

Bcl11b orchestrates a Treg cell transcription program by regulating functional recruitment of the master regulator Foxp3.
Abstract
Foxp3 and its protein partners establish a regulatory T (Treg) cell transcription profile and promote immunological tolerance. However, molecular features contributing to a Treg-specific gene expression program are still incompletely understood. We find that the transcription factor Bcl11b is a prominent Foxp3 cofactor with multifaceted functions in Treg biology. Optimal genomic recruitment of Foxp3 and Bcl11b is critically interdependent. Genome-wide occupancy studies coupled with gene expression profiling reveal that Bcl11b, in association with Foxp3, is primarily responsible in establishing a Treg-specific gene activation program. Furthermore, Bcl11b restricts misdirected recruitment of Foxp3 to sites, which would otherwise result in an altered Treg transcriptome profile. Consequently, Treg-specific ablation of Bcl11b results in marked breakdown of immune tolerance, leading to aggressive systemic autoimmunity. Our study provides previously underappreciated mechanistic insights into molecular events contributing to basic aspects of Treg function. Furthermore, it establishes a therapeutic target with potential implications in autoimmunity and cancer.
INTRODUCTION
The precautionary act of defending self from autoreactive lymphocytes, a process termed “immunological tolerance,” is largely accepted to be mediated by two nonredundant mechanisms. Elimination of autoreactive thymocytes triggered by high-affinity interaction with self-antigens includes a class of cell-intrinsic mechanism referred to as recessive tolerance. An additional layer of control of overexuberant immune response is mediated by specialized classes of immune cell types that contribute toward an essential trans-acting mechanism termed dominant tolerance. Most prominent among the cell types mediating dominant tolerance are the so-called regulatory T (Treg) cells, a subset of CD4+ T lymphocytes critical for suppressing autoimmunity, tissue homeostasis, and uncontrolled immune response against pathogens. The X chromosome–encoded transcription factor Foxp3, a hallmark of Treg cells, is responsible for establishing a unique transcriptional program that functionally and phenotypically distinguishes them from all other T cell lineages (1).
Despite its unequivocal role in Treg biology, our understanding of the molecular basis of Foxp3’s function is still evolving. To determine the molecular composition of Foxp3-associated macromolecular complexes and their physiological relevance in maintaining immune homeostasis, we previously performed unbiased proteomic identification of Foxp3-associated nuclear factors (2). One of the most prominent transcription factors that we confirmed to be strongly interacting with Foxp3 is the C2H2 zinc finger domain–containing transcription factor Bcl11b. Initially identified as a tumor suppressor in T cells, Bcl11b in recent years have been implicated in multiple aspects of immune-related processes of development and function. During hematopoietic development, Bcl11b acts as a checkpoint necessary to promote T cell lineage commitment while suppressing natural killer cell potential (3). In early stages of T cell development, it is necessary for survival of double-negative and double-positive thymocytes, β selection, and positive selection of CD4 and CD8 single-positive thymocytes (4). It has been shown to suppress T helper 1 (TH1) differentiation and restrict TH17 cell plasticity by blocking the TH2 effector program (5). Furthermore, extending its role beyond T cell biology, Bcl11b was also demonstrated to be a critical transcription factor required to maintain the identity and function of type 2 innate lymphoid cells (6).
In recent past, primarily using a pan T cell–specific conditional knockout model in combination with an adoptive transfer–based experimental system, Bcl11b has been implicated in Treg-dependent suppression of inflammatory bowel disease, at least part of which is mediated by Bcl11b-dependent maintenance of optimal gene expression of the anti-inflammatory cytokine IL10 and of Foxp3 itself (7). While these findings point toward a critical role of Bcl11b in Treg suppressive function, precise understanding of the Treg cell–specific role of Bcl11b remains unclear, due to delayed and inefficient deletion of the loxP-flanked Bcl11b conditional (Bcl11bf/f) allele when crossed to mice harboring a bacterial artificial chromosome (BAC) transgene encoding Foxp3–enhanced green fluorescent protein–Cre (7).
Because of the strong interaction between Bcl11b and Foxp3 observed in our earlier study (2) and the different lines of physiological implications discussed above, here we directly investigate the consequence of conditional ablation of Bcl11b in Treg cells. Notably, we find that in contrast to the relatively mild and delayed phenotype observed earlier, efficient deletion of Bcl11b at an early age results in drastic whole-body autoimmunity, characterized by an overtly activated immune system that is reminiscent of mice in which functional Treg cells are absent. In concert to these findings, mechanistic investigations strongly implicate Bcl11b to be an essential cofactor of Foxp3 and establish its diverse functions in configuring the gene expression network required for mediating Treg homeostasis and function.
RESULTS
Systemic and lethal autoimmunity upon Treg-specific deletion of Bcl11b
To investigate the consequence of conditional ablation of Bcl11b in Treg cells, we generated Bcl11bf/fFoxp3IRES-YFP-Cre mice [henceforth designated as “KO” and control littermates Bcl11b+/+Foxp3IRES-YFP-Cre or Bcl11bf/+Foxp3IRES-YFP-Cre, which are phenotypically indistinguishable, designated as “WT” (wild type)] by crossing mice harboring Bcl11bf/f allele with Foxp3IRES-YFP-Cre mice expressing yellow fluorescent protein (YFP)–Cre recombinase fusion protein under the control of Foxp3 regulatory elements. Of note, the Foxp3IRES-YFP-Cre knock-in mice were generated independently (8), different from a BAC transgene-encoded “Cre”-expressing mouse line used earlier to delete Bcl11b in Treg cells (7), which is routinely used for efficient Treg-specific depletion of target alleles. In these newly generated KO mice, we observed efficient Treg-specific deletion of Bcl11b, 3 weeks after birth, which was the earliest point that they were analyzed (fig. S1A). The KO mice were born in expected Mendelian ratios and, more importantly, displayed an overtly aggressive autoimmune phenotype characterized by extreme forms of lymphadenopathy, splenomegaly, and cellular infiltration in various tissues, which led to early mortality within 5 weeks after birth. Overall, the extent of autoimmunity in these mice is qualitatively similar to those harboring a Foxp3 reporter null allele (Foxp3GFPKO) that leads to complete lack of functional Treg population from birth (fig. S1, B to D) (9).
As expected, analysis of KO mice revealed markedly increased total cellularity and increased CD4+ and CD8+ T cell compartments within lymph nodes and spleen (Fig. 1, A and B). CD4+Foxp3− population in these mice exhibited enhanced expression of a panel of markers known to be associated with activation, including CD25, glucocorticoid-induced TNFR-related protein (GITR), inducible T-cell co-stimulator (ICOS), and cytotoxic T-lymphocyte associated protein 4 (CTLA4) (Fig. 1C and fig. S2A). The effector memory population, characterized by CD62LloCD44hi, was markedly increased in CD4+Foxp3− and CD8+ compartments, as was the percentage of Ki67+ proliferating T cells (Fig. 1, D and E). A similar increase was observed in cytokine production of both TH1 and TH2 effector types, characterized by interferon-γ (IFN-γ) and interleukin-4 (IL-4) and IL-13, respectively (Fig. 1F). Furthermore, serum concentrations of all the immunoglobulin subtypes examined, IgM, IgG, and IgE, were remarkably increased in KO mice compared to WT littermates (Fig. 1G). Together, these results demonstrated overt and systemic immune dysregulation upon Treg-specific deletion of Bcl11b.
Fig. 1. Marked immune dysregulation in Bcl11bf/fFoxp3IRES-YFP-cre mice.
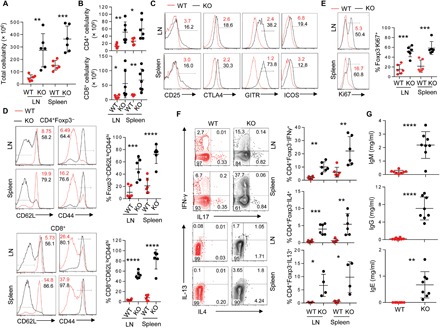
(A and B) Total (A) and CD4+ and CD8+ T cell compartment cellularity (B) within indicated secondary lymphoid organs in 3- to 5-week-old Bcl11bf/fFoxp3IRES-YFP-cre (KO) or littermate Bcl11b+/+Foxp3IRES-YFP-Cre (WT) mice. (C) Representative fluorescence-activated cell sorting (FACS) plots showing the expressions of indicated activation markers on CD4+Foxp3− cells in WT and KO mice. Statistical quantification is summarized in fig. S2A. (D to F) Representative FACS plots and quantification of CD4+ and CD8+ effector memory CD44hiCD62Llo T cells (D), Ki67+ proliferating CD4+ T cells (E), and cytokine-producing CD4+ T cells (F) within indicated lymphoid organs of WT and KO mice. (G) Concentrations of IgM, IgG, and IgE in serum of 3- to 5-week-old WT or KO mice, determined by enzyme-linked immunosorbent (ELISA) assay. Data are representative of six to eight mice from two to three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001(Student’s t test; error bars denote SD).
