

Abstract
Most of the natural schweinfurthins are potent and selective inhibitors of cell growth as measured by the National Cancer Institute’s 60-cell line screen. Due to the limited supply of these natural products, we have initiated a program aimed at their synthesis. To date, this effort has led to the preparation of three natural schweinfurthins and more than 40 analogues, and assays on these compounds have afforded some understanding of structure-activity relationships in this family. Further development of schweinfurthins as chemotherapeutic agents would benefit from characterization of their mechanism(s) of action. This perspective led to development of a fluorescent schweinfurthin analogue that retains the differential activity of the natural products, and yet has properties that facilitate its visualization within cells.
Graphical Abstract
Synthesis of a fluorescent schweinfurthin analogue has been accomplished, and its biological activity and sub-cellular localization have been studied.
1. Introduction
Higher plants have proven to be an excellent source of novel compounds that are useful in the treatment of cancer.1 Among the most notable successes in this area are the alkaloids vincristine and vinblastine isolated from Catharanthus roseus,2 paclitaxel isolated from Taxus brevifola,3,4 and camptothecin isolated from Camptothecin acuminate.5 As part of an ongoing National Cancer Institute (NCI) initiative to examine new plant extracts for potential chemotherapeutic agents, several closely related compounds named schweinfurthins have been discovered. After an extract from the Cameroonian plant Macaranga schweinfurthii displayed potent and cell type selective growth inhibition (GI) in the NCI’s 60-cell line screen,6 the more active components of this extract were isolated, characterized, and named schweinfurthin A and B7 (1 and 2, Figure 1). A less active analogue was named schweinfurthin C, and later studies yielded the closely related schweinfurthin D.8 Independent studies of Macaranga alnifolia later yielded schweinfurthins E–H.9
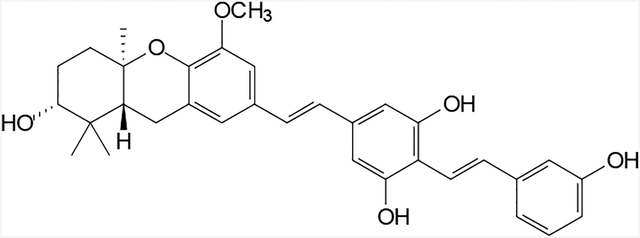
Figure 1.

Structures of some natural (1 and 2) and synthetic (3 and 4) schweinfurthins.
Further study of the schweinfurthins as potential cancer therapeutics is encouraged by the substantial differential activity seen among the cell lines of the NCI’s 60-cell line screen. In addition, the unique pattern of activity can be considered a “fingerprint” indicative of a possible novel mechanism of action.10 Finally, these agents demonstrated very potent antiproliferative activity against human-derived CNS cell lines such as SF-295, SF-539, and SNB-75 (GI50’s of 11 nM, 10 nM, and 15 nM respectively, for schweinfurthin A). However, development of schweinfurthins as cancer therapeutics has been hindered by limited material due to difficulties in isolation of significant additional quantities of the active compounds from the original plant source. Therefore, we have undertaken the total synthesis of these natural products. While installation of the vicinal diol on the A-ring has proven to be elusive, analogues without the C-3 hydroxyl group have been prepared and shown to maintain the growth inhibition and differential activity exhibited by the natural schweinfurthin diols.6–9 Furthermore, compounds lacking the C-3 hydroxyl group were isolated during the more recent studies of M. alnifolia,9 and already have been synthesized as single enantiomers.11
Two of the synthetic compounds, (R,R,R)-3-deoxyschweinfurthin A (3, 3dSA)12 and B (4, 3dSB),13 have excellent potency with sub-micromolar mean GI50’s, while maintaining differential activity across the NCI’s 60-cell cancer screen. Since the inception of the synthetic program, our group has prepared three natural schweinfurthins and more than 40 analogues. These compounds have allowed us to explore some structure-function relationships within the family, particularly as they pertain to the D-ring.14 To date however, these analogues have not clarified the mechanism of action, which remains essentially unknown.
