

We used 3D microcrystal electron diffraction (MicroED) to solve a new protein structure.
Abstract
Microcrystal electron diffraction (MicroED) has recently shown potential for structural biology. It enables the study of biomolecules from micrometer-sized 3D crystals that are too small to be studied by conventional x-ray crystallography. However, to date, MicroED has only been applied to redetermine protein structures that had already been solved previously by x-ray diffraction. Here, we present the first new protein structure—an R2lox enzyme—solved using MicroED. The structure was phased by molecular replacement using a search model of 35% sequence identity. The resulting electrostatic scattering potential map at 3.0-Å resolution was of sufficient quality to allow accurate model building and refinement. The dinuclear metal cofactor could be located in the map and was modeled as a heterodinuclear Mn/Fe center based on previous studies. Our results demonstrate that MicroED has the potential to become a widely applicable tool for revealing novel insights into protein structure and function.
INTRODUCTION
Electrons, similar to x-rays and neutrons, are a powerful source for diffraction experiments (1–3). Because of the strong interactions between electrons and matter, crystals that are considered as powder in x-ray crystallography can be treated as single crystals by microcrystal electron diffraction (MicroED) (4). This enables structure determination of molecules from micrometer- to nanometer-sized three-dimensional (3D) crystals that are too small for conventional x-ray diffraction (4–8). Furthermore, MicroED can be applied to study biomolecules of low molecular weight that are beyond what can be resolved by single-particle cryo–electron microscopy (cryo-EM) imaging (1, 9).
Over the past decades, 3D ED methods have been developed for structure determination of small inorganic compounds (10–12) and organic molecules (13–15). At the early stages of 3D ED method development, tilting of the crystal was done manually, while diffraction patterns were collected on negative film. It could take years before sufficient data were obtained and processed to determine the crystal structure (16). The computerization of transmission electron microscopes (TEMs) and the development of charge-coupled device detectors allowed software to be developed that can semi-automatically collect 3D ED data in less than an hour (11, 13). Owing to the recent advancement in complementary metal-oxide semiconductor and hybrid detector technology, it is now feasible to collect diffraction data in movie mode while continuously rotating the crystal (5, 12, 17). Benefiting from these technological advances, data collection and structure refinement can now be performed within hours (18, 19). Furthermore, peptide structures have been solved ab initio using high-resolution MicroED data (20, 21). Since 2013, several research groups have shown that it is feasible to redetermine already known protein structures using MicroED (table S1), and it is only very recently that a new polymorph of hen egg white lysozyme was unveiled by MicroED, but again phased using a previously determined structure of the identical protein (22).
The sample handling of MicroED is similar to that of cryo-EM, while the data collection and processing are similar to that used in x-ray crystallography, making the technique highly adaptable to existing cryo-EM and general TEM laboratories (4–8, 21). Therefore, it is of great benefit to further develop MicroED methods to meet the needs of scientists in a wide community.
Here, we report the next step in the development of MicroED by solving a novel protein structure, Sulfolobus acidocaldarius R2-like ligand-binding oxidase, SaR2lox. The R2lox metalloenzyme family was discovered a decade ago from its similarities with the ribonucleotide reductase R2 protein (23). Its physiological function is unknown to date, but the two crystal structures solved (23, 24) of proteins belonging to this family reveal a four-helix bundle core accommodating a dinuclear metal cluster, characterized by electron paramagnetic resonance as Mn(III)/Fe(III) (25), interacting with a long-chain fatty acid.
We demonstrate that MicroED data can be collected from SaR2lox 3D microcrystals by the continuous rotation method (17, 5). Conventional x-ray crystallography software can be used directly for processing ED data [XDS (26)], determining the phases using a homologous protein model of 35% sequence identity [Phaser (27)], and refining the model structure [phenix.refine (28)]. These results illustrate that MicroED is a powerful tool for determining novel protein structures with sample requirements complementing those of x-ray crystallography, single-particle cryo-EM, and x-ray free-electron lasers.
RESULTS
Micrometer-sized 3D crystals of SaR2lox were grown using conventional hanging drop vapor diffusion and are barely large enough to be distinguished under an optical microscope from precipitate and phase separation occurring in the drop (Fig. 1A). The mother liquor used for growing microcrystals contains 44% (v/v) polyethylene glycol (PEG) 400. The viscosity of the sample prevented preparation of thin vitrified cryo-EM samples using the traditional deposit-blot-plunge routine. Thus, we used manual backside blotting to remove excess liquid and vitrified the sample in liquid ethane (29). The plate-like crystals are triangular in shape and a few micrometers in size (Fig. 1B). The thickness of the crystals is estimated to be less than 0.5 μm. We note that it is crucial to reduce the thickness of the protective layer of vitrified ice as much as possible to collect ED data of high signal-to-noise ratio from the crystals. Even in the presence of 44% (v/v) PEG 400, the backside blotting approach provided good distribution of crystals in a thin layer of vitreous ice (Fig. 1, A and B). This ability makes MicroED a more generally applicable tool in structural biology as highly viscous solutions are common in crystallization of macromolecules.