Bcl11b-deficient Treg cells develop normally but are compromised for function and homeostasis
The extreme autoimmune phenotype and activated state of the effector T cell population observed in the KO mice strongly suggested an indispensable and nonredundant role of Bcl11b in Treg cell function and/or homeostasis. Analysis of the CD4+Foxp3+ compartments of WT and KO mice revealed that Treg cell frequency, while lightly reduced in the spleen, is largely unaffected in the lymph nodes of KO mice. Also, the mean fluorescence intensity (MFI) of Foxp3 remained unaltered in Bcl11b-deficient Treg cells (Fig. 2A). The effector Treg population characterized by CD62LloCD44hi was found to be marginally increased in KO mice, primarily in the spleen (fig. S2B). Furthermore, the Treg cells displayed higher frequencies of GITR, CTLA4, ICOS, and Ki67, indicating heightened activation and proliferation, presumably as a consequence secondary to the marked immune dysregulation ensuing in these mice (Fig. 2, B and C and fig. S2C). These overall characterizations of Treg cells suggested that the observed phenotype of the KO mice is unlikely a result of sheer reduction in Treg cell number or due to compromised expression of Foxp3 at a per-cell level. We therefore anticipated that Bcl11b might be directly associated with Treg cell function. In an in vitro suppression assay, Bcl11b-deficient Treg cells displayed an almost complete loss of suppressive capacity compared to their WT counterparts (Fig. 2D).
Fig. 2. Phenotypic analyses of Bcl11b-deficient Treg cells.
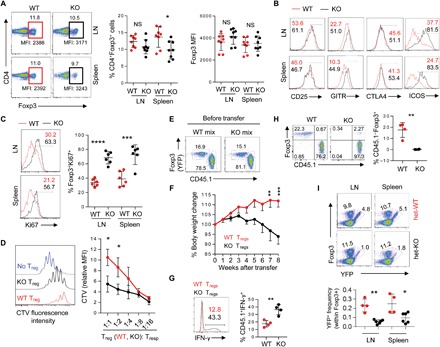
(A) Representative FACS plot (left), quantification of percentage CD4+Foxp3+ Treg cells (middle), and mean fluorescence intensity (MFI) of Foxp3 (right) from cells isolated from 3- to 5-week-old WT or KO mice. (B) Representative FACS plots showing the expressions of indicated activation markers on Treg cells in WT and KO mice. Statistical quantification is summarized in fig. S2C. (C) Representative FACS plots and quantification of Ki67+ proliferating CD4+Foxp3+ Treg cells isolated from indicated lymphoid organs. (D) In vitro suppression assay, illustrating suppression of T responder (Tresp) cell proliferation by Treg cells sorted from WT or KO mice. Histogram represents CTV dilution of labeled sorted Tnv cells alone (no Treg) or cocultured sorted WT- or KO-derived Treg cell populations at 1:2 Treg/Tresp ratio (left). Cumulative data expressed as CTV MFI of responder CD4+ cells for all the indicated Treg/Tresp coculture ratios normalized against CTV MFI of responder cells cultured alone are shown on the right. Data represent two to three independent experiments. *P < 0.05 (Student’s t test; error bars denote SD). (E) FACS plots representing mix of sorted YFP+ Treg cells from WT or KO mice, along with colitogenic Ly5.1+CD45RBhi T cells that were transferred in recipient RAG1−/− host mice, which were eventually monitored for weight loss, indicative of in vivo suppressive capacity of cotransferred Treg cell populations. (F) Percentage body weight change over time of RAG1−/− mice that were cotransferred with Ly5.1+CD45RBhi T cells along with WT- or KO-derived Treg cells. Data are representative of two independent experiments (n = 6 in each group). Mean ± SEM.**P < 0.01 and ***P < 0.001 [two way analysis of variance (ANOVA), Bonferroni posttest]. (G) Intracellular staining for IFN-γ in lymph nodes of representative recipient mice with WT or KO Treg cells that were viable at the end of the experiment. (H) FACS plots (left) and summary (right) of percentage of Treg cells remaining in the lymph nodes of representative recipient mice at the end of the experiment. (I) Analyses of Bcl11b-sufficient and Bcl11b-deficient Treg cells in heterozygous female Bcl11b+/+Foxp3IRES-YFP-Cre/+ (het-WT) and Bcl11bf/fFoxp3IRES-YFP-Cre/+ (het-KO) mice, in which half of the Treg compartment expresses YFP-cre. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.001; NS, not significant (Student’s t test; error bars denote SD).
Next, we employed a cell transfer model of colitis to assess Treg-specific functions of Bcl11b in vivo in an inflammatory setting. Allelically marked colitogenic CD4+Foxp3−CD45RBhi T cells were transferred in RAG1−/− (recombination activating gene 1-deficient) recipient mice along with Treg cells isolated from either WT or KO mice (Fig. 2E). As shown in Fig. 2F, while cotransfer with WT Treg cells could prevent body weight loss, that of Treg cells isolated from KO mice could not (Fig. 2F). This was associated with a significant increase in IFN-γ+ effector T cell population in mice harboring KO-derived Treg cells (Fig. 2G), suggesting severely compromised suppression in these mice. While the ratio between Treg and CD45.1+ effector T cells remained unperturbed in the mice that received WT Treg cells, we could hardly detect any Bcl11b-deficient Foxp3+ Treg population in transferred RAG1−/− recipients at the end of the experiment (Fig. 2H). These results suggest that under inflammatory settings created by the cell transfer model of colitis, Bcl11b deficiency results in severely compromised homeostasis in Treg cells, presumably due to enhanced cell death.
The above results raised the question whether homeostasis and maintenance of Bcl11b-deficient Treg cells are also affected in a competitive scenario. We therefore examined relative sizes of Bcl11b-sufficient and Bcl11b-deficient Treg cell subsets in heterozygous Bcl11bf/fFoxp3IRES-YFP-Cre/+ (designated as “het-KO”) and littermate Bcl11b+/+ or f/+ Foxp3IRES-YFP-Cre/+ (designated as “het-WT”) female mice. Notably, het-KO females are disease free, since owing to X chromosome inactivation, only half of the Treg cells express YFP-Cre, resulting in deletion of Bcl11b. Analyses of the YFP-Cre+ compartments of Treg cell populations in het-WT and het-KO mice revealed that the proportion of peripheral YFP-Cre+ Treg cells was markedly reduced in het-KO mice compared to that in het-WT littermates (Fig. 2I). Deletion of Bcl11b in the absence of inflammation therefore results in diminished competitive fitness of Treg cells when their WT counterparts are present. Together, these results strongly suggested that Bcl11b is critical for overall suppressive functions, as well as homeostasis and maintenance of Treg cells.
The Treg gene expression program is critically altered in the absence of Bcl11b
To acquire an in-depth understanding of the Treg-specific molecular functions of Bcl11b, we first wished to determine the transcriptional profile of Bcl11b-deficient Treg cells by high-throughput next-generation cDNA sequencing [RNA sequencing (RNA-seq)]. Comparing gene expression between WT and KO mice is not ideal due to bystander effects arising from overt autoimmunity upon Treg-specific deletion of Bcl11b. Therefore, to directly compare the cell-intrinsic gene expression profile from Bcl11b-deficient and Bcl11b-sufficient Treg cells, we decided to perform RNA-seq analyses from cells sorted from het-WT and het-KO mice, where these cells reside in comparable noninflammatory conditions. RNA-seq analysis was performed on CD4+YFP+ cells that were sorted at high purity from het-WT and het-KO mice (fig. S3A). In an initial phase of analysis, considering the marked loss of Treg cell function in the absence of Bcl11b, we examined the expression of several effector molecules that have been implicated in Treg suppressive activity. We found that the expression of most of these molecules was markedly reduced in het-KO–derived CD4+YFP+ cells compared to those from het-WT mice. In agreement with what was reported previously (7), IL10 was observed to be one of the most affected genes, with ~10-fold reduction in expression. Other affected genes implicated in Treg suppressive function were Itgae (encoding CD103), Lag3, Itgb8, Ebi3, and Tigit. Nt5e (encoding CD73), Icos, and Tgfb1 were also reduced, albeit to a lesser extent (Fig. 3A). Of note, we also observed significant down-regulation of genes encoding CD25 (Il2ra) and CTLA4 in het-KO compared to het-WT, although the expression of these gene products remained unchanged or even increased in KO mice (Fig. 2B and fig. S2C). This observation suggested that the overt autoimmune backdrop in KO mice resulted in up-regulation of these gene products, which are otherwise cell intrinsically repressed in the absence of Bcl11b under disease-free conditions. Overall, these findings are in concert with a gross defect in suppressive capacity of Bcl11b-deficient Treg cells and the marked autoimmune phenotype observed in KO mice.
Fig. 3. Treg genetic signature is critically dependent on the presence of Bcl11b.
