Leave photocrosslinked samples in molds for 5 min, and then carefully remove the PDMS molds from each sample using forceps (Fig. 1a4). Next, add 1 mL of BTB culture media to each sample well. Culture cell-encapsulated GelMA constructs in a humidified incubator with 5% CO2 at 37 °C for 1–14 days. Change media every 2–3 days.
Fig. 1.
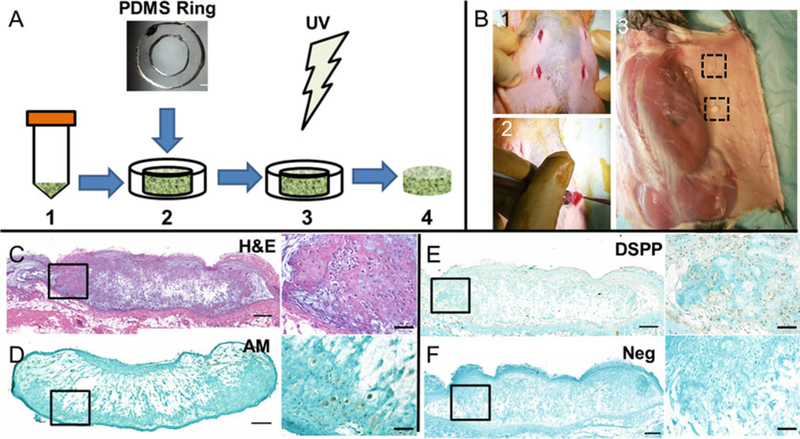
Fabrication and analysis of in vivo bioengineered GelMA tooth bud constructs. (a) Cultured dental cells are harvested, pelleted, and resuspended in GelMA hydrogel (a1). The GelMA/cell solution is then pipetted into PDMS ring molds that are placed within a 24-well plate (a2). UV exposure is used to photocrosslink the GelMA/ cell solution (a3). The PDMS molds are then removed and media is added for in vitro culture (a4). (b) Four incisions are made on the backs of immunocompromised rats (b1) to make subcutaneous pockets. The GelMA tooth bud constructs are carefully placed within the pockets (b2). To harvest, the skin and subcutaneous tissue are dissected to reveal the tooth bud constructs (b3). Formalin-fixed paraffin-embedded sections can be stained with various histological stains such as H&E (c). IF and IHC can be used to investigate the expression of various proteins including amelogenin (AM, d) and dentin sialophosphoprotein (DSPP, e) which will be detected by brown staining in contrast to a negative control (f). Scale bar: c-d 200 μm, insets 50 μm
3.3. Live/Dead Analysis of In Vitro Cultured GelMA Tooth Bud Constructs
Wash GelMA tooth bud constructs in the culture plate 3× in PBS to remove all media and serum.
Prepare 1 μM calcein-AM and 4.5 μM ethidium homodimer (EthD-1) solution in PBS. Add 2 mL of calcein/ethidium solution to each sample well, and incubate in a humidified incubator with 5% CO2 at 37 °C for 30 min.
Transfer stained samples from culture plate to a new plate containing 2 mL of PBS per well.
Immediately image the stained samples using confocal microscopy.
3.4. Subcutaneous Implantation, Harvest, and Fixation of GelMA Tooth Bud Constructs
After 1 week in vitro culture in osteogenic media, wash cultured tooth bud constructs 3× in 2 mL PBS. Carry washed samples in covered 24-well plates to animal facilities.
Create four subcutaneous implantation pockets (two on each side) on the back of each anesthetized rat host by making 1 cm incisions about 1.5 cm away from the midline (Fig. 1b1). Place one GelMA tooth bud construct in each subcutaneous pocket (Fig. 1b2) (see Note 6). Close each incision with wound clips. Remove wound clips after 2 weeks.
To harvest implanted tooth bud constructs, euthanize the rat host and make a ~6 cm incision at the midline. Next, make another incision at the top of and perpendicular to the midline incision, also ~6 cm long. Make a similar incision at the bottom of the midline incision. Use a scalpel and forceps to remove the skin and subcutaneous layer from the back of the host (Fig. 1b3).
Use a scalpel to cut a square around each implant. Use forceps to peel the implant and subcutaneous tissue from the skin, and place each sample in individual 5 mL sample collection bottle washed 3 × in PBS.
Immediately fix harvested samples in 10% formalin overnight at room temperature.
Wash samples 3 × in PBS and store in PBS at 4 °C.
3.5. Processing and Analyses of GelMA Tooth Bud Constructs
3.5.1. Decalcification and Processing of GelMA Tooth Bud Constructs
Characterize mineralized tissue formation in fixed GelMA constructs using X-ray or microCT. Begin decalcification of mineralized constructs by immersing in 5 mL of decalcification solution and gently rocking at room temperature. Change decalcification solution every 24–48 h.
