

Abstract
Purpose:
A subset of colorectal cancer (CRC) cases are attributable to Lynch syndrome (LS), a hereditary form of CRC. Effective evaluation for LS can be done on CRC tumors to guide diagnostic testing. Increased diagnosis of LS allows for surveillance and risk reduction, which can mitigate CRC-related burden and prevent cancer-related deaths.
Methods:
We evaluated participation in LS screening among newly diagnosed adult CRC patients. Some cases were referred for genetics evaluation prior to study recruitment (selective screening). Those not referred directly were randomized to the intervention or control (usual care) arms. Control cases were observed for one year, then given information about LS screening. Patients who declined participation were followed through the medical record.
Results:
Of 601 cases of CRC, 194 (32%) enrolled in our study and were offered LS screening, 43 (7%) were followed as a control group, 148 (25%) declined participation and 216 (36%) were ineligible (63 (10%) of which received prior selective screening). Six and nine cases of LS were identified through the intervention and selective screening groups, respectively. Overall, a higher proportion of PMS2 variants were identified in the intervention (3/6, 50%) vs. selective screening groups (2/9, 22%) (not statistically significant). Eighty-eight percent and 23% of intervention and control patients, respectively, received LS screening. No control patients were found to have LS.
Conclusion:
Systems-based approaches are needed to ensure we fully identify LS cases. The proportion of LS cases from this program was 4% of newly diagnosed cases of CRC, similar to other programs.
Keywords: Genetics, colorectal cancer (CRC), DNA mismatch repair (MMR) genes
INTRODUCTION
In the United States, 2–4% of colorectal cancer (CRC) cases are attributable to germline pathogenic variants in DNA mismatch repair (MMR) genes (EPCAM, MLH1, MSH2, MSH6 and PMS2) associated with Lynch syndrome (LS) [1, 2]. Patients with LS are at up to 80% risk of developing CRC without preventive measures [3–5], and are also at elevated risk of developing other kinds of cancer, including endometrial and ovarian cancer [6]. Diagnosing LS provides medically actionable information that can guide treatment decisions for cancer patients, as well as recommendations for intensive screening and risk-reducing surgery aimed at early detection or prevention of CRC, endometrial and other cancers, which has the potential to alleviate disease burden and prevent cancer-related deaths [7].
Diagnosis of LS in patients with cancer is often initiated by screening tumor tissue for the presence of MMR deficiency, using either microsatellite instability (MSI) or immunohistochemistry (IHC) testing [8]. Microsatellite unstable tumors are classified by 3 MSI phenotypes: MSI-high (MSI-H), indicating instability at two or more genetic loci, MSI-low (MSI-L), indicating instability at one genetic locus or MSI-stable (MSI-S), indicating no genetic loci with instability. Patients with an MMR-deficient tumor that lacks a somatic cause, such as a BRAF mutation or MLH1 promoter hypermethylation, can then be directed to genetic counseling and germline testing to confirm a diagnosis of LS [9].
Several strategies have been successfully used to reduce cancer incidence and burden of disease in patients diagnosed with LS. Surveillance by regular colonoscopy significantly reduces LS-attributable CRC incidence, enables earlier-stage CRC detection, and reduces CRC-associated mortality [10]. Accordingly, the United States Multi-Society Task Force on CRC (USMSTF) recommends that patients with LS should undergo colonoscopy every 1–2 years starting at age 20–25 years [11]. Further, a systematic review has demonstrated the effectiveness of cascade testing for LS (i.e., testing family members of patients who test positive) and conducting surveillance among patients with LS and their families [12]. Surveillance has been shown to significantly reduce CRC incidence and CRC-related mortality [12].
Because not all patients with LS meet high risk criteria and family history can be limited or unreliable, selective screening programs based on these variables and age at onset can miss cases of LS [13, 14]. Additionally, not every individual who meets the high-risk criteria will necessarily undergo evaluation for LS [13, 14]. For these reasons, universal tumor screening programs, which screen all newly diagnosed cases of CRC and/or endometrial cancer, have been recommended by the Evaluation of Genomic Applications in Practice and Prevention (EGAPP) and others and are increasingly being implemented by healthcare systems and hospitals [2, 15, 16–19].
Since not all institutions have incorporated this approach into routine care, examining the implementation and outcomes of systematic screening programs can help inform the future planning efforts to incorporate these programs into health systems. Here, we provide high-quality evidence from a randomized clinical trial (RCT) which evaluates systematic MSI screening for LS versus limiting evaluation of LS to patients referred outside the study (usual care). Our study population drew from patients who presented with new CRC and who underwent colorectal surgery. This study was implemented at Kaiser Permanente Northwest (KPNW), an integrated health-care system serving about 600,000 members in the Pacific Northwest. The overall goal of our RCT was to assess the effect of a systematic tumor screening strategy on genetic testing uptake, subsequent diagnosis of LS, and on the specific genetic variants detected compared with usual care.
METHODS
Population Description
The study population included a consecutive series of patients over 18 years of age with a new diagnosis of CRC who underwent surgery related to that cancer at KPNW between January 2012 and December 2015, regardless of family history of cancer or any other clinical characteristic. Patients were excluded if they had cognitive impairment (N=36), were in hospice (N=32), did not speak English (N=21), out of area (N=27), or other/unspecified reasons (N=10), e.g., LS screening performed elsewhere. In addition, 27 patients initially identified as eligible died prior to enrollment. Patients were also excluded if they had prior LS diagnosis or selective screening, which we defined as: a prior diagnosis of a hereditary cancer syndrome including LS (N=5), previous LS screening (N=24), or had already initiated contact with the Medical Genetics department to seek LS screening (N=34). Patients could actively decline participation in the study (Figure 1).