Fig. 1. Overview of the MicroED experiment.
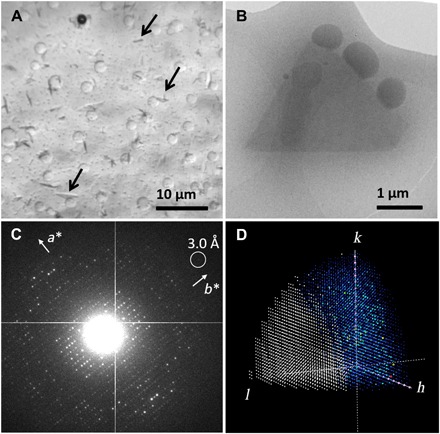
(A) SaR2lox microcrystals (pointed out by arrows) viewed under an optical microscope. (B) Overfocused TEM image of a typical diffracting SaR2lox crystal preserved in vitrified ice. The volume of this particular crystal is estimated to be approximately 2 μm3, which contains approximately 6 × 106 unit cells. (C) Typical ED pattern of the SaR2lox crystal. The frame was taken after an accumulated electron dose of 4.3 e−/Å2. (D) Reconstructed reciprocal lattice showing limited data completeness with predominately missing reflections in the direction of c* (fig. S1); missing observations are shown in white, observed reflections are in a rainbow color scheme, and systematic absences are shown in pink.
MicroED data were collected by continuously rotating a single crystal in the electron beam at a rate of 0.45°/s. The exposure time of each frame was 2 s, integrating over 0.90° of the reciprocal lattice. Data were typically collected over a total rotation range of 54° within 2 min. The crystals diffracted beyond a resolution of 3.0 Å (Fig. 1C). The electron dose rate applied was approximately 0.08 e−/Å2 per second, resulting in a total dose of less than 10 e−/Å2 per dataset. Because of their plate-like shape, a morphology commonly observed in protein crystallography, the SaR2lox crystals are dispersed on the TEM grid with a preferred orientation. A total of 35 ED datasets were collected from the plate-like SaR2lox crystals. Data were processed using crystallography software XDS (26). We could determine that the crystals are orthorhombic with a primitive unit cell using rotation ED processing software (REDp) (11). Two screw axes were identified from the reflection conditions (Fig. 1C). On the basis of unit cell consistency and cross-correlation, 21 of the 35 datasets were merged. Because of the preferred orientation of the crystals, the data completeness increased from 50.9% for a single dataset to only 62.8% (Fig. 1D and fig. S1). However, the multiplicity (~32) and overall I/σ(I) (6.12 up to a resolution of 3.0 Å) improved drastically (table S2). We demonstrated previously that the resulting structural model can be improved by merging data from a large number of crystals (8).
The processed MicroED data were of sufficient quality to solve and refine the structure of SaR2lox (Fig. 2A). The closest homolog to SaR2lox in the Protein Data Bank (PDB) is an R2lox protein from Mycobacterium tuberculosis (MtR2lox) sharing 33.8% identity of the full-length protein and 44% identity over the most conserved two-thirds of the protein sequence [PDB ID 3EE4 (23)] (fig. S2). Using a modified search model of 35% sequence identity, we were able to phase the merged MicroED data by molecular replacement using Phaser (27), obtaining a clear single solution in space group P21212. The structure was iteratively built and refined in COOT (30) and phenix.refine (28) (table S3). The refined electrostatic potential map provided sufficient detail to model side chains and allowed rebuilding of the main chain (Fig. 2, B and D), although certain parts of the maps were less well defined along the c* direction (owing to the incomplete data). The dinuclear metal cofactor was located in the map and modeled as a heterodinuclear Mn/Fe center based on previous studies on this protein family (23, 24). To assess the data quality and eliminate the influence of model bias, we generated a composite omit map covering the entire unit cell, confirming the correct interpretation for modeling the structure (Fig. 2, C and E).
Fig. 2. High-quality electrostatic potential maps allowing accurate model interpretation.