Development of schweinfurthins as cancer therapeutics might be accelerated by elucidation of their mechanism(s) of action. Fluorescence microscopy often has been used to determine the localization of drugs15 or proteins, and potential interaction with other cellular components.16 Synthesis of schweinfurthin analogues that incorporate fluorescence properties could advance our current understanding of schweinfurthin activity, as long as such compounds retain their anti-proliferative activity. Here we present the synthesis and biological activity of a novel fluorescent schweinfurthin analogue.
During the synthesis of 3dSB, it was determined that many of these stilbenes were fluorescent under UV light. For example, 3dSB itself has an absorption maximum of 331 nm and an emission maximum of 391 nm (data not shown). The possibility that these properties might provide information on the sub-cellular localization of schweinfurthins immediately was recognized. After treatment with 3dSB, SF-295 cells (a human-derived glioblastoma multiforme cell line) were examined for potential UV fluorescence. However, 3dSB failed to generate a significant fluorescent signal above the autofluorescence of control cells (data not shown).
The limited utility of 3dSB as a fluorescent probe prompted a review of our prior structure-activity data with the hope of finding a position amenable to change and consistent with improved fluorescent characteristics. Previous work has demonstrated the importance of at least one free D-ring phenol for biological activity.14 Consideration of the natural schweinfurthins indicates the para position of the D–ring as a point where the structure might be modified without loss of the desirable biological properties. For example, 3dSB bears a 10-carbon geranyl substituent at this position while schweinfurthin F carries only a 5-carbon prenyl substituent and both display comparable activity.11 Therefore, a literature review was performed with an eye toward identification of accessible substitutions that might be made at this position to afford new analogues with improved fluorescence properties.
Substituted stilbenes are among the most studied fluorescent compounds.17,18 For example, Maddy et al. synthesized the fluorescent stilbene 4-acetamido-4’-isothiocyanostilbene-2,2’-disulfonic acid in the mid 1960’s.19 This compound then was shown to be an excellent fluorescent label for the plasma membrane of erythrocytes, and was subsequently shown to penetrate the blood-brain barrier.20 More recently a bis-stilbene was demonstrated to penetrate mouse brain and fluorescently label myelin.21 The idea of attaching a second stilbene linkage at the para position of the D-ring also was attractive from a synthetic standpoint, because it fit well into our modular approach to the schweinfurthins. Therefore the novel bis-stilbene 13 was identified as the initial target.
2. Chemical Synthesis
Synthesis of compound 13 began with the known aryl bromide 5 (Scheme 1).22,23 Halogen-metal exchange on bromide 5 presumably afforded a lithiated intermediate, which was subsequently allowed to react with anhydrous dimethylformamide to afford the new aldehyde 6 in acceptable yield. This aldehyde was treated with the known phosphonate 714 under modified Horner-Wadsworth-Emmons (HWE) conditions to give the protected stilbene 8. Removal of the silyl protecting group under standard conditions gave the benzylic alcohol 9. A three step procedure then was used to convert the alcohol 9 into the benzylic phosphonate 10. Thus, treatment of the alcohol 9 with methanesulfonyl chloride and triethylamine affords the expected mesylate which can be smoothly transformed into the iodide upon reaction with NaI. Displacement of the iodide by the soft nucleophile triethyl phosphite gives the desired benzylic phosphonate 10 in high overall yield. A second HWE condensation with the nonracemic tricyclic aldehyde 1113 gives the protected schweinfurthin analogue 12. Final hydrolysis of the MOM protecting groups through treatment with camphorsulfonic acid or toluenesulfonic acid gives the desired bis-stilbene 13. This compound was highly fluorescent with an absorbance maximum of 330 nm, an emission maximum of 416 nm and an extinction coefficient ε of 31,200 (Figure 2).
Scheme 1.

Synthesis of fluorescent stilbene 13.
Figure 2.

Absorbtion and emission spectra of compound 13.