Figure 1.
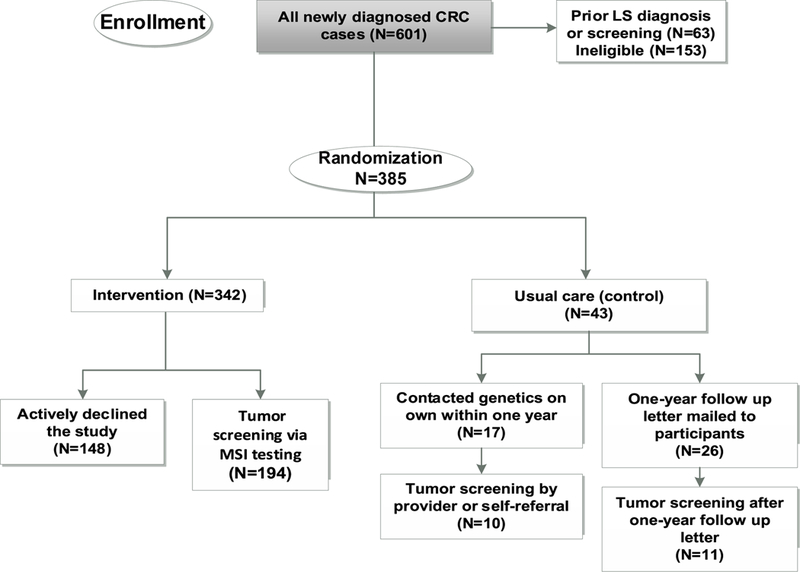
Flow chart of a RCT evaluating LS screening in patients newly diagnosed with CRC newly diagnosed with CRC who were randomized to study participation (N=385). To ensure adequate power for subsequent analyses in the intervention arm, we randomized 43 patients to the control arm and 342 to the intervention arm.
Eligible patients were randomized to either the intervention arm (tumor screening using MSI testing) or the control arm (observation of usual care) prior to being contacted by the study team. To ensure adequate power for subsequent analyses in the intervention arm that are reported elsewhere [20, 21], 10% of CRC cases were randomized to the control group and 90% randomized to the intervention arm. Patients in the control arm were observed for one year following randomization. According to our internal practice guidelines, providers or patients could refer directly to Medical Genetics for evaluation on the basis of personal or family history of LS-related cancers. Patients in the control arm were not informed of their participation in the study, which involved passive review of their electronic medical record, although they did have the opportunity to opt out of research, as described below. After one year, if they had not already sought LS screening as part of their usual care, they were sent a follow up letter to inform them of the availability of this screening and how to access it. Patients randomized to the intervention arm were contacted to introduce the study and were asked whether they would be interested in receiving LS tumor screening. Written informed consent was obtained from patients in the intervention arm who expressed interest. Those who actively declined were also followed through the medical record to observe LS related screening or testing and were included in the participant total. A waiver of informed consent was approved for patients in the control arm and those who declined participation, since their involvement was limited to a review of their electronic medical record (EMR) data to evaluate whether they pursued LS screening on their own outside of the study. Except for patients in the intervention arm, who gave consent for genetic research, all other patients were excluded from analysis if they are in databases maintained by the research center of patients who have opted out of any research (<0.001% of KPNW membership) or genetic research (approximately 5% of KPNW membership). This study was approved by the KPNW institutional review board.
Data Collection Procedures
Patients in the intervention arm completed a baseline survey that included demographic information not easily obtained through the EMR including marital status and income level. Other demographic characteristics (age, sex, race, ethnicity, insurance status, personal cancer history, and age at diagnosis with CRC) were obtained from the EMR. Patients in the intervention arm were contacted by a study team member and completed a phone interview to obtain an extended pedigree and detailed information on cancer types and age of diagnosis of patients and relatives using the Genetic Risk Easy Assessment Tool (GREAT) [22]. Details of tumor screening and germline testing were collected as part of a study database [23].
For patients who received LS screening outside of the intervention (e.g., those in the control group, those who declined participation, or were ineligible for the study because of prior LS screening or diagnosis), we performed manual chart reviews to abstract information on the details of tumor screening (e.g., MSI, IHC) and germline testing, if available. Some of these patients were offered germline testing and were referred for this testing based on an internal practice guideline that specified criteria to determine eligibility for testing. These criteria included the age of diagnosis, presence of other cancer(s) and family history of cancer. In addition, genetic counselors and/or medical geneticists may have offered testing even when the MSI/IHC was negative because of other clinical indicating factors. A Research Electronic Data Capture (REDCap) data entry form was created to support consistent data capture [23].