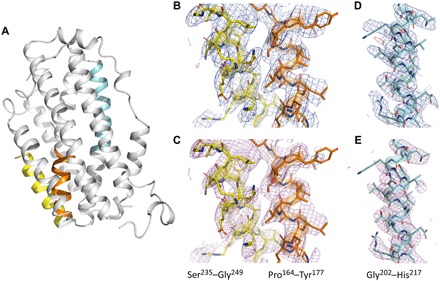
(A) Overall structure of SaR2lox solved by MicroED with three colored selections as examples to show electrostatic potential maps in (B) to (E). Electrostatic scattering potential maps 2Fo − Fc (contoured at 1σ; colored blue) and Fo − Fc (contoured at ±3σ; colored green and red for positive and negative peaks, respectively) and simulated annealing composite omit maps (contoured at 1σ; colored magenta) are shown for residues 164 to 177 (orange) and 235 to 249 (yellow) in (B) and (C), respectively, and for residues 202 to 217 (cyan) in (D) and (E), respectively. Simulated annealing composite omit electrostatic potential maps are calculated with sequential 5% fractions of the structure omitted. Only observed reflections were used for map calculations, i.e., no missing F(obs) were restored using a weighted F(calc). Despite the low completeness, the data produce high-quality well-resolved maps. Oxygen and nitrogen atoms are colored red and blue, respectively, and carbons are colored according to the selection previously mentioned.
The final model of SaR2lox shows a protein backbone Cα root mean square deviation of 0.94 Å compared to the structure of MtR2lox used for molecular replacement. This value is within the expected range from proteins of this size and sequence identity level. The structure presented here confirms the dimeric biological assembly and the ferritin-like helix bundle overall fold, previously seen in the R2lox protein family (fig. S3) (23, 24). Furthermore, the structure reveals new biochemically important features of the SaR2lox enzyme, differing from the two structures known in this family: The substrate-binding pocket is reshaped and shows an altered electrostatic potential distribution (Fig. 3). In addition, the sequence identity with existing structures is lower than 40%, which indicates a possibly divergent enzyme function (31). Thus, our results suggest that SaR2lox has a substrate specificity different from that of the enzymes previously structurally characterized. There are additional ongoing biochemical studies in our laboratory to confirm this hypothesis.
Fig. 3. Differences between substrate-binding pockets support substrate specificity divergences in the R2lox metalloenzyme family.
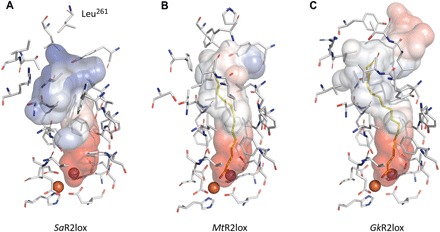
The molecular surface and identity of residues defining the substrate-binding pocket in the SaR2lox structure (A) are compared with the two already known structures of this protein family, (B) MtR2lox (PDB ID 3EE4) (23) and (C) GkR2lox (PDB ID 4HR0) (24). Although the Mn- and Fe-coordinating residues are fully conserved, the distal residues lining the cavity are remarkably divergent, drastically affecting the shape and electrostatic contact potentials of the pocket accommodating the putative substrate. This suggests that the substrate specificity could be different between these three enzymes, and thus, their function could be divergent. Electrostatic contact potentials plotted on molecular surfaces are colored in a gradient from red (negative) to blue (positive). Ligands modeled as myristic acid and palmitic acid for MtR2lox and GkR2lox, respectively, are colored yellow. A ligand was not modeled in the SaR2lox structure due to a very weak signal in the corresponding scattering potential map, but its presence cannot be excluded. Carbon, nitrogen, oxygen, manganese, and iron atoms are colored gray, blue, red, purple, and orange, respectively. We note that the missing stretch of unmodeled residues in SaR2lox (250 to 260) is not shared with MtR2lox or GkR2lox and is likely to affect distal closure of the cavity, further emphasizing differences between proteins.
DISCUSSION
MicroED data can be collected from crystals previously considered to be insufficient in size, which is its main advantage over x-ray crystallography. There are several aspects where ED can be further improved. For instance, by developing sample preparation methods such as cryogenic focused ion beam (29, 32–34), the orientation of the crystals may be precisely controlled to achieve near 100% data completeness. Furthermore, the MicroED data are not yet as accurate or as precise as generally achieved by x-ray crystallography, as indicated by poorer crystallographic quality indicators, e.g., R factors. Rapid development of data collection strategies, as well as improvements of TEM hardware, may further accelerate the development of MicroED. Our results show that MicroED can be used to determine novel protein structures, even under the circumstances frequently encountered in macromolecular crystallography, where a viscous sample environment, preferred orientation, and poor signal-to-noise ratio complicate data collection and structure determination.