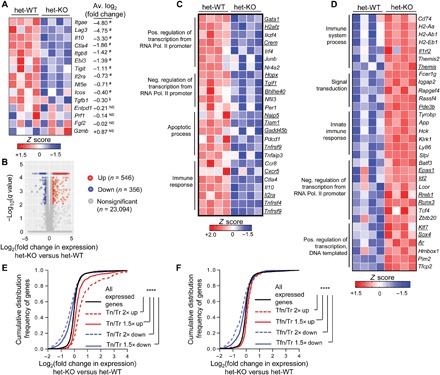
RNA-seq analysis was performed on YFP-Cre+ Treg cells sorted from het-WT or het-KO mice. (A) Expression of candidate Treg signature genes implicated in suppressive function. Each box represents one of four replicates per group. #P < 10−3 and $P < 10−2. P values are for het-KO versus het-WT Treg cells as determined by differential gene expression (DGE) analysis. (B) DGE analysis comparing combined gene expression in YFP-Cre+ Treg cells from het-KO versus het-WT. Up and Down signify genes whose expressions are 1.5-fold up-regulated or down-regulated, respectively. (C and D) Heatmaps of selective Down (C) or Up (D) genes that also represent bonafide Treg signature genes, clustered according to Gene Ontology analysis. The respective terms for each cluster under the “biological process” category are indicated on the left. Underlined genes represent Foxp3-dependent ones, whose expressions are more than 1.5-fold altered between wild-type (WT) Treg versus GFP+ Foxp3-null (Tfn) cells derived from heterozygous Foxp3GFPKO/WT females. (E and F) Cumulative distribution function (CDF) analyses of gene expression changes between Bcl11b-deficient versus Bcl11b-sufficient Treg cells for gene subsets 1.5 or 2-fold up- or down-regulated in Treg cells compared to T naïve (E) or Tfn (F). ****P < 10−16 (two-tailed Kolmogorov-Smirnov test).
We next performed differential gene expression (DGE) analysis of the RNA-seq data. Among all the expressed genes (10,349 among 23,996 genes present), 546 genes were up-regulated (“Up” genes) and 356 genes were down-regulated (“Down” genes) ≥1.5-fold in het-KO–derived CD4+YFP+ cells compared to that from het-WT mice, with a false discovery rate (FDR)–adjusted P value cutoff of 0.05 (Fig. 3B). Gene Ontology analysis [using Database for Annotation, Visualization and Integrated Discovery (DAVID 6.8) software package https://david.ncifcrf.gov/home.jsp] of the “biological-process” category on Bcl11b-dependent gene sets revealed, as expected, a number of genes implicated in immune-related functions (such as “Immune system process,” “Inflammatory response,” and “Innate immune response”) and processes contributing to cell survival and homeostasis (like “Apoptotic process”), as well as genes implicated in transcriptional control of gene expression (fig. S3B and table S1).
To determine whether the absence of Bcl11b in Treg cells affects the Treg-specific and/or Foxp3-dependent gene expression program, we next compared the above DGE dataset with Treg signature genes [differentially expressed genes (DEGs) in CD4+Foxp3+ Treg compared to CD4+Foxp3− naïve T (Tn) cells], as well as Foxp3-dependent signature genes [DEGs in CD4+Foxp3+ compared to GFP+ Foxp3-null (Tfn) cells derived from Foxp3GFPKO mice] (9, 10). It is well established that while many are Foxp3 dependent, the magnitude and pattern of expression for a large fraction of Treg signature genes are shaped independent of Foxp3 (9). We observed that a large proportion of the Bcl11b-dependent Up or Down genes are bonafide Treg signatures, whose expressions are negatively altered in the absence of Bcl11b. For example, Gata1, Ikzf4, and Irf4, which represent “activated” Treg signature, are Down in Bcl11b-deficient Treg cells, and genes encoding Pde3b, Tcf4, and Fcer1g, which represent “repressed” Treg signature, are Up in Bcl11b-deficient Treg cells (Fig. 3, C and D, and table S1).
As expected, a sizable fraction of Bcl11b-dependent genes representing Treg-signature are also Foxp3 dependent (Fig. 3, C and D, underlined genes, and table S1). However, such genes appeared to be overrepresented in the Bcl11b-dependent Down gene category (116 of 356 Down genes versus 54 of 546 Up genes are Foxp3 dependent). To achieve a more robust understanding, we therefore performed a cumulative distribution function (CDF) analysis of the difference in expression for gene subsets encoding Treg signature genes or Foxp3-dependent genes. A shift in cumulative gene expression curve to the left and right of the distribution for all expressed genes indicate that most of the genes within the respective gene sets analyzed were down-regulated or up-regulated, respectively. Quantitatively extending our inference above, we found very high and significant correlations between cumulative expressions of both activated and repressed Treg signature genes with Bcl11b-dependent global gene expression patterns (Fig. 3E and table S1). In contrast to this observation, when Foxp3-dependent gene sets were considered, specifically the Foxp3-dependent activated genes were found to be mainly down-regulated in Bcl11b-deficient Treg cells (Fig. 3F and table S1). In agreement with these findings, in an independently performed gene set enrichment analysis (GSEA) (11), while Bcl11b-dependent Down or Up genes were both found to be overrepresented in Treg signature genes when compared to Tn (fig. S3, C and D), only the Down genes were found to be significantly enriched within “GAVIN_FOXP3_TARGETS_CLUSTER” T4 and P4, which were implicated previously by Gavin et al. (9) as “Foxp3-enriched” (fig. S3E). Therefore, in terms of molecular functions, while Bcl11b was found to contribute to shaping up both activated and repressed Treg-specific gene signatures, it primarily appears to promote transcriptional activation as far as Foxp3-dependent gene expression is considered.
Collectively, these results strongly establish Bcl11b-dependent gene expression as a mainstay of Treg transcriptional program, the disruption of which results in catastrophic breakdown of systemic tolerance even at resting state. Furthermore, considering the extent of colossal alteration of global genetic signature, it seemed implausible that the functional incompetence of Bcl11b-deficient Treg cells is likely to be rectified by modulating the expression of any specific gene product.
Large overlap between Bcl11b- and Foxp3-bound sites in Treg cells
To identify Bcl11b-associated genes in Treg cells, we next performed genome-wide chromatin immunoprecipitation followed by next-generation sequencing (ChIP-seq). In addition to CD4+Foxp3+ Treg cells, we performed Bcl11b ChIP-seq analysis on CD4+Foxp3− Tn cells as well. Initial analysis identified 270 among 902 of the DEGs in Treg cells (37% among Down genes and 25% among Up genes) to be bound by Bcl11b within ± 20 kb of their respective transcription start sites (TSSs) (Fig. 4A and table S2), suggesting direct Bcl11b-mediated regulation. Furthermore, while majority of the Bcl11b-associated peaks in Treg cells were common to that in Tn (Tn/Tr-common), ~31% of the sites were found to be unique for Treg cells (Tr-specific) (Fig. 4, B and C). Many of the Tr-specific sites, upon manual inspection, appeared to be associated with prominent genes implicated in Treg homeostasis and function (Fig. 4C, right, and table S2). We therefore sorted to determine whether any correlation exists between Tn/Tr-common or Tr-specific Bcl11b-bound genes, with respect to the directionality of their corresponding gene expression changes. We found that Tr-specific sites were significantly enriched within genes whose expressions are down-regulated in the absence of Bcl11b (Fig. 4D, bottom). In contrast, Tn/Tr-common sites showed marginal enrichment on up-regulated genes (Fig. 4D, top). For both cases, however, the enrichment for Up or Down genes was significantly higher than that for randomly selected genes, suggesting that while there is significant biasness for Tr-specific genes to be down-regulated in the absence of Bcl11b, this is not a rule (Fig. 4D). In agreement with these results, a CDF analysis of gene expression changes for the genes exclusively bound by Bcl11b in a Tr-specific manner showed a significant shift toward the left (Fig. 4E and table S2). In line with these findings, transcription factor motif analysis also revealed differences in motif enrichment among the two categories of sites. For example, while the Tn/Tr-common sites are highly enriched for ETS (E-twenty-six) DNA-binding domain, with more than 44% of the sites having Elk1 (ETS) motif ±100 base pairs (bp) of Bcl11b peaks, the ETS motifs are significantly underrepresented (~14% targets) near the Bcl11b peaks in the Tr-specific sites. On the other hand, binding motifs for the CCAAT/enhancer-binding protein α (~23% targets) were found to be overrepresented near the Tr-specific Bcl11b peaks (fig. S4, A and B).
Fig. 4. Large overlap between Bcl11b- and Foxp3-occupied sites in Treg cells.