Collection of Samples and Clinical Data
LS screening for the intervention group was performed from formalin fixed paraffin embedded (FFPE) tissue from surgical specimens according to standard clinical procedures at the KPNW pathology department. Tumor specimens that were evaluated through the study were tested for microsatellite instability (MSI) at Oregon Health and Sciences University using seven markers (BAT-25, BAT-26, NR-21, NR-24, MONO-27, Penta C and Penta D). Samples with instability in two or more of these mononucleotide markers were designated MSI-high (MSI-H), whereas those with no detectable alterations were MSI-stable (MSI-S) and patients in the intervention arm with an insufficient sample (MSI-I) at this point were determined to be ineligible for the study and we did not include them in further analysis. These designations are in accordance with the National Cancer Institute’s Bethesda guidelines. Patients with MSI-H (but not MSI-L or MSI-S) results were contacted by a genetic counselor to offer genetic counseling and additional tumor screening including immunohistochemistry (IHC) testing for four MMR proteins (MLH1, MSH2, MSH6, and PMS2), which included reflex testing for MLH1 hypermethylation, and BRAF V600E, as indicated, to rule out sporadic cases of CRC [24]. Cases suggestive of LS were then offered an appointment in the Genetics clinic to facilitate germline testing (Online resource 1). Patients who received tumor screening outside of the study as part of routine clinical care that was ordered by their provider were tested at the Mayo Clinic or the Associated Regional and University Pathologists laboratories according to their usual clinical procedures.
Genetic Counseling
Participants with an MSI-H result were contacted by the study genetic counselor for discussion of the results to communicate information on LS and discuss next steps. They were then offered IHC as part of clinical care, followed by germline testing when appropriate based on IHC results. If LS was diagnosed, a genetic counselor reviewed the clinical implications of this diagnosis and surveillance recommendations were reviewed in detail. The patients were encouraged to discuss these results with their at-risk relatives.
For all study patients, a genetic counselor reviewed demographic characteristics, clinical characteristics, and detailed family history information collected using the GREAT tool to classify patients’ risk for LS according to three risk assessment procedures: (1) the Bethesda criteria [22], (2) the GREAT risk assessment based on modified Bethesda criteria, and (3) Prediction of Mutations in MLH1 and MSH2 (PREMM1,2,6) tool [25], a web-based decision-making tool that incorporates information on proband and family member cancer history to estimate an individual’s risk for pathogenic variants in the MMR genes MLH1, MSH2, and MSH6. This was done so we could determine how many patients would be considered high risk by these commonly used risk assessment tools.
Statistical Methods
We determined rates of LS evaluation for patients in the intervention arm and the control arm. We compared the proportion of patients in the intervention arm to the control arm who received LS screening within one year of randomization using a chi-square test. We defined LS evaluation as receiving either MSI or IHC testing, or both, to account for differences in testing strategy for the control arm. Differences in rates of germline testing to confirm LS diagnosis were evaluated separately. We evaluated the differences in demographic characteristics between the intervention arm, control arm, those who received prior LS diagnosis or screening and those who actively declined the study using a t-test for age and chi-square tests for the dichotomous variables (SAS Version 9.4). A threshold of p<.05 was used to indicate statistical significance.
RESULTS
Recruitment and Randomization
We identified 601 newly diagnosed cases of CRC at KPNW as initially eligible, 36% (216/601) of whom met exclusion criteria, including those who received prior LS diagnosis or tumor screening (10%, 63/601) (Figure 1). Of the remaining CRC cases, 43 were randomized to the control arm and 342 to the intervention arm. A total of 194 (57%) patients in the intervention arm consented to participate. Of the 148 patients who actively declined to participate and that we were able to reach, most expressed being concerned about their own health, not feeling well, and desiring to focus their energy solely on upcoming treatment, whereas only a small number of individuals had privacy concerns regarding participation.
Patient demographics are shown in Table 1. In the intervention arm, patients who were older or not white/non-Hispanic were significantly more likely to decline participation. Sex was not statistically associated with participation. Decliners were older than participants, (p=.0048), but did not differ from participants based on race or sex (p=.0535 and p=.3574, respectively).
Table 1.
Demographic characteristics.
| Characteristic | Participants | Decliners | P-valuea | |
|---|---|---|---|---|
| Intervention | Control | |||
| N | 194 | 43 | 148 | |
| Mean age, years (SD) | 66 (12) | 62 (12) | 69 (12) | 0.0048 |
| White, non-Hispanic (%) | 93 | 86 | 86 | 0.0535 |
| Sex (% male) | 59 | 60 | 55 | 0.3574 |
Comparison of participants (intervention + control) and decliners by Chi-Square test for all variables except age (t-test).
LS Evaluation Results
Overall, 88% (170/194) of patients randomized and consented to the intervention arm were evaluated for LS within one year of randomization compared with 23% (10/43) in the control arm (p<.0001). For the remaining 24 patients in the intervention arm, there was insufficient tumor tissue available to complete the testing, often because they were biopsy samples. Of the 170 patients evaluated, 129/170 (76%) were MSI-S, 40/170 (24%) were MSI-H and 1/170 (0.6%) was MSI-I. Of the MSI-H patients, 36/40 (90%) agreed to further testing and 35/36 (97%) of these had abnormal IHC results. Of these, 24/35 (69%) were positive for BRAF mutation or MLH1 hypermethylation (and thus not referred for germline testing), 1/35 (3%) was deceased and the remaining 10/35 (29%) underwent germline testing. When the total number of participants assigned to the intervention (i.e., consented participants plus those who declined) are compared to the control participants, 54% (186/342) of patients randomized to the intervention arm were evaluated for LS within one year of randomization compared with 23% (10/43) in the control arm (p<.0001). A total of 11% (16/148) of those who declined the study were evaluated for LS.