3. Biological Results and Discussion
As noted above, the schweinfurthins display potent growth inhibition and differential activity in the NCI’s 60-cell screen. In this assay, the SF-295 cell line is among the most sensitive to schweinfurthin-induced growth inhibition, while the human-derived lung adenocarcinoma A549 cells display only moderate growth inhibition upon parallel treatment. Due to this difference in sensitivity between SF-295 and A549 cells, these cell lines can be used to investigate the schweinfurthin-like activity of synthetic analogues such as compound 13. Indeed, investigation of cytotoxicity with compound 13 demonstrates a significant toxicity in SF-295 cells (IC50 ~800 nM) while A549 cells are much less sensitive.
In addition to their toxic effects, schweinfurthins induce drastic changes in cell morphology at time points beyond 24 hours. In culture, SF-295 cells are large polygonal shaped cells with multiple outstretched processes (Figure 3A). Treatment of SF-295 cells with schweinfurthin analogues such as 3dSB causes morphologic changes congruent with those induced by the natural products. These alterations are characterized by a decrease in cell area and a reduction in F-actin, which results in spindle-shaped cells with F-actin staining only at the periphery of the treated cells.
Figure 3-.

SF-295 (A) and A549 (B) cells treated with indicated concentrations and intervals of 3dSB and compound 13 were stained with fluorescent phalloidin as an F-actin marker (green) and DAPI as nuclear stain (blue).
When SF-295 cells were treated for 24 hours with compound 13 (500 nM) or 3dSB (500 nM), they exhibited morphologic characteristics somewhat similar to control cells (Figure 3A). However at 48 hours cells treated with 3dSB (500 nM) or compound 13 (500 nM) display altered morphology. These changes appear to differ from those induced by the Rho kinase (or ROCK) inhibitor Y-27632 (10 μM),24 which caused jagged F-actin at the periphery and long slender projections to form. Inhibition of ROCK disrupts RhoA signaling, which is critical in the maintenance of cell shape and motility.25 In addition, ROCK inhibition induces changes readily detected at 24 hours. At equivalent concentrations compound 13 displays similar activity to 3dSB in SF-295 cells, yet both compound 13 and 3dSB differ from the activity of Y-27632.
In comparison to the SF-295 cells, the schweinfurthins are much less active against the A549 cells. In control experiments, the A549 cells are rectangular and cover less surface area in comparison to SF-295 cells (Figure 3B). Treatment with 3dSB (500 nM) or compound 13 (500 nM) does not induce a morphologic change apparent at either 24 hours or 48 hours (Figure 3B). The absence of change is consistent with previous studies which indicate that A549 cells are less sensitive to schweinfurthin treatment. Similar to the effects in SF-295 cells, Y-27632 (10 μM) induces ruffled F-actin at the edges of the A549 cells (Figure 3B). These findings further suggest that the structural changes introduced in compound 13 do not alter the differential activity observed with the schweinfurthins.
The bis-stilbene 13 represents the first compound that might further understanding of the schweinfurthin localization through fluorescence microscopy. Indeed, SF-295 cells treated with concentrations of compound 13 as low as 100 nM could be visualized at all time points tested (Figure 4). Unlike previous experiments with other schweinfurthins, the fluorescence in treated cells was significantly greater than the background of control cells. As expected, treatment of SF-295 cells with increasing concentrations of compound 13 increased fluorescence intensity at all time points tested (Figure 3). The localization of compound 13 within cells appears largely cytosolic with the most intense fluorescence in the perinuclear region. The fluorescent properties of compound 13 allow its visualization above the background autofluorescence in control cells.26
Figure 4-.

Fluorescence of compound 13 in SF-295 cells at indicated concentrations and time points.