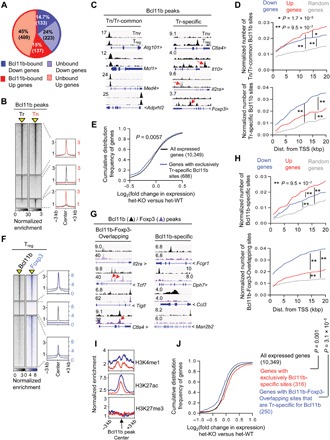
ChIP-seq was performed for Bcl11b on sorted CD4+Foxp3+ Treg (Tr) and CD4+Foxp3−CD62lhi T naïve (Tn) cells. (A) Overview of the percentage of Bcl11b-dependent Up or Down genes bound by Bcl11b within ±20 kb of their transcription start sites (TSSs). (B) Genome-wide occupancy of Bcl11b in Tr and Tn cells. Each column depicts Bcl11b binding in Tr or Tn cells within a window ±3 kb centered around Bcl11b-bound sites (indicated as a yellow triangle). Normalized enrichments of indicated Bcl11b-bound peak sets are shown on the right as histogram plots. (C) Representative tracks, as viewed in Integrative Genome Viewer (IGV), of Bcl11b ChIP-seq reads aligned to the corresponding gene loci. Y scale is normalized tag intensity (reads per million mapped reads). Red arrows point toward peaks in each category that are otherwise not obvious. (D) Plots depicting enrichment of Tn/Tr-common (top) or Tr-specific (bottom) sites within a given distance to promoter of randomly selected genes, or genes that are down-regulated or up-regulated in Bcl11b-deficient Treg cells. Wilcoxon rank-sum test P values are shown. (E) CDF analysis of gene expression changes between Bcl11b-deficient versus Bcl11b-sufficient Treg cells for gene subsets that “exclusively” contain Tr-specific Bcl11b-specific sites. Of note, there was a subcategory of genes prebound by Bcl11b in Tn and gained new Bcl11b peaks at different site(s) in Tr. While these genes are also categorized as Tr-specific site–containing genes, they are not included in this analysis. Two-tailed Kolmogorov-Smirnov test P value is shown. (F) Genome-wide occupancy of Bcl11b and Foxp3 in Treg cells. Each column depicts Bcl11b or Foxp3 binding within a window ±3 kb centered on peaks identified by ChIP-seq analyses (indicated as a yellow triangle). Normalized enrichments of indicated Bcl11b- and Foxp3-bound peak sets are shown on the right as histogram plots. (G) Examples of Bcl11b- and Foxp3-bound peaks representing common sites (Bcl11b-Foxp3-Overlapping) or sites where Bcl11b binds in the absence of Foxp3 (Bcl11b-specific). (H) Plots depicting enrichment of Bcl11b-specific (top) or Bcl11b-Foxp3-Overlapping (bottom) sites within a given distance to promoter of randomly selected genes, or genes that are down-regulated or up-regulated in Treg cells in the absence of Bcl11b. (I) Normalized enrichment of indicated chromatin marks around Bcl11b binding sites within ±20 kb of TSS for the gene subsets analyzed. The blue line represents genes with Bcl11b-Foxp3-Overlapping sites that are Tr-specific for Bcl11b. The red line represents genes that are only bound by Bcl11b in Treg cells. (J) CDF analysis of gene expression changes between Bcl11b-deficient versus Bcl11b-sufficient Treg cells for indicated gene subsets. Genes that are bound by both Bcl11b and Foxp3, but at two different sites (nonoverlapping), are excluded from this analysis. Two-tailed Kolmogorov-Smirnov test P values are shown.
Provided the large overlap between Treg and Foxp3-dependent signatures with gene expression changes in Bcl11b-deficient Treg cells, we next performed Foxp3 ChIP-seq to determine whether Bcl11b and Foxp3 bind to similar sites. We found that ~65% of Bcl11b-bound sites were also bound by Foxp3. We termed them “Bcl11b-Foxp3-Overlapping” and the sites bound by Bcl11b alone as “Bcl11b-specific” sites (Fig. 4, F and G, and table S3). For Foxp3-bound sites, relatively less than ~33% were found to have high enrichment for Bcl11b peaks. The remaining sites where Bcl11b peaks were not called for still showed slight but detectable enrichment for Bcl11b binding (Fig. 4F), suggesting loose or transient association. As observed for Tr-specific Bcl11b-bound genes, Bcl11b-Foxp3-Overlapping sites were found to be highly enriched on Bcl11b-dependent Down genes (Fig. 4H, bottom, and table S3). Bcl11b-specific sites, on the other hand, were overrepresented on Up genes (Fig. 4H, top, and table S3).
Together, these results collectively led to the following conclusions. While most of the Bcl11b-associated sites are carried over from precursor Tn to Treg, a fraction of Bcl11b is “redistributed” to specific sites in Treg cells. Furthermore, in light of the observed expression changes of the genes that are cobound by Bcl11b and Foxp3 [with Bcl11b-Foxp3-Overlapping sites (Fig. 4H)], it seemed likely that in these Tr-specific sites, Bcl11b is recruited, presumably by interacting with Foxp3, to enhance the expression of genes relevant for Treg function and homeostasis. In agreement with this notion, upon reexamining previously published ChIP-seq data (12), we observed higher enhancer activity indicated by higher enrichment of H3K27 acetylated (H3K27ac) and H3K4 monomethyl (H3K4me1) chromatin modification marks associated with genes harboring Bcl11b-Foxp3-Overlapping sites that are Tr specifically bound by Bcl11b, compared to genes that are exclusively bound only by Bcl11b (with Bcl11b-specific sites) (Fig. 2I, middle and top). H3K27 trimethyl (H3K27me3) chromatin marks, on the other hand, showed no difference among the two groups (Fig. 4I, bottom). This was associated with a highly significant negative shift in cumulative expression for genes harboring Tr-specific Bcl11b-Foxp3-Overlapping sites. In contrast, genes exclusively harboring Bcl11b-specific sites were significantly up-regulated upon its deletion, suggesting a primarily repressive role of Bcl11b, when it is bound in a Foxp3-independent manner (Fig. 4J and table S3).
Optimal recruitment of Bcl11b in Treg cells is dependent on Foxp3
The large overlap between Foxp3 and Bcl11b binding sites in Treg cells raised the possibility that the genomic occupancy of these two factors may be mutually dependent on each other. We first asked whether Bcl11b’s binding to its genomic sites is dependent on Foxp3. To this end, we performed ChIP-seq analysis for Bcl11b binding in Tfn cells derived from Foxp3GFPKO mice. Compared to WT Treg cells, Bcl11b’s binding to majority (~60%) of its sites was found to be markedly reduced in Tfn cells (Fig. 5A and table S4), suggesting direct or indirect dependence on Foxp3. Upon manual inspection of randomly selected representative Tn/Tr-common and Tr-specific Bcl11b binding sites, we observed that, to a large extent, the peaks that were otherwise gained by Bcl11b in Treg cells compared to Tn (Tr-specific sites) were the ones that were significantly reduced in the absence of Foxp3. For example, while Tr-specific Bcl11b binding sites on Ctla4, Il2ra, Tigit, Foxp3, and Mrpl22 are lost (Fig. 5B, right), among the Tn/Tr-common sites tested, only the site corresponding to Ifnar1 was reduced. On the other hand, Bcl11b sites on Med4, Wdr70, Uba5, and Gins4 were still retained in Tfn (Fig. 5B, left). In agreement with these observations, quantitative analysis revealed that 84% of Tr-specific sites, as opposed to 39% of Tn/Tr-common sites corresponding to genes expressed in Treg cells, were lost in Tfn compared to WT (Fig. 5C and table S4).
Fig. 5. Foxp3 promotes functional recruitment of Bcl11b in Treg cells.
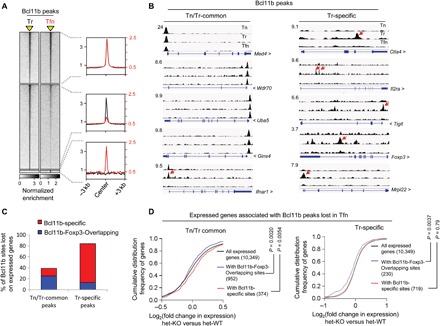
(A) Genome-wide occupancy of Bcl11b in WT Treg (Tr) and Tfn cells derived from Foxp3GFPKO mice. Bcl11b binding within a window ±3 kb centered on peaks identified by ChIP-seq analyses (indicated as a yellow triangle). Normalized enrichments of indicated Bcl11b peak sets are shown on the right as histogram plots. (B) Examples of Bcl11b peaks in Tn, Tr, and Tfn cells as viewed in IGV, representing the indicated categories. Red arrows indicate peaks that are significantly reduced in Tfn cells compared to Tr. (C) Graphical representation summarizing the subcategories to which the Bcl11b peaks lost in Tfn cells belong. (D) CDF analysis of gene expression changes between Bcl11b-deficient versus Bcl11b-sufficient Treg cells for indicated gene subsets. Two-tailed Kolmogorov-Smirnov test P values are shown.