MATERIALS AND METHODS
Cloning, expression, and protein purification
A construct encoding full-length S. acidocaldarius R2lox (accession number WP_011278976) was polymerase chain reaction–amplified from genomic DNA [DSM number 639, obtained from DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen), Braunschweig, Germany] and inserted into pET-46 Ek/LIC (Novagen) using the following primers: GACGACGACAAGATGAAGGAAAAATTACTTGAATTCAGAAGT and GAGGAGAAGCCCGGTTATTTGTCCAGCTTAATCTCCTCTATGAC.
Expression was carried out in Escherichia coli BL21(DE3) (Novagen). Cells were cultured at 37°C in terrific broth medium (Formedium) supplemented with ampicillin (50 μg/ml) in a benchtop bioreactor system (Harbinger). When an optical density at 600 nm of 0.7 was reached, expression was induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside and allowed to continue for 3 hours. Then, cells were harvested by centrifugation and stored at −80°C. The N-terminal His6-tagged SaR2lox protein was purified via immobilized metal-ion affinity chromatography and size exclusion chromatography. Cells were resuspended in buffer A [25 mM Hepes-Na (pH 7.0), 300 mM NaCl, and 20 mM imidazole] and disrupted by high-pressure homogenization. The lysate was cleared by centrifugation and applied to a nickel–nitrilotriacetic acid agarose (Protino) gravity flow column. The beads were washed extensively with buffer B (buffer A containing 40 mM imidazole). Protein was then eluted using buffer C (buffer A containing 250 mM imidazole), concentrated using Vivaspin 20 centrifugal concentrators with a 30,000 molecular weight cutoff polyethersulfone membrane (Sartorius), and applied to a HiLoad 16/60 Superdex 200 prep grade size exclusion column (GE Healthcare) equilibrated in a final buffer of 25 mM Hepes-Na (pH 7.0) and 50 mM NaCl. Fractions corresponding to the pure SaR2lox protein were pooled, concentrated to 16 mg/ml, aliquoted, flash-frozen in liquid nitrogen, and stored at −80°C. The protein concentration was obtained using a calculated molecular weight for this construct of 38,006 and an experimentally determined extinction coefficient at 280 nm for metal-bound protein of 52.13 mM−1 cm−1.
Crystallization
SaR2lox protein was crystallized using the hanging drop vapor diffusion method. A volume of 2 μl of a protein solution (8 mg/ml) was mixed with 2 μl of reservoir solution consisting of 44% (v/v) PEG 400, 0.2 M lithium sulfate, and 0.1 M sodium acetate (pH 3.4). Plate-like crystals grew within 48 hours at 21°C.
Sample preparation
A cryo-EM sample of SaR2lox was prepared by freezing the crystals in a thin layer of vitrified ice. A thin and uniform vitrified ice layer is crucial for obtaining MicroED of high signal-to-noise ratio. Meanwhile, the ice layer has to protect the crystals from being dehydrated under vacuum inside a TEM. The 4-μl hanging drop was deposited onto a QUANTIFOIL R 3.5/1 (300 mesh) Cu holy carbon TEM grid. The excessive liquid was removed by manual back-side blotting. The grid was then rapidly plunge-frozen in liquid ethane. We note that the automated blotting and vitrification routine using a FEI Vitrobot Mark IV was not efficient in removing the viscous mother liquid while leaving a sufficient number of crystals on the TEM grid.
Data collection
MicroED data were collected on a JEOL JEM-2100 (LaB6 filament) TEM operated at 200 kV, with a Gatan 914 cryo-transfer holder. Before searching for suitable crystals, the electron beam was aligned, while the center of the TEM grid was brought to the mechanical eucentric height. During crystal searching, by inserting a 50-μm condenser lens aperture, the size of the electron beam was set to be slightly larger than the field of view (6 μm) on the side entry Orius detector. As soon as a suitable crystal was found, the beam was blanked while we set up for MicroED data collection. These measures were taken to avoid unnecessary electron dose on surrounding crystals. An area with a diameter of 2 μm as defined by a selected area aperture was used to select the region of interest on the crystal. ED data were collected by continuously rotating the SaR2lox crystal under the electron beam while simultaneously collecting the diffraction patterns on a fast Timepix hybrid pixel detector (Amsterdam Scientific Instruments). Data were collected at a sample-to-detector distance of 1830 mm, equivalent to 0.001198 Å−1 per pixel.