Overall, a total of 6% (10/170) of patients in the intervention arm and 5% (2/43) in the control group underwent germline testing within one year of randomization (p=.0186). In the intervention arm, 6/10 (60%) were positive for a germline pathogenic variant compared to none in the control group. Three of the patients in the intervention arm who received germline testing were MSI-S. The testing was provided to these patients based on young age (< 50 years) and/or family history. Online resource 1 displays detailed a screening flow for the intervention group.
We found that 63 patients were in the process of being screened for LS or other hereditary cancer syndromes prior to the study recruitment. To obtain a more comprehensive picture of the entire population, we evaluated the outcomes of LS screening among these patients. In general, these patients had a younger age at CRC diagnosis (Table 1) and a stronger family history (Table 3) of LS-associated cancers. Three cases were already known to have LS (one MSH2, one MSH6, one PMS2). Two patients had a different genetic disorder and six cases did not receive any LS-related tests. The remaining 83% (52/63) of patients were screened for LS.
Table 3.
Characteristics of patients with LS identified from either a universal tumor screening program or from prior LS diagnosis or screening.a
| Patient | Age | MMR Gene | Variant | Bethesda | PREMM | GREAT | Personal and Family | |
|---|---|---|---|---|---|---|---|---|
| Criteria | ≥5% | Genetic Risk | History | |||||
| Intervention | 1 | 42 | MLH1 | c.676C>T | Y | Y | N | None |
| Arm | 2 | 70 | MSH2 | C.1618delA | Y | Y | Y | 1° Relatives: CRC (2)b |
| 3 | 57 | MSH6 | C.3939_3957dup | Y | Y | Y | 1° Relatives: CRC (1) | |
| 4 | 63 | PMS2 | C.137G>T | N | N | N | None | |
| 5 | 67 | PMS2 | ex9_10del | N | N | Y | 1° Relatives: Ovarian (1) 2° Relatives: Stomach (1) |
|
| 6 | 76 | PMS2 | C.2239A>T | N | N | N | None | |
| Prior LS diagnosis or screening | 1 | 51 | MLH1 | del of exons 16–19 | Y | Y | N/A | Previously diagnosed with CRC (1) 1° Relatives: CRC (2) |
| 2 | 31 | MLH1 | c.885–2A>G and c.80G>A | Y | Y | N/A | 1° Relatives: Unknown (1) | |
| 3 | 56 | MSH2 | del exon 8 | Y | Y | N/A | Previously diagnosed with CRC (1) | |
| 1° Relatives: Ovarian (1), Pancreatic (1) | ||||||||
| 2° Relatives: CRC (2), Breast (2), Uterine (1), Stomach (1), Brain (1), Prostate (1) | ||||||||
| 4 | 51 | MSH2 | C.2461_2462delGT | Y | Y | N/A | Previously diagnosed with Uterine Cancer (1) | |
| 1° Relatives: CRC (2), Uterine (1) | ||||||||
| 2° Relatives: CRC (2) | ||||||||
| 5 | 59 | MSH2 | c. 1968C>A | Y | Y | N/A | Previously diagnosed with Uterine Cancer (1) | |
| 1° Relatives: CRC (2), Uterine (2), Renal (1) | ||||||||
| 2° Relatives: Pancreatic (2), Uterine (1), Stomach (1) | ||||||||
| 6 | 53 | MSH2 | Not specified | Y | Y | N/A | Previously diagnosed with Rectal (1) | |
| 7 | 64 | MSH2, EPCAM | EPCAM 3’ terminal deletion | Y | Y | N/A | 1° Relatives: CRC (2), Small Intestine (1), Bile Duct (1), Pancreatic (1) 2° Relatives: Gastric (2), CRC (2) | |
| 8 | 51 | MSH6 | C.3203C>T | Y | Y | N/A | 1° Relatives: CRC (1) 2° Relatives: CRC (1), Gastric (1) |
|
| 9 | 50 | MSH6 | C.3203C>T | Y | Y | N/A | 1° Relatives: CRC (1), Renal Pelvis (1) 2° Relatives: CRC (1), Stomach (1) |
|
| 10 | 64 | PMS2 | C.631C>T | Y | Y | N/A | 2° Relatives: Pancreatic (2) | |
| 11 | 76 | PMS2 | C.736_741del6 | Y | Y | N/A | 1° Relatives: CRC (1) | |
| 12 | 76 | PMS2 | c.736_741del6ins11 | Y | Y | N/A | 1° Relatives: CRC (1) |
N/A = not available.
Family size was similar between LS cases in intervention arm and those with prior LS diagnosis or selective screening.
The number in parentheses indicates the number of relatives with the indicated type of cancer and degree of relationship.
DISCUSSION
In this study, a systematic LS screening intervention led to led to very high rates (88%) of appropriate evaluation for LS among patients with newly diagnosed CRC, compared to only 23% in the control arm. The majority of patients in the control arm did not receive screening for LS, even after they were sent a letter informing them about how to receive this screening. The fact that there was not uptake in the control group during this period, despite substantial risk for “contamination” of the study arms, further supports the need for systematic approaches to screening that do not rely on clinicians or patients to make referrals to the Medical Genetics Department.