Somewhat counterintuitively, among the Tr-specific sites lost, only ~15% were found to be sites that were cobound by Foxp3 and Bcl11b in WT Treg cells (Bcl11b-Foxp3-Overlapping) (Fig. 5C). Underscoring their functional relevance, however, a CDF analysis of the corresponding genes harboring such sites showed that these genes are significantly down-regulated in Bcl11b-deficient Treg cells (Fig. 5D, right, and table S4). Therefore, in line with the conclusions from results obtained so far, Bcl11b appears to be recruited to a relatively small fraction of newly occupied Tr-specific sites by virtue of direct interaction with Foxp3 to promote active gene expression. These results also suggest that Bcl11b occupies the remaining 85% of Tr-specific sites by mechanisms that are indirectly dependent on Foxp3, for example, by interacting with transcription factors whose optimal expression and genomic occupancy in Treg cells are, in turn, dependent on Foxp3. However, unlike the Bcl11b-Foxp3-Overlapping sites, the functional relevance of Bcl11b binding to these Bcl11b-specific sites is not clear, since a CDF analysis did not reveal any biasness for Bcl11b-dependent active or repressed gene expression state for the corresponding gene set (Fig. 5D, right, and table S4).
In contrast to Tr-specific peaks, a significantly larger proportion (~64%) of the Tn/Tr-common Bcl11b peaks lost in Tfn cells were found to be co-occupied by Bcl11b and Foxp3 in WT Treg (Fig. 5C) and collectively showed a negative shift in CDF analysis (Fig. 5D, left, and table S4). The remaining 36%, on the other hand, displayed a shift toward the right (Fig. 5D, left, and table S4). Therefore, similar to Tr-specific sites, even for the Bcl11b sites carried over in Treg cells from Tn, Bcl11b appears to gain stability in genomic association and promote gene activation in association with Foxp3. On the other hand, in agreement with previous results, the Bcl11b carried over sites where it is bound independent of Foxp3; it most likely acts as a repressor.
Genomic association of Foxp3 is altered in the absence of Bcl11b
Finally, to ask whether genome-wide occupancy of Foxp3 is affected in the absence of Bcl11b, we performed ChIP-seq analysis for Foxp3 binding in Bcl11b-deficient Treg cells. While ~68% of Foxp3 occupied sites were found to be retained (termed “Unaltered”), the binding intensities of the remaining 32% of Foxp3 peaks were significantly reduced in KO compared to WT Treg (termed “Lost”). In addition to this, we also observed a large number of newly formed sites where Foxp3 binding is markedly increased only in Bcl11b-deficient Treg cells (termed “Gained”) (Fig. 6, A and B; fig. S5A; and table S5).
Fig. 6. Bcl11b promotes intensity and assures fidelity of Foxp3-bound sites in Tregcells.
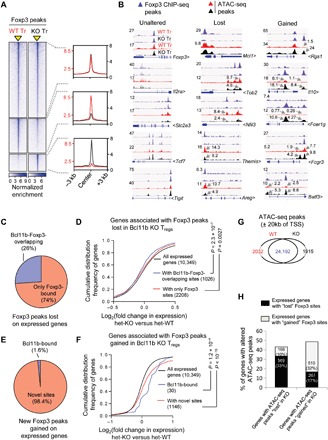
(A) Genome-wide binding intensity of Foxp3 in WT and Bcl11b-deficient (KO) Treg cells. Foxp3 binding within a window ±3 kb centered on peaks identified by ChIP-seq analyses (indicated as a yellow triangle). Normalized enrichments of indicated Foxp3-bound peak sets are shown on the right as histogram plots. (B) IGV tracks of Foxp3-bound peaks and corresponding ATAC-seq peaks of the indicated categories in WT- and KO-derived Treg cells. (C and E) Graphical summary demonstrating Bcl11b or Foxp3 binding preference in WT Treg cells, for genes harboring Foxp3 peaks lost (C) or gained (E) in KO. (D and F) CDF analysis summarizing gene expression changes between Bcl11b-deficient and Bcl11b-sufficient Treg cells for indicated gene subsets categorized in (C) and (E), respectively. Two-tailed Kolmogorov-Smirnov test P values are shown. (G) Summary of ATAC-seq peaks in WT and KO Treg cells. (H) Graphical representation summarizing the subcategories to which the ATAC-seq peaks lost or gained in Bcl11b-deficient Treg cells belong.
To determine the functional relevance of these sites, we first focused on the Lost sites. Twenty-six percent of the Lost sites were found to be co-occupied by Bcl11b and Foxp3 in WT Treg cells (Fig. 6C), suggesting that optimal Foxp3 binding to these sites are directly dependent on Bcl11b. In agreement with previous findings, a CDF analysis revealed that the expressions of genes corresponding to these sites are negatively affected in the absence of Bcl11b (Fig. 6D, blue line, and table S5). The expressions of the genes corresponding to the remaining 74% of the Lost sites were also found to be dependent on Bcl11b for their optimal activation, albeit to a lesser extent (Fig. 6D, red line, and table S5). For these gene subsets, therefore, Bcl11b mediates optimal Foxp3 binding either by directly associating with it or by indirectly promoting its recruitment, thereby positively affecting the expression of corresponding gene products.
Among the 1863 genes harboring a total of 2312 Gained sites, 1176 were found to be expressed genes. We considered the possibility that Bcl11b might be competitively restricting Foxp3’s binding by itself being associated to these sites. However, only a very tiny fraction of them (1.6%) were found to be Bcl11b-bound in WT Treg cells (Fig. 6E). The remaining 98.4% are sites that are not associated with either Bcl11b or Foxp3 in WT Treg cells. Notably, by CDF analysis, we found that most of these genes are up-regulated in Bcl11b-deficient Treg cells (Fig. 6F and table S5). To get a better understanding of their biological functions, we curated the candidate genes that gained Foxp3 sites, as well as 1.5-fold Up in Bcl11b KO Treg cells (table S5). Many of these genes were found to encode factors implicated in signal transduction and innate immune response (fig. S5B and table S5). For example, Fcer1g, the gene encoding the Fc fragment of the IgE receptor, was found to be more than 200-fold up-regulated in Bcl11b-deficient Treg cells (Fig. 6B, right, and table S5). Chemokine receptors Ccr3 and Cxcr6 were up-regulated two- to fivefold. Major histocompatibility complex II α subunit H2-Aa was found to be up-regulated more than fivefold. Expression of Batf3, a transcription factor implicated in CD8α+ dendritic cell generation and functions (13), was up-regulated eightfold (Fig. 6B, right, and table S5). These results therefore reveal an interesting function of Bcl11b whereby it restricts misdirected recruitment of Foxp3 to genomic sites, for majority of which it would otherwise promote erroneous expression of genes irrelevant for Treg function. As opposed to these Up genes, among the relatively fewer candidates that gained Foxp3 binding sites and were down-regulated in a Bcl11b-dependent manner, we found important genes implicated in Treg functions like Il10, Ctla4, Dusp4, Tigit, and Lag3 (fig. S5C) (8, 14–18). Therefore, for this subcategory, erroneous binding of Foxp3 to these gene loci in the absence of Bcl11b appears to contribute to their repressive state.
Last, to determine whether Bcl11b affects Foxp3’s recruitment to the Lost or Gained sites by influencing chromatin accessibility, we performed assay for transposase-accessible chromatin using sequencing (ATAC-seq) on Treg cells sorted from WT and KO mice. Analysis of candidate genes revealed a notable correlation between the consequences of Bcl11b-dependent Foxp3’s binding, with the chromatin accessibility of associated sites. The Lost sites were primarily associated with reduced chromatin accessibility, whereas Gained sites were associated with increased chromatin accessibility (Fig. 6B, middle and right, and fig. S5, A and C). On the other hand, accessibility of the candidate sites analyzed, where Foxp3 binding remained unaltered, were mostly unchanged (Fig. 6B, left). Furthermore, global analysis of ATAC-seq peaks altered in Bcl11b-deficient Treg cells substantiated this observation genome-wide (Fig. 6, G and H, and table S5). Together, these results strongly suggest that, to a large extent, genomic association of Foxp3 is governed as well as regulated by Bcl11b by its ability to modulate the accessibility of corresponding sites. In some cases, accessibility to chromatin enables Foxp3’s binding, whereas in other cases, Foxp3’s association to inappropriate regions is restricted by actively maintaining the closed chromatin conformation of these sites. Bcl11b appears to critically influence both the scenarios.
DISCUSSION
Despite being the key component critical for establishing a Treg transcription program, it is now clear that like most lineage-specifying factors, Foxp3 exerts its effects by forming molecular complexes with protein partners it interacts with (2). Provided the current knowledge on the diversity in functions and localization of Treg cells (19), as well as the transcriptional modalities that appear to fine-tune their genetic makeup (20), it is understandable that, in many cases, such interactions are context dependent and presumably transient. In stark contrast, the data presented here establish Bcl11b as a major transcription factor that either independently, or to a large extent by serving as a critical cofactor of Foxp3, sets up key functional aspects of the Treg-mediated suppressive program.