Data processing
The native format of collected data is tagged image file (TIF). The data were converted to the Super Marty View (SMV) format using a python script developed in-house. The important metadata of the experiment was written automatically to the headers of each SMV frame. Data were processed using XDS (26). The data were scaled and merged based on unit cell consistency, correlation coefficients between the datasets (analyzed with XDS nonisomorphism) (35), I/σ(I), and resolution using XSCALE (26). Data were converted to MTZ format using POINTLESS (36) and merged with AIMLESS (37), and structure factor amplitudes were calculated using TRUNCATE (38). Figure 1D and fig. S1 were prepared with the Reflection Data Viewer in Phenix (39).
Structure solution and refinement
The structure was solved by molecular replacement using Phaser (27) with atomic scattering factors for electrons. A truncated search model was created using the atomic coordinates of the R2-like ligand-binding oxidase from M. tuberculosis (PDB ID 3EE4) (23) using Sculptor (39). A well-contrasted solution was obtained with one molecule per asymmetric unit in the space group P21212 [log-likelihood gain (LLG) = 161, translation function Z-score (TFZ) = 14.0]. A solution in the alternative space group P212121 was substantially worse (LLG = 48, TFZ = 5.6), confirming the absence of the screw axis along c*. The structure model was refined using rigid body refinement directly after molecular replacement in phenix.refine (28). The structure was iteratively built using COOT (30) and refined using phenix.refine (28) with atomic scattering factors for electrons, automatic weighting of the geometry term, and group B factors per residue. Structure solution and refinement were performed using the merged intensities.
Certain regions of the map were less well resolved, particularly residues 250 to 260. As the map was difficult to interpret, we did not attempt to place those residues. Furthermore, a segment of an α helix, residues 235 to 249, appeared to be shifted judging from the difference potential map after attempting to fit the corresponding side chains. This part was corrected and refined by real-space refinement in COOT (30) using geometrical restraints. Simulated annealing composite omit maps were generated using phenix.composite_omit_map (28), calculated with sequential 5% fractions of the structure omitted.
The model was validated using MolProbity (40). Table S2 lists the crystallographic statistics in which the test set represents 5% of the reflections. The core root mean square deviation values between structures were calculated by the secondary-structure matching tool (41). Figures 2 and 3 and fig. S3 were prepared using the PyMOL Molecular Graphics System, version 2.2.3, Schrödinger LLC. Electrostatic protein contact potentials were generated with the vacuum electrostatics tool in PyMOL, using the default values for cavity detection radius and cutoff and using a level range of ±50.
Supplementary Material
Acknowledgments
H.L. thanks P. Stenmark for discussions about this manuscript. Funding: We acknowledge financial support from the Knut and Alice Wallenberg Foundation through the project grants 3DEM-NATUR (no. 2012.0112 to X.Z.) and Wallenberg Academy Fellows (no. 2017.0275 to M.H.), the Science for Life Laboratory through the pilot project grant Electron Nanocrystallography, the European Research Council (HIGH-GEAR 724394 to M.H.), and the Swedish Research Council (2017-04018 to M.H. and 2017-05333 to H.X.). Author contributions: H.X. contributed to project design, conception, ED data collection, ED data analysis, manuscript writing, and figure creation. H.L contributed to project design, crystal growth, structure determination, manuscript writing, and figure creation. M.T.B.C. contributed to ED data analysis, structure determination, manuscript writing, and figure creation. J.Z. contributed to ED data collection. J.J.G. contributed to cloning. X.Z. and M.H. contributed to project design, data analysis, conception, and manuscript writing. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The atomic coordinates of the SaR2lox structure are deposited in the PDB under accession code 6QRZ. Raw MicroED data are available upon request. The python script used for converting diffraction frames from TIF to MRC and IMG (SMV format) is available upon request. REDp can be downloaded at mmk.su.se/zou. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaax4621/DC1
Fig. S1. Views of reciprocal space showing the merged intensities of SaR2lox.
Fig. S2. Sequence alignment between SaR2lox (UniProt identifier Q4J6V7) and R2lox from M. tuberculosis MtR2lox (UniProt identifier P9WH69) used as molecular replacement search model for the structure presented in this study (PDB ID 3EE4) (23).
Fig. S3. Overall superimposition of crystal structures of R2lox dimers.
Table S1. Overview of protein structures in the PDB determined by MicroED.
Table S2. ED merging statistics for a single-crystal dataset and merging of 11, 14 and 21 crystal datasets.
Table S3. Data collection and refinement statistics of SaR2lox.