4. Conclusions
In conclusion, these studies have led to the preparation of the first schweinfurthin analogue with fluorescence properties that allow its cellular localization to be tracked by microscopy. This compound maintains the activity of schweinfurthins in SF-295 cells, and results in characteristic changes in cell morphology over time. While the morphologic changes induced by schweinfurthins differ from those induced by the ROCK inhibitor Y-27632, these findings do not preclude the involvement of RhoA through other downstream mediators or other Rho family members such as Rac1 and Cdc 42, which are also crucial for proper cell structure and movement.27 The localization of compound 13 appears be cytosolic as well as perinuclear. Unfortunately, compound 13 undergoes rapid photobleaching and thus did not allow co-localization studies to determine its potential concentration in organelles. Nevertheless, incorporation of such a sizeable substituent at the para position of the D-ring suggests that other substituents might be placed at this site without significant loss of biological activity, and a series of fluorescent analogues can be readily envisioned. Other useful probes might include compounds linked to moieties such as biotin or polyethylene glycol to aide in the examination of mechanism.28–30 Such probes may allow determination of binding partners, signaling cascades, and/or localization. Studies in this vein are ongoing and will be reported in due course.
5. Experimental Procedures and Methods
5.1. General experimental conditions.
Tetrahydrofuran (THF) was distilled from sodium/benzophenone immediately prior to use, CH2Cl2 was freshly distilled from CaH2, and DMF was dried over CaH2. All reactions in non-aqueous solvents were conducted in oven-dried glassware under a positive pressure of argon and with magnetic stirring. NMR spectra were recorded at 300 MHz for 1H, and 75 MHz for 13C with CDCl3 as solvent and (CH3)4Si (1H) or CDCl3 (13C, 77.2 ppm) as internal standards unless otherwise noted. Silica gel (60 Å, 0.040–0.063 mm) was used for flash chromatography. Yields refer to pure compounds after chromatography. High resolution mass spectra were obtained at the University of Iowa Mass Spectrometry Facility.
5.2. Aldehyde 6.
To a solution of the known bromide 522,23 (1.25 g, 3.0 mmol) in THF (10 mL) at −78 °C was added n-BuLi (1.6 mL, 2.0 M in hexanes) dropwise over 2 min. After 30 min, DMF was added (0.5 mL, 6.5 mmol, containing some CaH2 as a drying agent). The reaction was allowed to progress for an additional 3 hr, and quenched by addition of saturated aq. NH4Cl. The aqueous phase was extracted with EtOAc, and the combined organic phases were washed with brine, dried (MgSO4), and concentrated in vacuo to afford a yellow oil. Final purification by column chromatography afforded aldehyde 6 (1.09 g, 99%) as a clear oil: 1H NMR (CDCl3) δ 10.5 (s, 1H), 6.84 (s, 2H), 5.26 (s, 4H), 4.73 (s, 2H), 3.50 (s, 6H), 0.95 (s, 9H), 0.12 (s, 6H); 13C NMR (CDCl3) δ 188.9, 159.6 (2C), 151.0, 114.7, 105.6 (2C), 94.8 (2C), 64.5, 56.4 (2C), 25.8 (3C), 18.3, −5.4 (2C); HRMS (EI) calcd for C18H30O6Si (M+) 370.1812; found 370.1822.
5.3. Stilbene 8.
A suspension of NaH (290 mg, 7.8 mmol, 60% in oil), and 15-crown-5 (1 drop, cat.) in THF (10 mL) was cooled to 0 °C. To this was added a solution of aldehyde 6 (168 mg, 0.45 mmol) and phosphonate 7 (185 mg, 0.64 mmol) in THF (10 mL). The mixture was allowed to warm to rt and stirred a total of 10 hr. Water was added dropwise, and the solution was extracted with EtOAc. The resulting organic phase was washed with brine, dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography (6:1 to 2:1 hexanes:ethyl acetate) gave the stilbene 8 (228 mg, 100%) as a clear oil: 1H NMR (CDCl3) δ 7.61 (d, J = 17 Hz, 1H), 7.50 (d, J = 17 Hz, 1H), 7.38–7.29 (m, 3H),7.03–7.00 (m, 1H), 6.93 (s, 2H), 5.34 (s, 4H), 5.30 (s, 2H), 4.80 (s, 2H), 3.60 (s, 6H), 3.59 (s, 3H), 1.05 (s, 9H), 0.02 (s, 6H); 13C NMR (CDCl3) δ 157.4, 156.1 (2C), 142.3, 140.7, 132.0, 129.4, 120.4, 120.1, 115.1, 114.6, 114.1, 106.0 (2C), 94.8 (2C), 94.3, 64.7, 56.1 (2C), 55.9, 25.8 (3C), 18.3, −5.3 (2C). Anal. Calcd for C27H40O7Si : C, 64.30; H, 7.99. Found: C, 64.09; H, 8.02.