Conditional deletion of many Foxp3-interacting transcription factors have been shown to result in specific alterations in Treg functions, leading to distinct, in some cases subtle or redundant phenotypic consequences. For example, compromising transcriptional activity of the Runt-related transcription factor family of RUNX proteins by depleting their non–DNA-binding subunit Cbfβ led to relatively moderate lymphoproliferative disorder, splenomegaly, and increased activation of effector T cells, largely resulting from defective maintenance of Foxp3 itself (21). While the phenotype was significantly more pronounced in BALB/c background, which developed gastritis and displayed elevated cytokine production, the overall extent of autoimmunity was considerably less than mice devoid of functional Treg population (22). As opposed to Runx proteins, in accordance with an effector response-specific functional heterogeneity among Treg cells, ablation of the interferon regulatory factor Irf4, the transcription factor Stat3, or the TH1 cell lineage-specifying factor T-bet in Treg cells resulted in mice with selective TH2, TH17, and TH1 pathologies, respectively (23–25). In line with these findings, Treg-specific deletion of Bcl6 resulted in compromised suppression specifically of follicular helper T cells (26). Extending the context-dependent functional significance of Foxp3-interacting factors, peroxisome proliferator–activated receptor γ, a transcription factor well known for its role in adipocyte differentiation, was implicated specifically in adipose tissue–resident Treg homeostasis and function (27). Another example underscoring functional redundancy among transcription factors in Treg function is the finding that while Treg-specific deletion of T-bet or Gata3 independently results in relatively subtle phenotype, combined deletion of both in Treg cells resulted in enhanced autoimmunity, albeit relatively less than observed in mice with nonfunctional Foxp3 protein (28). In contrast to these findings, however, efficient deletion of Bcl11b in our experiments resulted in systemic disruption of Treg function, leading to catastrophic autoimmune phenotype almost indistinguishable to Foxp3GFPKO mice. Looking at the extreme phenotypic consequence, it seems unlikely that the devastating breakdown of tolerance in Bcl11bf/fFoxp3IRES-YFP-cre mice is a consequence of alteration in any single Treg-mediated suppression mechanism. We propose that Treg-specific deletion of Bcl11b results in near-complete crash down of its genetic signature, leading to disruption of multiple suppressive modalities that is, to an extent, almost similar to that resulting from functional ablation of Foxp3 itself.
In terms of physical distribution of Bcl11b to its binding sites, while majority of sites were carried over from naïve T cells, almost a third were found to be Treg cell specific. The underlying mechanism of such redistribution of Bcl11b is not entirely clear. However, it is not unlikely that three-dimensional rearrangements of higher-order genomic architecture during Treg development results in efficient binding of Bcl11b to these sites, where otherwise it is loosely or transiently associated in precursor cells. In this regard, a recent study reported that during thymic development of T cells, the Bcl11b gene locus itself, via a non-coding RNA termed ThymoD, is repositioned from nuclear lamina to its interior, resulting in chromatin folding and compartmentalization, eventually leading to its transcriptional activation (29). The affinity of Bcl11b for a large proportion of its associated sites was found to be greatly reduced in the absence of Foxp3. While majority of them represent Treg-specific binding sites of Bcl11b, many of them also included sites where Bcl11b was prebound in naïve T cells. These results strongly indicate that, to a large extent, the association of Bcl11b to its binding sites in Treg cells is stabilized directly or indirectly in a Foxp3-dependent manner.
Our results put forward the following molecular contributions of Bcl11b with regard to its role in shaping up the Treg cell transcriptional profile. We found a large overlap between Foxp3 and Bcl11b binding sites, for majority of which RNA-seq data largely correlate with down-regulation of associated genes in the absence of Bcl11b. These observations suggest that the Bcl11b-Foxp3 gene regulatory axis is a key determinant of the gene activation program in Treg cells. On the other hand, in most cases, Bcl11b’s binding in the absence of Foxp3 correlates with repressive states of the corresponding genes. Provided that Bcl11b has been previously mainly implicated in repressing gene expression by being associated with the nucleosome remodeling and deacetylase corepressor complex (30), it appears that in association with Foxp3, extensive rearrangements take place in the molecular composition of its associated factors.
The absence of Bcl11b in Treg cells resulted in interesting consequences as far as Foxp3’s genome-wide association is concerned. First, the optimal binding of Foxp3 is reduced in a subset of its associated sites. In accordance to a mechanism suggesting the direct involvement of Bcl11b in stabilizing functionally relevant binding of Foxp3, a sizable fraction of these sites are co-occupied by Foxp3 and Bcl11b in WT Treg cells. Furthermore, these sites display reduction in ATAC-seq peaks in Bcl11b-deficient Treg cells compared to WT, suggesting Bcl11b’s role in promoting chromatin accessibility. These sites also cumulatively correlate with reduced expression of the corresponding gene products upon depletion of Bcl11b. Second, the absence of Bcl11b further results in ectopic association of Foxp3 to sites where it is otherwise not bound in WT Treg cells. Such newly formed sites, in turn, correlate with altered expressions of associated genes, as well as enhanced chromatin accessibility in Bcl11b-deficient Treg cells compared to WT, thereby suggesting a novel role of Bcl11b in restricting erroneous genomic association of Foxp3 to maintain Treg-specific steady-state gene expression profile. This phenomenon is reminiscent of a recent finding where the transcription factor PU.1 was found to redirect partner transcription factor bindings from sites, which the latter would otherwise prefer if PU.1 was absent (31). Together, these results strongly establish that Bcl11b, either directly by recruiting Foxp3 or indirectly by positively or negatively influencing chromatin accessibility, may contribute to Foxp3’s optimal occupancy to its binding sites. In this regard, whether Bcl11b-dependent changes in chromatin accessibility are also influenced by its potential contribution toward short- or long-range chromatin looping remains to be investigated.
In conclusion, this study establishes Bcl11b as a cardinal factor defining molecular signatures of Treg cells. We propose that both by serving as a “core” component of the otherwise heterogeneous Foxp3 interactome and by exerting its repressing effect on transcription in a Foxp3-independent manner, Bcl11b is critically involved in shaping up key functional features of Treg transcriptome. In the future, further refinements of mechanisms involved and identification of its Treg-specific molecular partners would be instrumental in deciphering novel components contributing to immune homeostasis. Furthermore, developing unique strategies to disrupt interaction between Foxp3 and Bcl11b, ideally in a tumor-specific manner, might prove to be of great therapeutic relevance in cancer immunotherapy.
MATERIALS AND METHODS
Mice
All mice were generated on or backcrossed to C57BL/6 background. The Bcl11bf/f mice line (32) was a gift from P. Liu (then at Wellcome Sanger Institute). Foxp3IRES-YFP-Cre mice (8) and Foxp3GFPKO mice (9) are described elsewhere. All animals were maintained in specific pathogen–free barrier facilities and were used in accordance with Institutional Animal Care and Use Committee protocols approved by the POSTECH Institutional Animal Care and Use Committee.
FACS staining and antibodies
For surface staining, fluorochrome-conjugated antibodies were purchased from eBioscience, BioLegend, Tonbo Biosciences, and R&D Biosciences. Anti-CD4 (GK1.5), anti-CD8 (53-6.7), anti-CD25 (PC61.5), anti-GITR (DTA-1), anti-ICOS (C398.4A), anti-CTLA4 (UC10-4B9), anti-Ki67 (16A8), anti-CD62L (MEL-14), and anti-CD44 (IM7) were used in the study. For intracellular staining, surface-stained cells were fixed and permeabilized with a Foxp3 staining kit (eBioscience) according to the manufacturer’s instructions and were stained with the following antibodies: anti-Foxp3 (FJK-16s), anti-IFNg (XMG1.2), anti–IL-17A (eBio17B7), anti–IL-4 (11B11), and anti–IL-13 (eBio13A) wherever applicable. Stained cells were analyzed using an LSRFortessa SORP flow cytometer with Data-Interpolating Variational Analysis software (BD Biosciences) and FlowJo (TreeStar).
Histology
Mouse tissues were fixed in 10% formalin, buffered neutrally, and were processed with hematoxylin and eosin staining. Blinded assessment of at least three sections of each tissue per mouse was performed by an expert pathologist.
Adoptive transfer–induced colitis
Splenic CD4+Foxp3−CD45RBhi cells were sorted from Foxp3GFPCD45.1+ mice and mixed with Treg cells sorted from lymph nodes and spleens of Bcl11b+/+Foxp3IRES-YFP-Cre or Bcl11bfl/flFoxp3IRES-YFP-Cre mice. Colitis was induced in RAG1-deficient hosts by intravenous injection of 8 × 105 CD45RBhi + 2 × 105 Treg cells. Mice weight was recorded blindly once a week for 8 weeks.
RNA-seq library preparation
CD4+YFP+ cells from heterozygous female Bcl11b f/fFoxp3YFP-Cre/+ (het-KO) or littermate Bcl11bf/+ or +/+Foxp3YFP-Cre/wt (het-WT) mice were sorted at >97% purity using a BD Moflo-XDP cell sorter. Total RNA was extracted using the RNeasy Micro Kit (Qiagen-74004). Integrity and concentration of purified RNA were checked using the Agilent RNA 6000 Nano Kit (5067-1511) in Agilent BioAnalyzer and Nanodrop, respectively. Library was prepared from total RNA using the TruSeq Stranded Total RNA kit (Illumina-RS-122-2001) following the manufacturer’s protocols. Briefly, cDNA was made from total RNA followed by adenylation, barcoded adaptor ligation, and polymerase chain reaction (PCR) amplification (15 cycles). Libraries were size-selected for 200- to 300-bp fragments using Agencourt AMPure XP beads (A63880), and size distribution was confirmed using the Agilent High Sensitivity DNA Kit (5067-4626) in Agilent BioAnalyzer. Equivalent amounts of barcoded libraries were pooled and sequenced using HiSeq 2500 or MiSeq (Illumina) instruments.