REFERENCES AND NOTES
- 1.Henderson R., The potential and limitations of neutrons, electrons and X-rays for atomic resolution microscopy of unstained biological molecules. Q. Rev. Biophys. 28, 171–193 (1995). [DOI] [PubMed] [Google Scholar]
- 2.Glaeser R. M., Downing K. H., High-resolution electron crystallography of protein molecules. Ultramicroscopy 52, 478–486 (1993). [DOI] [PubMed] [Google Scholar]
- 3.Subramanian G., Basu S., Liu H., Zuo J. M., Spence J. C. H., Solving protein nanocrystals by cryo-EM diffraction: Multiple scattering artifacts. Ultramicroscopy 148, 87–93 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Shi D., Nannenga B. L., Iadanza M. G., Gonen T., Three-dimensional electron crystallography of protein microcrystals. eLife 2, e01345 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nannenga B. L., Shi D., Leslie A. G. W., Gonen T., High-resolution structure determination by continuous-rotation data collection in MicroED. Nat. Methods 11, 927–930 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yonekura K., Kato K., Ogasawara M., Tomita M., Toyoshima C., Electron crystallography of ultrathin 3D protein crystals: Atomic model with charges. Proc. Natl. Acad. Sci. U.S.A. 112, 3368–3373 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clabbers M. T. B., van Genderen E., Wan W., Wiegers E. L., Gruene T., Abrahams J. P., Protein structure determination by electron diffraction using a single three-dimensional nanocrystal. Acta Crystallogr., Sect. D: Sect. Struct. Biol. 73, 738–748 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu H., Lebrette H., Yang T., Srinivas V., Hovmöller S., Högbom M., Zou X., A rare lysozyme crystal form solved using highly redundant multiple electron diffraction datasets from micron-sized crystals. Structure 26, 667–675.e3 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Khoshouei M., Radjainia M., Baumeister W., Danev R., Cryo-EM structure of haemoglobin at 3.2 Å determined with the Volta phase plate. Nat. Commun. 8, 16099 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mugnaioli E., Gorelik T., Kolb U., “Ab initio” structure solution from electron diffraction data obtained by a combination of automated diffraction tomography and precession technique. Ultramicroscopy 109, 758–765 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Wan W., Sun J., Su J., Hovmöller S., Zou X., Three-dimensional rotation electron diffraction: Software RED for automated data collection and data processing. J. Appl. Crystallogr. 46, 1863–1873 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gemmi M., La Placa M. G. I., Galanis A. S., Rauch E. F., Nicolopoulos S., Fast electron diffraction tomography. J. Appl. Crystallogr. 48, 718–727 (2015). [Google Scholar]
- 13.Kolb U., Gorelik T. E., Mugnaioli E., Stewart A., Structural characterization of organics using manual and automated electron diffraction. Polym. Rev. 50, 385–409 (2010). [Google Scholar]
- 14.van Genderen E., Clabbers M. T. B., Das P. P., Stewart A., Nederlof I., Barentsen K. C., Portillo Q., Pannu N. S., Nicolopoulos S., Gruene T., Abrahams J. P., Ab initio structure determination of nanocrystals of organic pharmaceutical compounds by electron diffraction at room temperature using a Timepix quantum area direct electron detector. Acta Crystallogr., Sect. A: Found. Adv. 72, 236–242 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palatinus L., Brázda P., Boullay P., Perez O., Klementová M., Petit S., Eigner V., Zaarour M., Mintova S., Hydrogen positions in single nanocrystals revealed by electron diffraction. Science 355, 166–169 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Zou X. D., Mo Z. M., Hovmöller S., Li X. Z., Kuo K. H., Three-dimensional reconstruction of the ν-AlCrFe phase by electron crystallography. Acta Crystallogr. A 59, 526–539 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Nederlof I., van Genderen E., Li Y.-W., Abrahams J. P., A Medipix quantum area detector allows rotation electron diffraction data collection from submicrometre three-dimensional protein crystals. Acta Crystallogr. D Biol. Crystallogr. 