5.4. Benzylic Alcohol 9.
Silyl ether 8 (227 mg, 0.45 mmol) was dissolved in THF (10 mL) and the solution was cooled to 0 °C. To this solution was added TBAF (0.6 mL, 1.00 M in THF), after 5 minutes the ice bath was removed, and after 4 hr the reaction was quenched by addition of sat. NH4Cl. After extraction with ethyl acetate, the combined organic extracts were washed with water and brine, dried (MgSO4), and concentrated in vacuo to give the desired benzylic alcohol 9 (176 mg, 100%) as a clear oil: 1H NMR (CDCl3) δ 7.52 (d, J = 17 Hz, 1H), 7.40 (d, J = 17 Hz, 1H), 7.29–7.19 (m, 3H), 6.94–6.91 (m,1 H), 6.82 (s, 2H), 5.24 (s, 4H), 5.21 (s, 2H), 4.62 (s, 2H), 3.49 (s, 9H), 2.51 (br s, 1H); 13C NMR (CDCl3) δ 157.4, 156.1 (2C), 141.9, 140.5, 132.3, 129.4, 120.1, 120.1, 115.7, 114.7, 114.1, 106.7 (2C), 94.6 (2C), 94.3, 64.9, 56.2 (2C), 55.9.
5.5. Phosphonate 10.
Methanesulfonyl chloride (0.15 mL, 1.9 mmol) was added to a solution of benzylic alcohol 9 (176 mg, 0.45 mmol) and Et3N (0.2 mL 1.4 mmol) in CH2Cl2 (10 mL). The reaction mixture was allowed to warm to room temperature over 1 h, quenched by addition of H2O, and extracted with CH2Cl2. The combined organic layers were washed with NH4Cl (sat), brine, dried (MgSO4), and concentrated in vacuo. The resulting residue and NaI (150 mg, 1.0 mmol) were stirred in acetone (10 mL) for 8 h. The reaction mixture was concentrated in vacuo to afford a red solid, which was dissolved in EtOAc. After the resulting yellow solution was washed with Na2S2O3 until the color faded, it was washed with brine, dried (MgSO4) and concentrated in vacuo. The resulting yellow oil was added to triethyl phosphite (2 mL) and the reaction mixture was heated at 80 °C for 24 h. After the solution was allowed to cool to rt, the excess phosphite was removed at high vacuum. The initial yellow oil was purified by flash chromatography (1:2 hexanes:EtOAc to 100% EtOAc) to afford phosphonate 10 (201 mg, 88%) as a clear oil: 1H NMR (CDCl3) δ 7.53 (d, J = 16.7 Hz, 1H), 7.40 (d, J = 16.7 Hz, 1H), 7.28 – 7.21 (m, 3H), 6.97 – 6.94 (m, 1H), 6.80 – 6.79 (m, 2H), 5.26 (s, 4H), 5.23 (s, 2H), 4.12 – 4.02 (m, 4H), 3.52 (s, 9H), 3.13 (d, JHP = 21.8 Hz, 2H), 1.30 (t, J = 7.0 Hz, 6H); 13C NMR (CDCl3) δ 157.4, 155.9 (d, JCP = 3.7 Hz, 2C), 140.6, 132.3 (d, JCP = 1.6 Hz), 132.0 (d, JCP = 8.0 Hz), 129.4, 120.1, 120.1, 115.3 (d, JCP =4.1 Hz), 114.7, 114.1, 110.2 (d, JCP = 6.7 Hz, 2C), 94.8 (2C), 94.4, 62.1 (d, JCP = 6.8 Hz, 2C), 56.2 (2C), 55.9, 33.5 (d, JCP = 138 Hz), 16.3 (d, JCP = 6.0 Hz, 2C), HRMS (EI) calcd for C25H35O9P (M+) 510.2019; found 510.2018.