The evaluations in the intervention arm led to the overall diagnosis of 6 cases of LS in that could have escaped detection under usual care practices of self- and provider-referral to the Medical Genetics department. These cases represent 4% of participants in the intervention arm, a figure that is consistent with prior studies of screening programs, which have reported rates of LS among those screened of between 0.5% (18/500) – 4% (8/214) [2, 9, 16, 17, 26–29].
This study provided us with the opportunity to examine screening uptake and results among all patients newly diagnosed with CRC during the study period, including those who received prior LS diagnosis or screening and those who declined participation in the study. All patients who were referred to Medical Genetics prior to study enrollment met traditional criteria based on family history, whereas only half of the patients identified through our program met traditional screening criteria. However, other differences also emerged between the two groups: LS patients who sought testing on their own were clearly younger (Table 1) with a stronger personal or family history of LS (Table 3) and had higher proportions of pathogenic variants in MLH1 and MSH2 (Tables 2 and 3) than LS patients in the intervention group. As such, patients with prior LS diagnosis or screening were more similar to LS patients identified in selective screening approaches than those identified through our systematic screening intervention [2, 9, 16, 17, 26–29].
Table 2.
Patients with a molecular diagnosis of LS in MMR genes MLH1, MLH2, MLH6, and PMS2 among those who underwent germline testing in the Intervention arm (N=170) and among those with prior LS diagnosis or screening (N=63). No LS cases were identified in the Usual Care (N=43) or Study Decliner (N=148) arms.
| Number of patients with a pathogenic variant who underwent germline testing | ||
|---|---|---|
| MMR gene | Intervention (N=14) | Prior LS diagnosis or screening (N=16) |
| MLH1 | 1 | 2 |
| MSH2 | 1 | 4 |
| MSH6 | 1 | 1 |
| PMS2 | 3 | 2 |
| No pathogenic variant | 8 | 7 |
While our study was not designed to specifically evaluate cascade genetic testing in family members, this is an important outcome for LS universal screening programs. Patients diagnosed with LS in this study worked with a genetic counselor to discuss sharing information with at-risk relatives. Of the six LS cases identified through the intervention arm, five informed some or all of their first-degree relatives. Six of these patients’ relatives were tested for LS, and one underwent a colonoscopy, but none of these tests resulted in new LS diagnoses.[21] These data demonstrate that systematic screening programs are essential in promoting cascade screening, because the delivery of results to probands offers a teachable moment to convey the importance of genetic testing for at-risk relatives and potentially preventing future diagnoses of CRC or other cancers.
A total of 50% (3/6) of patients diagnosed with LS in the intervention arm had pathogenic variants in PMS2, in contrast to 22% (2/9) among LS patients who received prior LS diagnosis or screening. This difference was not statistically significant (p=.33, Fisher’s exact test, 2-sided probability). In general, other systematic LS screening programs have reported PMS2 variants less frequently (0 – 17%) than this study [2, 9, 16, 17, 26–29], though a recent Swiss study also identified a PMS2 variant frequency of 50% (2 of 4 LS cases) [30]. These differences may be due in part to more limited evaluation of PMS2 in previous studies. For example, the MMRpredict and PREMM5 prediction models can be less effective at identifying PMS2 carriers even though they predict whether an individual is likely to have LS [31]. In addition, population selection criteria may have also contributed to findings of fewer PMS2 variants in past research. For example, studies that required participant consent may be more likely to include younger cases with a stronger family history compared with the entire population, similar to the patients with prior LS diagnosis or selective screening. PMS2 variants have lower penetrance compared with other genes, and PMS2 pathogenic variants are associated with lower risk and older age of CRC onset than variants in other LS-associated genes [32, 33]. These results suggest that PMS2 may represent a larger proportion of LS cases than previously believed.
Limitations
Requiring participant consent limited participation in the intervention arm. However, when those who declined the study are considered, 54% (186/342) of patients randomized to the intervention arm received screening, which is more than double than the proportion who were screened in the control arm (23%; 10/43). When patients who declined research study participation or who were randomized to the control arm were told about the availability of LS screening and were provided with information on how to obtain screening, most did not elect to receive the screening. Thus, lack of consent for this study may reflect disinterest in screening for LS rather than disinterest in participation in research.
We chose MSI as the initial screening test because there was not clear consensus on the optimal screening strategy at the time we started this study. We also considered IHC screening to meet our criteria for LS evaluation. Many other universal LS screening programs either use IHC or both tests [2, 9, 16, 17, 26–29]. however, the performance characteristics of MSI and IHC are highly correlated [34, 35]. Thus, it is unlikely that the results of our screening program were substantially impacted by a different choice of the initial screening test. As the costs of sequencing continue to decline, direct sequencing may become a cost-effective strategy in the future [34, 36]. While the optimal screening method continues to be refined, our results are nevertheless relevant regarding a community population that receives universal screening.
A limitation of tumor screening programs in general is that they only identify patients to screen for LS after they are diagnosed with cancer. Nevertheless, such screening programs are an important tool for improving the ongoing surveillance of LS patients for additional cancer diagnoses, as well as for identifying other family members at risk for hereditary cancer susceptibility, particularly when combined with cascade testing in family members. Our study demonstrates that a systematic screening program increases the diagnosis of LS, which suggests that there is a role for LS screening and other universal screening programs in mitigating CRC sequelae and disease severity.