Monitor decalcification every 24–48 h via ammonium oxalatecalcium precipitation assay; remove 5 mL of harvested decalcification solution, place in a small glass specimen bottle, and add 1 mL of saturated ammonium oxalate. Watch for precipitate formation after 20 min at room temperature. Continue to decalcify sample if precipitate forms. If precipitation does not form within 20 min, wash the demineralized sample 3 × in PBS and store in fresh PBS at 4 °C.
To process harvested and demineralized constructs, place each tooth bud construct between two tissue processing sponges, and secure within tissue cassettes labeled with pencil. Immediately immerse in graded ethanol (50, 70, 80,90, and 100%) for 2–4 h each. Then immerse samples 2 × in 100% ethanol for 2 h each and then 3× in 100% xylenes 1–2 h each.
3.5.2. Paraffin Embedding and Sectioning
Immediately after processing, incubate tooth bud constructs in molten paraffin for 12–16 h, 2×. Once fully infiltrated with paraffin, embed tooth bud constructs in molten paraffin using a plastic mold, orienting with pre-warmed forceps (see Note 7).
Once embedded, place each construct on a cold plate or cryo console (e.g., Microm EC 350–2, Thermo Fisher Scientific) for 1 h to solidify, and then allow to set at room temperature overnight. Place at 4 °C for long-term storage.
Use a microtome to section paraffin block containing samples into 6 μm sections.
Float sections onto a microscope slide using a tissue floating bath set at 45 °C. Allow the slides to dry for at least 15 min at room temperature. Place section-mounted slides on a hot plate at 45 °C for 1 h and then at 55 °C overnight. Store mounted sections in slide boxes at room temperature.
3.5.3. Hematoxylin and Eosin Staining
Deparaffinize sections in 100% xylenes 2× for 5 min each.
Rehydrate deparaffinized sections in graded ethanol (100, 95, 70, and 50%) 2 min each, and then rinse in tap H2O for 2 min.
Stain sections with Mayor’s hematoxylin working solution for 1 min, followed by rinsing with tap water for 2.5 min.
Dip hematoxylin-stained sections once into diluted hydrochloric acid. Next, dip 3× into ammonium hydroxide working solution.
Rinse sections in H2O for 2 min, and then place in eosin working solution for 20 s.
Dehydrate H&E-stained sections by dipping 6× each in 4 different ethanol baths (95, 95, 100, and 100%).
Clear in 100% xylenes, 2× 5 min each.
Mount coverslips over stained sections using Permount mounting media, and let set overnight.
3.5.4. Immunohistochemical Staining Using Vectastain Kit and DAB Detection
Deparaffinize sections in 100% xylenes 2× 5 min.
Rehydrate sections in graded ethanol (100, 95, 70, and 50%) min each and then in H2O for 5 min.
Block endogenous peroxidase activity by incubating sections in 3% H2O2 for 20 min at room temperature in the dark (e.g., in an opaque slide container). Wash 3 × in PBS for 10 min each.
Place slides in warmed TE buffer in a steamer for 20 min (see Note 8). Cool slides in TE buffer on benchtop for 30 min. Wash 3 × in PBS for 5 min each.
Block sections with 10% donkey serum for 15 min at 37 °C in a humidified chamber (see Note 9).
Remove blocking solution, and incubate sections with primary antibody diluted in 2% donkey serum for 1 h at room temperature in a humidified chamber (see Note 10). Then wash 3× in PBS for 5 min each.
Incubate with secondary antibody diluted in 2% donkey serum for 45 min at room temperature in a humidified chamber. Then wash 3× in PBS for 5 min each.
Incubate sections in the dark with ABC reagent at room temperature for 45 min in a humidified chamber. Then wash slides 3× in PBS for 5 min each.
In the light, incubate with DAB substrate at room temperature for 5 min. Incubate in H2O for 5 min.
Counterstain sections in 0.2% Fast Green for 30–120 s (see Note 11).
Next, dehydrate sections by dipping slides into graded ethanol baths (95, 95, 100 and 100%) 3× each, and clear by dipping 3× in each 100% xylenes 2×.
Mount coverslips over sections using Permount mounting media, and let set overnight.
Image with Zeiss Axiophot Imager compound microscope (Carl Zeiss, Zi) equipped with Axiophot digital camera (Carl Zeiss AG, HRC) and AxioVision Rel 4.7 software (Fig. 1c).
3.5.5. Immunofluorescence Staining
Deparaffinize sections in 100% xylene 2× 5 min each.
Rehydrate sections in a graded ethanol series (100,95, 70, and 50%) 2 min each and then in H2O for 5 min.
Transfer slides to a plastic holder filled with pre-warmed 10 mM citric acid solution, and incubate in a steamer for 20 min (see Note 8). Cool at room temperature citric acid solution on benchtop for 30 min. Wash 3× in PBS for 5 min each.
Place in a humidified chamber, and block with 5% BSA for 20 min at 37 °C (see Note 9).