5.6. Protected bis-stilbene 12.
A suspension of NaH (80 mg, 20 mmol, 60% in oil), and 15-crown-5 (1 drop, cat.) in THF (5 mL) was cooled to 0 °C. To this mixture was added a solution of aldehyde 11 (60 mg, 0.20 mmol) and phosphonate 10 (95 mg, 0.19 mmol) in THF (5 mL). The resulting mixture was allowed to warm to rt and stirred a total of 30 hr. Water was added dropwise, and the solution was extracted with EtOAc. The resulting organic phase was washed with brine, dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography (3:1 to 1:1 hexanes:ethyl acetate) gave the stilbene 12 (71 mg, 56%) as a green waxy solid oil: 1H NMR (CDCl3) δ 7.55 (d, J = 16.6 Hz, 1H), 7.43 (d, J = 16.6 Hz, 1H), 7.32 – 7.20 (m, 4H), 7.04 – 6.87 (m, 6H), 5.32 (s, 4H), 5.23 (s, 2H), 3.91 (s, 3H), 3.55 (s, 6H), 3.52 (s, 3H), 3.47 – 3.42 (m, 1H), 2.74 – 2.71 (m, 2H), 2.20 – 2.12 (m, 1H), 1.90 – 1.47 (m, 5H), 1.27 (s, 3H), 1.14 (s, 3H), 0.90 (s, 3H); 13C NMR (CDCl3) δ 157.4, 156.3 (2C), 148.9, 142.7, 140.7, 137.8, 132.1, 129.5, 129.1, 128.6, 125.9, 122.6, 120.7, 120.3, 120.1, 125.7, 114.7, 114.2, 106.7, 106.4 (2C), 94.8 (2C), 94.4, 77.9, 77.1, 60.3, 56.3 (2C), 56.0, 46.7, 38.3, 37.6, 28.2, 27.3, 23.1, 19.8, 14.1; HRMS (EI) calcd for C39H48O9 (M+) 660.3298, found 660.3301.
5.7. Bis-stilbene 13.
To a solution of stilbene 12 (16 mg, 0.024 mmol) in MeOH (5 mL) was added p-toluenesulfonic acid (3 mg, 0.014 mmol). The resulting solution was heated 60 °C for 5 hr. The reaction was quenched by addition of sat. NaHCO3, extracted with ethyl acetate, and the organic phase was washed with brine and dried (MgSO4. Concentration in vacuo, followed by final purification by column chromatography (2:1 to 1:1 hexanes:ethyl acetate) afforded the bis-stilbene 13 (10 mg, 79%) as a slightly yellow oil: 1H NMR ((CD3)2CO) δ 7.19 (d, J = 17 Hz, 1H), 7.55 (d, J = 17 Hz, 1H), 7.18 (t, J = 7.9 Hz, 1H), 7.04 – 6.98 (m, 4H), 6.95 (d, J = 6.9 Hz, 2H), 6.90 – 6.88 (m, 1H), 6.73 – 6.70 (m, 2H), 3.82 (s, 3H), 3.43 – 3.38 (m, 1H), 2.76 – 2.73 (m, 2 H), 1.85 – 1.63 (m, 6 H), 1.23 (s, 3H), 1.13 (s, 3H), 0.90 (s, 3H); 13C NMR ((CD3)2CO) δ 158.5, 157.6 (2C), 150.2, 144.1, 142.1, 138.6, 131.5, 130.4, 129.7, 129.6, 126.5, 123.7, 121.8, 121.5, 118.7, 114.7, 113.0, 112.3, 108.3 (2C), 77.7, 77.6, 56.1, 47.2, 39.2, 38.7, 29.2, 27.9, 23.8, 20.4, 14.9; HRMS (EI) calcd for C33H36O6 (M+) 528.2512; found 528.2523.
5.8. Cell Culture.
The SF-295 cells were maintained in RPMI 1640 media supplemented with 10% bovine fetal calf serum and penstreptamycin. The A-549 cells were maintained in F-12 media with 10% fetal calf serum and penstreptamycin. Both cell lines were incubated at 37° C and 5% CO2.