Conclusions
This study shows that system-wide implementation of systematic tumor screening of colorectal tumors can identify previously unsuspected cases of LS, ensuring appropriate surveillance measures for patients and family members. The low rates of screening in our control arm, in which patients received usual care, demonstrates that without systems in place to ensure complete capture of the population, the condition will continue to be underdiagnosed. Since the conclusion of this study, our region has implemented universal LS screening given that this approach identifies cases that would otherwise be missed.
Future work should investigate the effectiveness of screening all adults with other types of LS-associated cancer in addition to CRC (e.g., endometrial and ovarian cancers), as has been done in several recently implemented universal screening programs [37–39]. Universal screening enables identification of pathogenic variants less likely to be associated with strong family history of CRC (i.e., PMS2) and provides opportunities for appropriate interventions to target LS patients for disease mitigation and possibly prevention.
Supplementary Material
Acknowledgments
Compliance with Ethical Standards
This work was supported through grants by the National Cancer Institute: R01CA140377 (Goddard) and R01CA132829 (Syngal). The funding body had no role in the design of the study, collection and analysis of data or decision to publish. Dr. Syngal reports being a consultant for Myriad Genetics, Inc.
Footnotes
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent was obtained from all individual participants included in the study.
REFERENCES
- 1.Carethers JM, Stoffel EM (2015) Lynch syndrome and Lynch syndrome mimics: The growing complex landscape of hereditary colon cancer. World J Gastroenterol 21 (31):9253–9261. doi: 10.3748/wjg.v21.i31.9253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Nakagawa H, Sotamaa K, Prior TW, Westman J, Panescu J, Fix D, Lockman J, Comeras I, de la CA (2005) Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 352 (18):1851–1860 [DOI] [PubMed] [Google Scholar]
- 3.Kohlmann W, Gruber SB (1993) Lynch Syndrome. In: Pagon RA, Adam MP, Ardinger HH et al. (eds) GeneReviews(R) University of Washington, Seattle; All rights reserved., Seattle (WA), [Google Scholar]
- 4.Chen S, Wang W, Lee S, Nafa K, Lee J, Romans K, Watson P, Gruber SB, Euhus D, Kinzler KW, Jass J, Gallinger S, Lindor NM, Casey G, Ellis N, Giardiello FM, Offit K, Parmigiani G (2006) Prediction of germline mutations and cancer risk in the Lynch syndrome. Jama 296 (12):1479–1487. doi: 10.1001/jama.296.12.1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindor NM, Petersen GM, Hadley DW, Kinney AY, Miesfeldt S, Lu KH, Lynch P, Burke W, Press N (2006) Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA 296 (12):1507–1517. doi:296/12/1507[pii]; 10.1001/jama.296.12.1507 [doi] [DOI] [PubMed] [Google Scholar]
- 6.Kastrinos F, Uno H, Ukaegbu C, Alvero C, McFarland A, Yurgelun MB, Kulke MH, Schrag D, Meyerhardt JA, Fuchs CS, Mayer RJ, Ng K, Steyerberg EW, Syngal S (2017) Development and Validation of the PREMM5 Model for Comprehensive Risk Assessment of Lynch Syndrome. J Clin Oncol 35 (19):2165–2172. doi: 10.1200/jco.2016.69.6120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mange S, Bellcross C, Cragun D, Duquette D, Gorman L, Hampel H, Jasperson K (2015) Creation of a network to promote universal screening for Lynch syndrome: the LynchSyndrome Screening Network. J Genet Couns 24 (3):421–427. doi: 10.1007/s10897-014-9770-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giardiello FM, Allen JI, Axilbund JE, Boland CR, Burke CA, Burt RW, Church JM, Dominitz JA, Johnson DA, Kaltenbach T, Levin TR, Lieberman DA, Robertson DJ, Syngal S, Rex DK (2014) Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-society Task Force on colorectal cancer. Am J Gastroenterol 109 (8):1159–1179. doi: 10.1038/ajg.2014.186 [DOI] [PubMed] [Google Scholar]
- 9.Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Clendenning M, Sotamaa K, Prior T, Westman JA, Panescu J, Fix D, Lockman J, LaJeunesse J, Comeras I, de la Chapelle A (2008) Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol 26 (35):5783–5788. doi:JCO.2008.17.5950[pii]; 10.1200/JCO.2008.17.5950 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.P. D. Q. Cancer Genetics Editorial Board (2002) Genetics of Colorectal Cancer (PDQ(R)): Health Professional Version. In: PDQ Cancer Information Summaries National Cancer Institute (US), Bethesda (MD), [Google Scholar]
- 11.Giardiello FM, Allen JI, Axilbund JE, Boland CR, Burke CA, Burt RW, Church JM, Dominitz JA, Johnson DA, Kaltenbach T, Levin TR, Lieberman DA, Robertson DJ, Syngal S, Rex DK (2014) Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-Society Task Force on colorectal cancer. Gastroenterology 147 (2):502–526. doi: 10.1053/j.gastro.2014.04.001 [DOI] [PubMed] [Google Scholar]