RNA-seq analysis
The quality of sequenced reads (50 bp; paired-end) was measured using FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/), and low-quality portions of the sequenced reads were trimmed using Trim Galore (version 0.4.2; www.bioinformatics.babraham.ac.uk/projects/trim:galore/) with Cutadapt (version 1.12) (MARTIN, Marcel. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal, [S.l.], v. 17, n. 1, p. pp. 10-12, May 2011. ISSN 2226-6089. Available at: https://journal.embnet.org/index.php/embnetjournal/article/view/200/479. Date accessed: 28 Sept. 2018. doi: https://doi.org/10.14806/ej.17.1.200). The trimmed reads were mapped to the mouse genome (reference assembly mm10) using STAR (version 2.5.1) (33). The abundances of reference genes were estimated using Cuffnorm (version 2.2.1) and DEGs between conditions were identified using Cuffdiff (version 2.2.1) with an FDR-adjusted P value cutoff of 0.05 (34). The fragments per kilobase of transcript per million mapped reads values were used to compare gene expression levels.
ChIP-seq library preparation
For Bcl11b ChIP-seq in WT Treg and WT Tn or Tfn cells, 1 × 107 Treg and Tn or CD4+GFP+ Tfn cells were sorted from Foxp3IRES-YFP-Cre or Foxp3GFPKO mice, respectively. For Foxp3 ChIP-seq in WT or KO Treg cells, 1 × 107 Treg cells were sorted from Foxp3IRES-YFP-Cre or Bcl11bf/fFoxp3IRES-YFP-Cre mice, respectively. Cells were cross-linked with 1% formaldehyde for 10 min, followed by addition of glycine to a final concentration of 0.125 M and washing two times with cold phosphate-buffered saline. Fixed cells were lysed for 10 min on ice in cell lysis buffer [25 mM Hepes (pH 7.8), 1.5 mM MgCl2, 10 mM KCl, 0.1% NP-40, and 1 mM dithiothreitol supplemented with complete protease inhibitor cocktail (Roche 05892970001)]. Chromatin was sheared using a digital sonifier (Branson 450) for 5 s every 15-s interval for 10 cycles at 10% intensity, and the sheared material was cleared by a 10-min centrifugation at 14,000 rpm at 4°C. The cleared chromatin was immunoprecipitated overnight on a rotating platform at 4°C with anti-Bcl11b (Bethyl Laboratories A300-385A) or anti-Foxp3 antibody [described in (35)] and magnetic protein A beads complex (Invitrogen, Dynabeads-10002D), which were preconjugated for at least 6 hours, followed by extensive washing of the beads with ice-cold wash buffer 1 (WB1) [50 mM Hepes (pH 7.9) 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and 1 mM phenylmethylsulfonyl fluoride (PMSF) (added before use)], WB2 [50 mM Hepes (pH 7.9), 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate, 0.1% SDS, and 1 mM PMSF (added before use)], WB3 [20 mM tris (pH 8.0), 1 mM EDTA, 250 mM LiCl, 0.5% NP-40, 0.5% Na-deoxycholate, and 1 mM PMSF (added before use)], and TE [10 mM tris-HCl (pH 8.0) and 1 mM EDTA (pH 8.0)]. Antibody/protein/DNA complexes were eluted from the beads, reverse cross-linked at 65°C for 4 to 5 hours, and treated with proteinase K for 1 to 2 hours at 37°C (each sample). DNA from reverse cross-linked materials was purified using the PCR purification kit (Biofactory, GP104-100).
Purified immunoprecipitated DNA were end-repaired and size-selected for 200- to 400-bp fragments with Agencourt AMPure XP beads (A63880) followed by adenylation, barcoded adaptor ligation, and PCR amplification (15 cycles). ChIP-seq library for each sample was prepared according to the manufacturer’s (NEXTFLEX ChIP-Seq Kit 5143-01) protocols. Size distribution and concentration of each library were checked using Agilent BioAnalyzer. Equivalent amounts of barcoded libraries were pooled and sequenced using HiSeq 2500 (Illumina) instruments. To control for background noise, DNA was prepared from the input of each sheared chromatin sample, and ChIP-seq libraries were prepared and sequenced as described above.
Assay for transposase-accessible chromatin using sequencing
CD4+Foxp3+ Treg cells (1 × 105) from Bcl11b+/+Foxp3IRES-YFP-Cre or Bcl11bfl/flFoxp3IRES-YFP-Cre mice were used for preparation of ATAC-seq libraries, adapting a published protocol (36). Briefly, sorted cells were centrifuged and suspended in 50 μl of cold hypotonic lysis buffer [10 mM tris-HCl (pH 7.5), 10 mM NaCl, 3 mM MgCl2, and 0.1% NP-40], followed by immediate centrifugation at 800g for 10 min. The pellet was resuspended in 50 μl of transposition reaction mix [5 μl of Tagment DNA enzyme, 25 μl of Tagment DNA buffer from the Nextera DNA Library Prep Kit (Illumina-15028212), and 20 μl H2O] and was incubated for 10 min at 55°C for DNA to be Tagmented (fragmented and tagged). Tagmented DNA was cleaned up and purified using the Zymo Research DNA Clean & Concentrator kit (D4003). For library preparation, two sequential seven cycles of PCR were performed to enrich small tagmented DNA fragments. After the first PCR, the libraries were size-selected for smaller fragments (around 200 to 400 bp) using Agencourt AMPure XP beads (A63880) followed by a second round of PCR under the same conditions in order to obtain the final library. Size distribution and concentration of each library were checked using the Agilent High Sensitivity DNA Kit (5067-4626) in Agilent BioAnalyzer. Equivalent amounts of barcoded libraries were pooled and sequenced using HiSeq 2500 (Illumina) instruments to generate paired-end short reads (100 bp).
ChIP-seq and ATAC-seq analyses
The quality of sequenced reads (50 bp, paired-end for ChIP-seq; 100 bp, paired-end for ATAC-seq) was measured using FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/), and low-quality portions of the sequenced reads were trimmed using Trim Galore (version 0.4.2; www.bioinformatics.babraham.ac.uk/projects/trim:galore/) with Cutadapt (version 1.12) (MARTIN, Marcel. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal, [S.l.], v. 17, n. 1, p. pp. 10–12, May 2011. ISSN 2226–6089. Available at: http://journal.embnet.org/index.php/embnetjournal/article/view/200/479. Date accessed: 28 Sept. 2018. doi: https://doi.org/10.14806/ej.17.1.200). The trimmed reads were mapped to the mouse genome (reference assembly mm10) using HISAT2 (version 2.1.0) (37). The mapped reads were sorted by genomic positions using SAMtools (version 1.3) (38), and duplicated mapped reads were removed using Sambamba (version 0.6.5) (39). Biological replicates for each condition were merged, and the genomic binding sites (peaks) of a given protein were identified using HOMER (version 4.10.1) with an FDR-adjusted P value cutoff of 0.001 (40). The peaks that overlapped with the blacklisted regions that show anomalous, unstructured, and high signal/read counts (www.encodeproject.org/annotations/ENCSR636HFF/) were discarded. To identify reliable target genes of a given protein, we further removed peaks located 20 kb outside of the gene’s TSSs. To identify common or specific peaks between given conditions, the identified peaks in each condition were merged. Then, the merged peaks were used to define common or specific peaks using HOMER (getDifferentialPeaks) with default parameters, which were further adjusted upon manual inspection. deepTools (version 2.5.3-2-503c71b) (41) was used to draw heatmaps.
Enzyme-linked immunosorbent assay
Approximately 0.1 ml of serum was collected from each mouse. Serum IgM and total IgG were measured using the Mouse IgM ELISA Ready-SET-Go! Kit (Invitrogen eBioscience-50246323) and Invitrogen eBioscience Mouse IgG total ELISA Ready-SET-Go! Kit (50246294), respectively, according to the manufacturer’s protocol. Serum IgE was measured using Mouse IgE ELISA MAX Standard (BioLegend-432402) according to the manufacturer’s protocol.
In vitro suppression assay
Sorted CD4+YFP+ cells from Bcl11b+/+lFoxp3IRES-YFP-Cre or Bcl11bfl/flFoxp3IRES-YFP-Cre mice were incubated with CD4+Foxp3−CD62Lhi responder cells that were prepulsed with CTV (Cell Tracking Violet) for 10 min at 37°C. T cell–depleted splenocytes (1 × 105 cells) were mixed with CTV-pulsed responder cells (5 × 104) and indicated ratios of Treg cells along with anti-CD3 (0.3 μg/ml) and plated in round-bottom 96-well plates. Cells were cultured for 5 days, and their proliferation was analyzed by flow cytometry to determine the dilution and MFI of CTV intensity. CTV MFIs for responder cells were normalized against those for the Te-only group, where responder cells were plated in the absence of Treg cells.