Remove blocking solution, and incubate sections with primary antibody diluted with 1% BSA for 1 h at room temperature in a humidified chamber (see Note 10). Wash slide 3× in PBS for 5 min each.
Incubate with secondary antibody diluted with 1% BSA for 1 h at room temperature in a humidified chamber. Wash slides 3× in PBS for 5 min each.
Add approximately 10 μL of Hard Set Mounting Medium with DAPI over each section, and mount with cover slips. Store at 4 °C.
Image within 24 h with Zeiss Axiophot Imager compound microscope (Carl Zeiss, Zi) equipped with Axiophot digital camera (Carl Zeiss AG, MRM) and AxioVision Rel 4.7 software.
Acknowledgments
All members of the Yelick Tissue Engineering Lab have contributed to optimize and validate these techniques. This work was supported by NIH/NIDCR R01 DE16132 (PCY) and NIH/NIDCR F31 DE026361 (EES).
4 Notes
When calculating the final GelMA concentration and volume, be sure to account for the final photoinitiator (PI) concentration and volume that will be added at a later step.
Filter GelMA/PI solution using a Steriflip Vacuum Filtration System (Millipore, SCGP00525).
Human umbilical vein endothelial cells (HUVECs) [9] or other cell types can be co-encapsulated with the dental cells. If incorporating other cells with the dental cells, be sure to supplement the odontogenic media with cell type-specific appropriate media. Also remember to include acellular control GelMA control samples to monitor GelMA over time in culture and for comparison with cell-encapsulated GelMA constructs.
Place a PDMS mold in every other well of a 24-well plate, totaling 12 molds per plate. This minimizes UV exposure from spot curing of adjacent samples.
The UV photocrosslinking parameters were optimized for the OmniCure S2000 (Lumen Dynamics Group) equipped with a 5 mm spot curer and 320–500 nm filter and calibrated to 1.8 W.
For in vivo subcutaneous implantation of constructs, use a randomization program to pre-assign samples to rat implantation pockets.
To limit the number of embedded sample blocks, include replicate samples in a single paraffin block.
Refer to manufacturer’s antibody data sheet for specific conditions for recommended antigen retrieval/unmasking and dilution ranges.
Prior to blocking, use a PAP pen to create a hydrophobic barrier around each section to minimize the volume of block solution and diluted antibody needed.
Include a no primary control to test for non-specific binding of the secondary antibody. Add 2% donkey serum in place of the primary antibody.
Refilter 0.2% Fast Green counterstain prior to use to remove any precipitate.
References
- 1.Greenstein G, Cavallaro J, Romanos G, Tar- now D (2008) Clinical recommendations for avoiding and managing surgical complications associated with implant dentistry: a review. J Periodontol 79:1317–1329 [DOI] [PubMed] [Google Scholar]
- 2.Jung RE, Pjetursson BE, Glauser R, Zembic A, Zwahlen M, Lang NP (2008) A systematic review of the 5-year survival and complication rates of implant-supported single crowns. Clin Oral Implants Res 19:119–130 [DOI] [PubMed] [Google Scholar]
- 3.Chrcanovic BR, Albrektsson T, Wennerberg A (2014) Reasons for failures of oral implants. J Oral Rehabil 41:443–476 [DOI] [PubMed] [Google Scholar]
- 4.Chrcanovic BR, Kisch J, Albrektsson T, Wen- nerberg A (2016) Factors influencing early dental implant failures. J Dent Res 95:995–1002 [DOI] [PubMed] [Google Scholar]
- 5.Yen AH, Yelick PC (2011) Dental tissue regeneration - a mini-review. Gerontology 57:85–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith EE, Yelick PC (2016) Progress in bioengineered whole tooth research: from bench to dental patient chair. Curr Oral Health Rep 3 (4):302–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai W-F, Lee J- M, Jung H- S (2014) Molecular and engineering approaches to regenerate and repair teeth in mammals. Cell Mol Life Sci 71:1691–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monteiro N, Smith EE, Angstadt S, Zhang W, Khademhosseini A, Yelick PC (2016) Dental cell sheet biomimetic tooth bud model. Biomaterials 106:167–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith EE, Zhang W, Schiele NR, Khademhosseini A, Kuo CK, Yelick PC (2017) Developing a biomimetic tooth bud model. J Tissue Eng Regen Med 11 (12):3326–3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monteiro N, Yelick PC (2016) Advances and perspectives in tooth tissue engineering. J Tissue Eng Regen Med 11(9):2443–2461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yue K, Santiago GT-d, Alvarez MM, Tamayol A, Annabi N, Khademhosseini A (2015) Synthesis, properties, and biomedical applications of gelatin methacryloyl (GelMA) hydrogels. Biomaterials 73:254–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nichol JW, Koshy ST, Bae H, Hwang CM, Yamanlar S, Khademhosseini A (2010) Cell-laden microengineered gelatin methacrylate hydrogels. Biomaterials 31:5536–5544 [DOI] [PMC free article] [PubMed] [Google Scholar]