69, 1223–1230 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gruene T., Wennmacher J. T. C., Zaubitzer C., Holstein J. J., Heidler J., Fecteau-Lefebvre A., De Carlo S., Müller E., Goldie K. N., Regeni I., Li T., Santiso-Quinones G., Steinfeld G., Handschin S., van Genderen E., van Bokhoven J. A., Clever G. H., Pantelic R., Rapid structure determination of microcrystalline molecular compounds using electron diffraction. Angew. Chem. Int. Ed. 57, 16313–16317 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones C. G., Martynowycz M. W., Hattne J., Fulton T. J., Stoltz B. M., Rodriguez J. A., Nelson H. M., Gonen T., The cryoEM method MicroED as a powerful tool for small molecule structure determination. ACS Cent. Sci. 4, 1587–1592 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodriguez J. A., Ivanova M. I., Sawaya M. R., Cascio D., Reyes F. E., Shi D., Sangwan S., Guenther E. L., Johnson L. M., Zhang M., Jiang L., Arbing M. A., Nannenga B. L., Hattne J., Whitelegge J., Brewster A. S., Messerschmidt M., Boutet S., Sauter N. K., Gonen T., Eisenberg D. S., Structure of the toxic core of α-synuclein from invisible crystals. Nature 525, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de la Cruz M. J., Hattne J., Shi D., Seidler P., Rodriguez J., Reyes F. E., Sawaya M. R., Cascio D., Weiss S. C., Kim S. K., Hinck C. S., Hinck A. P., Calero G., Eisenberg D., Gonen T., Atomic-resolution structures from fragmented protein crystals with the cryoEM method MicroED. Nat. Methods 14, 399–402 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lanza A., Margheritis E., Mugnaioli E., Cappello V., Garau G., Gemmi M., Nanobeam precession-assisted 3D electron diffraction reveals a new polymorph of hen egg-white lysozyme. IUCrJ 6, 178–188 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andersson C. S., Högbom M., A Mycobacterium tuberculosis ligand-binding Mn/Fe protein reveals a new cofactor in a remodeled R2-protein scaffold. Proc. Natl. Acad. Sci. U.S.A. 106, 5633–5638 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griese J. J., Roos K., Cox N., Shafaat H. S., Branca R. M. M., Lehtiö J., Gräslund A., Lubitz W., Siegbahn P. E. M., Högbom M., Direct observation of structurally encoded metal discrimination and ether bond formation in a heterodinuclear metalloprotein. Proc. Natl. Acad. Sci. U.S.A. 110, 17189–17194 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shafaat H. S., Griese J. J., Pantazis D. A., Roos K., Andersson C. S., Popović-Bijelić A., Gräslund A., Siegbahn P. E. M., Neese F., Lubitz W., Högbom M., Cox N., Electronic structural flexibility of heterobimetallic Mn/Fe cofactors: R2lox and R2c proteins. J. Am. Chem. Soc. 136, 13399–13409 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Kabsch W., XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D., Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duyvesteyn H. M. E., Kotecha A., Ginn H. M., Hecksel C. W., Beale E. V., de Haas F., Evans G., Zhang P., Chiu W., Stuart D. I., Machining protein microcrystals for structure determination by electron diffraction. Proc. Natl. Acad. Sci. U.S.A. 115, 9569–9573 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Tian W., Skolnick J., How well is enzyme function conserved as a function of pairwise sequence identity? J. Mol. Biol. 333, 863–882 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Martynowycz M. W., Zhao W., Hattne J., Jensen G. J., Gonen T., Collection of continuous rotation microED data from ion beam-milled crystals of any size. Structure 27, 545–548.e2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou H., Luo Z., Li X., Using focus ion beam to prepare crystal lamella for electron diffraction. J. Struct. Biol. 205, 59–64 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Li X., Zhang S., Zhang J., Sun F., In situ protein micro-crystal fabrication by cryo-FIB for electron diffraction. Biophys. Rep. 4, 339–347 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diederichs K., Dissecting random and systematic differences between noisy composite data sets. Acta Crystallogr., Sect. D: Struct. Biol. 73, 286–293 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evans P. R., Murshudov G. N., How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans P., Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 (2006). [DOI] [PubMed] [Google Scholar]
- 38.French S., Wilson K., On the treatment of negative intensity observations. Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr. 34, 517–525 (1978). [Google Scholar]
- 39.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen V. B., Arendall W. B. III, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krissinel E., Henrick K., Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Sauter C., Otálora F., Gavira J.-A., Vidal O., Giegé R., García-Ruiz J. M., Structure of tetragonal hen egg-white lysozyme at 0.94 Å from crystals grown by the counter-diffusion method. Acta Crystallogr. D Biol. Crystallogr. 57, 1119–1126 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Sharma P., Verma N., Singh P. K., Korpole S., Ashish, Characterization of heat induced spherulites of lysozyme reveals new insight on amyloid initiation. Sci. Rep. 6, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nannenga B. L., Shi D., Hattne J., Reyes F. E., Gonen T., Structure of catalase determined by MicroED. eLife 3, e03600 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Foroughi L. M., Kang Y.-N., Matzger A. J., Polymer-induced heteronucleation for protein single crystal growth: Structural elucidation of bovine liver catalase and concanavalin a forms. Cryst. Growth Des. 11, 1294–1298 (2011). [Google Scholar]