5.9. Cytotoxicity.
SF-295 and A549 cells were plated in 24-well plates. Upon 65% confluency, cells were treated with 3dSB, compound 9 or Y-27632. To measure cytotoxicity, the MTT assay was preformed. Briefly, thiazolyl blue tetrazolium bromide (Sigma, St. Louis, MO, USA) was added to all wells 44 hours after compound treatment. At 48 hours solubilization solution (HCl, triton X-100, Isopropyl alcohol) was added to each well and incubated overnight with gentle agitation at 37° C. To quantify toxicity, the absorbance of each well at 540 nm and 630 nM (reference λ) was measured spectrophotometrically.
5.10. Cellular Morphology.
Cells were plated on sterilized coverslips in 6-well plates and allowed to reach 65% confluency before treatment with the indicated compounds. Rock inhibitor Y-27632 was purchased from Calbiochem San Diego, CA. At the conclusion of the treatment interval, cells were rinsed and fixed with 3.7% formaldehyde for 15 minutes. After subsequent rinses in PBS, cells were permeabilized with 0.2% triton X-100 for 4 minutes and rinsed in PBS. Cells were then incubated with 1% BSA for 30 minutes before incubation with phalloidin stain (Molecular Probes, Eugene, OR) for 20 minutes. At the completion of the staining interval, cells were rinsed and mounted using Vectashield mounting media containing DAPI (Vector laboratories, Inc., Burlingame, CA). Slides were imaged using a Bio-Rad Multi-photon microscope and processed using Image-J software.
5.11. Fluorescence Microscopy.
SF-295 cells were plated on sterilized coverslips in 6-well plates and allowed to reach 65% confluency and then treated for indicated intervals. At the conclusion of the treatment interval cells were washed three times in complete media to remove residual compound. Immediately following rinsing, cells were mounted onto microscope slides and imaged using a Bio-Rad Multi-photon microscope at the University of Iowa Central Microscopy facility. Images were further processed using Image-J software.
Acknowledegements
This project was supported by the Roy. J. Carver Charitable Trust as a Research Program of Excellence, the Roland W. Holden Family Program for Experimental Cancer Therapeutics and The Iowa Centers for Enterprise.
References
- (1).Cragg GM; Newman DJ J. Ethnopharm 2005, 100, 72–79. [DOI] [PubMed] [Google Scholar]
- (2).Noble RL; Beer CT; Cutts JH Biochem. Pharmacol 1958, 1, 347–348. [Google Scholar]
- (3).Wani MC; Taylor HL; Wall ME; Coggon P; McPhail AT J. Am. Chem. Soc 1971, 93, 2325–2327. [DOI] [PubMed] [Google Scholar]
- (4).Schiff PB; Fant J; Horwitz SB Nature 1979, 277, 665–667. [DOI] [PubMed] [Google Scholar]
- (5).Hsiang YH; Hertzberg R; Hecht S; Liu LF J. Biol. Chem 1985, 260, 14873–14878. [PubMed] [Google Scholar]
- (6).Beutler JA; Jato JG; Cragg G; Wiemer DF; Neighbors JD; Salnikova MS; Hollingshead M; Scudiero DA; McCloud TG In Medicinal and Aromatic Plants; Bogers RJ, Ed.; Springer: 2005, p 301–309. [Google Scholar]
- (7).Beutler JA; Shoemaker RH; Johnson T; Boyd MR J. Nat. Prod 1998, 61, 1509–1512. [DOI] [PubMed] [Google Scholar]