- 12.Barrow P, Khan M, Lalloo F, Evans DG, Hill J (2013) Systematic review of the impact of registration and screening on colorectal cancer incidence and mortality in familial adenomatous polyposis and Lynch syndrome. The British journal of surgery 100 (13):1719–1731. doi: 10.1002/bjs.9316 [DOI] [PubMed] [Google Scholar]
- 13.Hampel H, Stephens JA, Pukkala E, Sankila R, Aaltonen LA, Mecklin JP, de la CA (2005) Cancer risk in hereditary nonpolyposis colorectal cancer syndrome: Later age of onset. Gastroenterology 129 (2):415–421 [DOI] [PubMed] [Google Scholar]
- 14.Stoffel E, Mukherjee B, Raymond VM, Tayob N, Kastrinos F, Sparr J, Wang F, Bandipalliam P, Syngal S, Gruber SB (2009) Calculation of risk of colorectal and endometrial cancer among patients with Lynch syndrome. Gastroenterology 137 (5):1621–1627. doi: 10.1053/j.gastro.2009.07.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cross DS, Rahm AK, Kauffman TL, Webster J, Le AQ, Spencer Feigelson H, Alexander G, Meier P, Onitilo AA, Pawloski PA, Williams AE, Honda S, Daida Y, McCarty CA, Goddard KA (2013) Underutilization of Lynch syndrome screening in a multisite study of patients with colorectal cancer. Genet Med 15 (12):933–940. doi: 10.1038/gim.2013.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Julie C, Tresallet C, Brouquet A, Vallot C, Zimmermann U, Mitry E, Radvanyi F, Rouleau E, Lidereau R, Coulet F, Olschwang S, Frebourg T, Rougier P, Nordlinger B, Laurent-Puig P, Penna C, Boileau C, Franc B, Muti C, Hofmann-Radvanyi H (2008) Identification in daily practice of patients with Lynch syndrome (hereditary nonpolyposis colorectal cancer): revised Bethesda guidelines-based approach versus molecular screening. Am J Gastroenterol 103 (11):2825–2835. doi:AJG2084 [pii]; 10.1111/j.1572-0241.2008.02084.x [doi] [DOI] [PubMed] [Google Scholar]
- 17.Perez-Carbonell L, Ruiz-Ponte C, Guarinos C, Alenda C, Paya A, Brea A, Egoavil CM, Castillejo A, Barbera VM, Bessa X, Xicola RM, Rodriguez-Soler M, Sanchez-Fortun C, Acame N, Castellvi-Bel S, Pinol V, Balaguer F, Bujanda L, De-Castro ML, Llor X, Andreu M, Carracedo A, Soto JL, Castells A, Jover R (2012) Comparison between universal molecular screening for Lynch syndrome and revised Bethesda guidelines in a large population-based cohort of patients with colorectal cancer. Gut 61 (6):865–872. doi: 10.1136/gutjnl-2011-300041 [DOI] [PubMed] [Google Scholar]
- 18.Van Lier MG, De Wilt JH, Wagemakers JJ, Dinjens WN, Damhuis RA, Wagner A, Kuipers EJ, Van Leerdam ME (2009) Underutilization of microsatellite instability analysis in colorectal cancer patients at high risk for Lynch syndrome. Scand J Gastroenterol 44 (5):600–604 [DOI] [PubMed] [Google Scholar]
- 19.Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives (2009). Genet Med 11 (1):35–41. doi:10.1097GIM.0b013e31818fa2ff [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunter JE, Zepp JM, Gilmore MJ, Davis JV, Esterberg EJ, Muessig KR, Peterson SK, Syngal S, Acheson LS, Wiesner GL, Reiss JA, Goddard KA (2015) Universal tumor screening for Lynch syndrome: Assessment of the perspectives of patients with colorectal cancer regarding benefits and barriers. Cancer 121 (18):3281–3289. doi: 10.1002/cncr.29470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hunter JE, Arnold KA, Cook JE, Zepp J, Gilmore MJ, Rope AF, Davis JV, Bergen KM, Esterberg E, Muessig KR, Peterson SK, Syngal S, Acheson L, Wiesner G, Reiss J, Goddard KAB (2017) Universal screening for Lynch syndrome among patients with colorectal cancer: patient perspectives on screening and sharing results with at-risk relatives. Fam Cancer 16 (3):377–387. doi: 10.1007/s10689-017-9972-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Acheson LS, Zyzanski SJ, Stange KC, Deptowicz A, Wiesner GL (2006) Validation of a self-administered, computerized tool for collecting and displaying the family history of cancer. J Clin Oncol 24 (34):5395–5402 [DOI] [PubMed] [Google Scholar]
- 23.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG (2009) A metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 42 (2):377–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vilar E, Gruber SB (2010) Microsatellite instability in colorectal cancer-the stable evidence. Nature reviews Clinical oncology 7 (3):153–162. doi: 10.1038/nrclinonc.2009.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kastrinos F, Steyerberg EW, Mercado R, Balmana J, Holter S, Gallinger S, Siegmund KD, Church JM, Jenkins MA, Lindor NM, Thibodeau SN, Burbidge LA, Wenstrup RJ, Syngal S (2011) The PREMM(1,2,6) model predicts risk of MLH1, MSH2, and MSH6 germline mutations based on cancer history. Gastroenterology 140 (1):73–81. doi:S0016–5085(10)01239–4 [pii]; 10.1053/j.gastro.2010.08.021 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ward RL, Hicks S, Hawkins NJ (2013) Population-based molecular screening for Lynch syndrome: implications for personalized medicine. J Clin Oncol 31 (20):2554–2562. doi: 10.1200/jco.2012.46.8454 [DOI] [PubMed] [Google Scholar]