- 46.Yonekura K., Maki-Yonekura S., Refinement of cryo-EM structures using scattering factors of charged atoms. J. Appl. Cryst. 49, 1517–1523 (2016). [Google Scholar]
- 47.Yonekura K., Ishikawa T., Maki-Yonekura S., A new cryo-EM system for electron 3D crystallography by eEFD. J. Struct. Biol. 206, 243–253 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Toyoshima C., Nakasako M., Nomura H., Ogawa H., Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature 405, 647–655 (2000). [DOI] [PubMed] [Google Scholar]
- 49.Hattne J., Shi D., de la Cruz M. J., Reyes F. E., Gonen T., Modeling truncated pixel values of faint reflections in MicroED images. J. Appl. Cryst. 49, 1029–1034 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo F., Zhou W., Li P., Mao Z., Yennawar N. H., French J. B., Huang T. J., Precise manipulation and patterning of protein crystals for macromolecular crystallography using surface acoustic waves. Small 11, 2733–2737 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hattne J., Shi D., Glynn C., Zee C.-T., Gallagher-Jones M., Martynowycz M. W., Rodriguez J. A., Gonen T., Analysis of global and site-specific radiation damage in cryo-EM. Structure 26, 759–766.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Watanabe N., From phasing to structure refinement in-house: Cr/Cu dual-wavelength system and a loopless free crystal-mounting method. Acta Crystallogr. D Biol. Crystallogr. 62, 891–896 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Warkentin M., Badeau R., Hopkins J. B., Thorne R. E., Spatial distribution of radiation damage to crystalline proteins at 25–300 K. Acta Crystallogr. D Biol. Crystallogr. 68, 1108–1117 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walter J., Steigemann W., Singh T. P., Bartunik H., Bode W., Huber R., On the disordered activation domain in trypsinogen: Chemical labelling and low-temperature crystallography. Acta Crystallogr. B 38, 1462–1472 (1982). [Google Scholar]
- 55.English A. C., Done S. H., Caves L. S. D., Groom C. R., Hubbard R. E., Locating interaction sites on proteins: The crystal structure of thermolysin soaked in 2% to 100% isopropanol. Proteins 37, 628–640 (1999). [PubMed] [Google Scholar]
- 56.Kim S. K., Barron L., Hinck C. S., Petrunak E. M., Cano K. E., Thangirala A., Iskra B., Brothers M., Vonberg M., Leal B., Richter B., Kodali R., Taylor A. B., Du S., Barnes C. O., Sulea T., Calero G., Hart P. J., Hart M. J., Demeler B., Hinck A. P., An engineered transforming growth factor β (TGF-β) monomer that functions as a dominant negative to block TGF-β signaling. J. Biol. Chem. 292, 7173–7188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu S., Gonen T., MicroED structure of the NaK ion channel reveals a Na+ partition process into the selectivity filter. Commun. Biol. 1, 38 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alam A., Jiang Y., Structural analysis of ion selectivity in the NaK channel. Nat. Struct. Mol. Biol. 16, 35–41 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Purdy M. D., Shi D., Chrustowicz J., Hattne J., Gonen T., Yeager M., MicroED structures of HIV-1 Gag CTD-SP1 reveal binding interactions with the maturation inhibitor bevirimat. Proc. Natl. Acad. Sci. U.S.A. 115, 13258–13263 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wagner J. M., Zadrozny K. K., Chrustowicz J., Purdy M. D., Yeager M., Ganser-Pornillos B. K., Pornillos O., Crystal structure of an HIV assembly and maturation switch. eLife 5, e17063 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karplus P. A., Diederichs K., Linking crystallographic model and data quality. Science 336, 1030–1033 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaax4621/DC1
Fig. S1. Views of reciprocal space showing the merged intensities of SaR2lox.
Fig. S2. Sequence alignment between SaR2lox (UniProt identifier Q4J6V7) and R2lox from M. tuberculosis MtR2lox (UniProt identifier P9WH69) used as molecular replacement search model for the structure presented in this study (PDB ID 3EE4) (23).
Fig. S3. Overall superimposition of crystal structures of R2lox dimers.
Table S1. Overview of protein structures in the PDB determined by MicroED.
Table S2. ED merging statistics for a single-crystal dataset and merging of 11, 14 and 21 crystal datasets.
Table S3. Data collection and refinement statistics of SaR2lox.