- (8).Beutler JA; Jato J; Cragg GM; Boyd MR Nat. Prod. Lett 2000, 14, 399–404. [Google Scholar]
- (9).Yoder BJ; Cao SG; Norris A; Miller JS; Ratovoson F; Razafitsalama J; Andriantsiferana R; Rasamison VE; Kingston DGI J. Nat. Prod 2007, 70, 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Paull KD; Shoemaker RH; Hodes L; Monks A; Scudiero DA; Rubinstein L; Plowman J; Boyd MR J. Nat. Cancer Inst 1989, 81, 1088–1092. [DOI] [PubMed] [Google Scholar]
- (11).Mente NR; Wiemer AJ; Neighbors JD; Beutler JA; Hohl RJ; Wiemer DF Biorg. Med. Chem. Lett 2007, 17, 911–915. [DOI] [PubMed] [Google Scholar]
- (12).Mente NR; Neighbors JD; Wiemer DF J. Org. Chem 2008, 73, 7963–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Neighbors JD; Beutler JA; Wiemer DF J. Org. Chem 2005, 70, 925–931. [DOI] [PubMed] [Google Scholar]
- (14).Neighbors JD; Salnikova MS; Beutler JA; Wiemer DF Biorg. Med. Chem 2006, 14, 1771–1784. [DOI] [PubMed] [Google Scholar]
- (15).Kashemirov BA; Bala JLF; Chen X; Ebetino FH; Xia Z; Russell RGG; Coxon FP; Roelofs AJ; Rogers MJ; McKenna CE Bioconjugate Chem. 2008, 19 2308–2310. [DOI] [PubMed] [Google Scholar]
- (16).Day RN Mol. Cell. Endocrinol 2005, 230, 1–6. [DOI] [PubMed] [Google Scholar]
- (17).Papper V; Likhtenshtein GI J. Photochem. Photobiol., A 2001, 140, 39–52. [Google Scholar]
- (18).Mazzucato U; Momicchioli F Chem. Rev 1991, 91, 1679–1719. [Google Scholar]
- (19).Maddy AH Biochim. Biophys. Acta 1964, 88, 390–399. [DOI] [PubMed] [Google Scholar]
- (20).Schmued LC; Fallon JH Brain Res. 1985, 346, 124–129. [DOI] [PubMed] [Google Scholar]
- (21).Wu CY; Tian DH; Feng Y; Polak P; Wei JJ; Sharp A; Stankoff B; Lubetzki C; Zalc B; Mufson EJ; Gould RM; Feinstein DL; Wang YM J. Histochem. Cytochem 2006, 54, 997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Furstner A; Stelzer F; Rumbo A; Krause H Chem. Eur. J 2002, 8, 1856–1871. [DOI] [PubMed] [Google Scholar]
- (23).Wang QL; Huang QG; Chen B; Lu JP; Wang H; She XG; Pan XF Angew. Chem. Int. Ed 2006, 45, 3651–3653. [DOI] [PubMed] [Google Scholar]
- (24).Uehata M; Ishizaki T; Satoh H; Ono T; Kawahara T; Morishita T; Tamakawa H; Yamagami K; Inui J; Maekawa M; Narumiya S Nature 1997, 389, 990–994. [DOI] [PubMed] [Google Scholar]
- (25).Ridley AJ Trends Cell Biol. 2006, 16, 522–529. [DOI] [PubMed] [Google Scholar]
- (26).Schneckenburger H; Wagner M; Weber P; Strauss WSL; Sailer RJ Fluoresc. 2004, 14, 649–654. [DOI] [PubMed] [Google Scholar]
- (27).Heasman SJ; Ridley AJ Nat. Rev. Mol. Cell Biol 2008, 9, 690–701. [DOI] [PubMed] [Google Scholar]
- (28).Bach S; Knockaert M; Reinhardt J; Lozach O; Schmitt S; Baratte B; Koken M; Coburn SP; Tang L; Jiang T; Liang D.-c.; Galons H; Dierick J-F; Pinna LA; Meggio F; Totzke F; Schachtele C; Lerman AS; Carnero A; Wan Y; Gray N; Meijer L J. Biol. Chem 2005, 280, 31208–31219. [DOI] [PubMed] [Google Scholar]
- (29).McPherson M; Yang Y; Hammond PW; Kreider BL Chem. Biol 2002, 9, 691–698. [DOI] [PubMed] [Google Scholar]
- (30).Sato S; Kwon Y; Kamisuki S; Srivastava N; Mao QA; Kawazoe Y; Uesugi M J. Am. Chem. Soc 2007, 129, 873–880. [DOI] [PubMed] [Google Scholar]