- 27.Kidambi TD, Blanco A, Myers M, Conrad P, Loranger K, Terdiman JP (2015) Selective Versus Universal Screening for Lynch Syndrome: A Six-Year Clinical Experience. Digestive diseases and sciences 60 (8):2463–2469. doi: 10.1007/s10620-014-3234-z [DOI] [PubMed] [Google Scholar]
- 28.Aaltonen LA, Salovaara R, Kristo P, Canzian F, Hemminki A, Peltomaki P, Chadwick RB, Kaariainen H, Eskelinen M, Jarvinen H, Mecklin JP, de la CA (1998) Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med 338 (21):1481–1487 [DOI] [PubMed] [Google Scholar]
- 29.Salovaara R, Loukola A, Kristo P, Kaariainen H, Ahtola H, Eskelinen M, Harkonen N, Julkunen R, Kangas E, Ojala S, Tulikoura J, Valkamo E, Jarvinen H, Mecklin JP, Aaltonen LA, de la Chapelle A (2000) Population-based molecular detection of hereditary nonpolyposis colorectal cancer. J Clin Oncol 18 (11):2193–2200 [DOI] [PubMed] [Google Scholar]
- 30.Zumstein V, Vinzens F, Zettl A, Heinimann K, Koeberle D, von Flue M, Bolli M (2016) Systematic immunohistochemical screening for Lynch syndrome in colorectal cancer: a single centre experience of 486 patients. Swiss medical weekly 146:w14315. doi: 10.4414/smw.2016.14315 [DOI] [PubMed] [Google Scholar]
- 31.Goverde A, Spaander MCW, Nieboer D, van den Ouweland AMW, Dinjens WNM, Dubbink HJ, Tops CJ, Ten Broeke SW, Bruno MJ, Hofstra RMW, Steyerberg EW, Wagner A (2017) Evaluation of current prediction models for Lynch syndrome: updating the PREMM5 model to identify PMS2 mutation carriers. Fam Cancer doi: 10.1007/s10689-017-0039-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Senter L, Clendenning M, Sotamaa K, Hampel H, Green J, Potter JD, Lindblom A, Lagerstedt K, Thibodeau SN, Lindor NM, Young J, Winship I, Dowty JG, White DM, Hopper JL, Baglietto L, Jenkins MA, de la Chapelle A (2008) The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology 135 (2):419–428. doi: 10.1053/j.gastro.2008.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moller P, Seppala T, Bernstein I, Holinski-Feder E, Sala P, Evans DG, Lindblom A, Macrae F, Blanco I, Sijmons R, Jeffries J, Vasen H, Burn J, Nakken S, Hovig E, Rodland EA, Tharmaratnam K, de Vos Tot Nederveen Cappel WH, Hill J, Wijnen J, Green K, Lalloo F, Sunde L, Mints M, Bertario L, Pineda M, Navarro M, Morak M, Renkonen-Sinisalo L, Frayling IM, Plazzer JP, Pylvanainen K, Sampson JR, Capella G, Mecklin JP, Moslein G (2017) Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut 66 (3):464–472. doi: 10.1136/gutjnl-2015-309675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mvundura M, Grosse S, Hampel H, Palomaki GE (2010) The cost-effectiveness of genetic testing strategies for Lynch syndrome among newly diagnosed patients with colorectal cancer. Genetics in Medicine 12 (2):93–104 [DOI] [PubMed] [Google Scholar]
- 35.Shia J (2008) Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J Mol Diagn 10 (4):293–300. doi: 10.2353/jmoldx.2008.080031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hampel H, Pearlman R, Beightol M, Zhao W, Jones D, Frankel WL, Goodfellow PJ, Yilmaz A, Miller K, Bacher J, Jacobson A, Paskett E, Shields PG, Goldberg RM, de la Chapelle A, Shirts BH, Pritchard CC, Ohio Colorectal Cancer Prevention Initiative Study G (2018) Assessment of Tumor Sequencing as a Replacement for Lynch Syndrome Screening and Current Molecular Tests for Patients With Colorectal Cancer. JAMA Oncol doi: 10.1001/jamaoncol.2018.0104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mills AM, Longacre TA (2016) Lynch Syndrome Screening in the Gynecologic Tract: Current State of the Art. The American journal of surgical pathology 40 (4):e35–44. doi: 10.1097/pas.0000000000000608 [DOI] [PubMed] [Google Scholar]
- 38.Batte BA, Bruegl AS, Daniels MS, Ring KL, Dempsey KM, Djordjevic B, Luthra R, Fellman BM, Lu KH, Broaddus RR (2014) Consequences of universal MSI/IHC in screening ENDOMETRIAL cancer patients for Lynch syndrome. Gynecol Oncol 134 (2):319–325. doi: 10.1016/j.ygyno.2014.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mills AM, Sloan EA, Thomas M, Modesitt SC, Stoler MH, Atkins KA, Moskaluk CA (2016) Clinicopathologic Comparison of Lynch Syndrome-associated and “Lynch-like” Endometrial Carcinomas Identified on Universal Screening Using Mismatch Repair Protein Immunohistochemistry. The American journal of surgical pathology 40 (2):155–165. doi: 10.1097/pas.0000000000000544 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.