Bioinformatics analyses
CDF curves were generated in the following way. For each gene set plotted, corresponding log2(fold change) values in gene expression for individual genes were looked up within “all genes” using “vlookup” function in Excel. The corresponding values were used to generate CDF curves in Prism 7 with the following workflow: Column analysis (Frequency distribution) > Create (Cumulative frequency distribution) > Tabulate [Relative frequency (fractions)] > Bin range (Auto) > Bin width (No beans. Tabulate exact Cumulative frequency) > New graph (Create a new graph of the results). Histogram of the data was plotted in the final graph. Some representative CDF plots were also cross-checked using the program “R”.
Plots depicting enrichment of ChIP-seq sites on Bcl11b-dependent Up or Down genes were generated by normalizing the number of ChIP-seq peaks within the indicated distance, every 1 kbp upstream or downstream of TSS of genes that fall in each category, against the total number of Tn/Tr-common and Tr-specific Bcl11b peaks (Fig. 4D) or Bcl11b-specific and Bcl11b-Foxp3-Overlapping sites (Fig. 4H).
For motif analysis, Bcl11b binding sites (peaks) located near the TSSs of given genes (±20 kb of TSS) were used to identify transcription factor binding sites (motifs) using HOMER (findMotifsGenome.pl). The length of the peak size was set to ±100 bp of the center, and repeat sequences in the sequences were masked (-mask option).
Statistics
Statistical analysis was performed with GraphPad Prism 7.0 (GraphPad software, La Jolla, CA, USA) using unpaired two-tailed Student’s t test, Wilcoxon rank-sum test, or Kolmogorov-Smirnov test where applicable. Statistically significant differences in mean values were considered at P < 0.05.
Supplementary Material
Acknowledgments
We thank N. Mullapudi, Scientific Officer, Hong Kong University of Science and Technology, for technical suggestions in ChIP-seq. We also thank H. Jung for technical assistance in cell sorting. Funding: This work was supported by Project IBS-R005 from the Institute for Basic Science, Korean Ministry of Science and ICT. Author contributions: S.N.H. designed and performed the experiments, analyzed data, and wrote the manuscript. A.S. and S.G. performed animal experiments. S.-W.H. prepared library for RNA-seq. S.R-C. performed histopathology analyses. K.K. performed computational analysis of ChIP-seq, RNA-seq, and ATAC-seq data. S.-H. I. provided intellectual suggestions during the course of the study and edited the manuscript. D.R. directed the project, designed and analyzed experiments, and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. RNA-seq, ChIP-seq, and ATAC-seq data were deposited in the Gene Expression Omnibus (NCBI) data repository under accession number GEO: GSE120948. ChIP-seq data for H3K4me1, H3K27ac, and H3k27me3 were obtained from NCBI SRA database under accession number DRP003376 (12).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaaw0706/DC1
Fig. S1. Treg-specific deletion of Bcl11b results in systemic autoimmunity comparable to that observed in mouse lacking functional Foxp3 expression.
Fig. S2. Marked immune activation in KO mice.
Fig. S3. Functional annotation and GSEA of Bcl11b-dependent up-regulated (Up) or down-regulated (Down) genes.
Fig. S4. Motif analysis to identify potential transcription factor binding sites.
Fig. S5. Characterization of genes harboring Lost or Gained Foxp3 peaks in Bcl11b-deficient Treg cells.
Table S1. Data tables related to Fig. 3.
Table S2. Data tables related to Fig. 4 (A to E).
Table S3. Data tables related to Fig. 4F to I.
Table S4. Data tables related to Fig. 5.
Table S5. Data tables related to Fig. 6.
REFERENCES AND NOTES
- 1.Ramsdell F., Ziegler S. F., FOXP3 and scurfy: How it all began. Nat. Rev. Immunol. 14, 343–349 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Rudra D., deRoos P., Chaudhry A., Niec R. E., Arvey A., Samstein R. M., Leslie C., Shaffer S. A., Goodlett D. R., Rudensky A. Y., Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat. Immunol. 13, 1010–1019 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Di Santo J. P., Immunology. A guardian of T cell fate. Science 329, 44–45 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Albu D. I., Feng D., Bhattacharya D., Jenkins N. A., Copeland N. G., Liu P., Avram D., BCL11B is required for positive selection and survival of double-positive thymocytes. J. Exp. Med. 204, 3003–3015 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Califano D., Sweeney K. J., Le H., VanValkenburgh J., Yager E., O’Connor W. Jr., Kennedy J. S., Jones D. M., Avram D., Diverting T helper cell trafficking through increased plasticity attenuates autoimmune encephalomyelitis. J. Clin. Invest. 124, 174–187 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Califano D., Cho J. J., Uddin M. N., Lorentsen K. J., Yang Q., Bhandoola A., Li H., Avram D., Transcription factor Bcl11b controls identity and function of mature type 2 innate lymphoid cells. Immunity 43, 354–368 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vanvalkenburgh J., Albu D. I., Bapanpally C., Casanova S., Califano D., Jones D. M., Ignatowicz L., Kawamoto S., Fagarasan S., Jenkins N. A., Copeland N. G., Liu P., Avram D., Critical role of Bcl11b in suppressor function of T regulatory cells and prevention of inflammatory bowel disease. J. Exp. Med. 208, 2069–2081 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rubtsov Y. P., Rasmussen J. P., Chi E. Y., Fontenot J., Castelli L., Ye X., Treuting P., Siewe L., Roers A., Henderson W. R. Jr., Muller W., Rudensky A. Y., Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Gavin M. A., Rasmussen J. P., Fontenot J. D., Vasta V., Manganiello V. C., Beavo J. A., Rudensky A. Y., Foxp3-dependent programme of regulatory T-cell differentiation. Nature 445, 771–775 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Samstein R. M., Arvey A., Josefowicz S. Z., Peng X., Reynolds A., Sandstrom R., Neph S., Sabo P., Kim J. M., Liao W., Li M. O., Leslie C., Stamatoyannopoulos J. A., Rudensky A. Y., Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 151, 153–166 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitagawa Y., Ohkura N., Kidani Y., Vandenbon A., Hirota K., Kawakami R., Yasuda K., Motooka D., Nakamura S., Kondo M., Taniuchi I., Kohwi-Shigematsu T., Sakaguchi S., Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nat. Immunol. 18, 173–183 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hildner K., Edelson B. T., Purtha W. E., Diamond M., Matsushita H., Kohyama M., Calderon B., Schraml B. U., Unanue E. R., Diamond M. S., Schreiber R. D., Murphy T. L., Murphy K. M., Batf3 deficiency reveals a critical role for CD8α+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wing K., Onishi Y., Prieto-Martin P., Yamaguchi T., Miyara M., Fehervari Z., Nomura T., Sakaguchi S., CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Yan D., Farache J., Mingueneau M., Mathis D., Benoist C., Imbalanced signal transduction in regulatory T cells expressing the transcription factor FoxP3. Proc. Natl. Acad. Sci. U.S.A. 112, 14942–14947 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joller N., Lozano E., Burkett P. R., Patel B., Xiao S., Zhu C., Xia J., Tan T. G., Sefik E., Yajnik V., Sharpe A. H., Quintana F. J., Mathis D., Benoist C., Hafler D. A., Kuchroo V. K., Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 40, 569–581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurtulus S., Sakuishi K., Ngiow S.-F., Joller N., Tan D. J., Teng M. W. L., Smyth M. J., Kuchroo V. K., Anderson A. C., TIGIT predominantly regulates the immune response via regulatory T cells. J. Clin. Invest. 125, 4053–4062 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Huang C. T., Workman C. J., Flies D., Pan X., Marson A. L., Zhou G., Hipkiss E. L., Ravi S., Kowalski J., Levitsky H. I., Powell J. D., Pardoll D. M., Drake C. G., Vignali D. A., Role of LAG-3 in regulatory T cells. Immunity 21, 503–513 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Sharma A., Rudra D., Emerging functions of regulatory T cells in tissue homeostasis. Front. Immunol. 9, 883 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaudhry A., Rudensky A. Y., Control of inflammation by integration of environmental cues by regulatory T cells. J. Clin. Invest. 123, 939–944 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rudra D., Egawa T., Chong M. M. W., Treuting P., Littman D. R., Rudensky A. Y., Runx-CBFbeta complexes control expression of the transcription factor Foxp3 in regulatory T cells. Nat. Immunol. 10, 1170–1177 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kitoh A., Ono M., Naoe Y., Ohkura N., Yamaguchi T., Yaguchi H., Kitabayashi I., Tsukada T., Nomura T., Miyachi Y., Taniuchi I., Sakaguchi S., Indispensable role of the Runx1-Cbfβ transcription complex for in vivo-suppressive function of FoxP3+ regulatory T cells. Immunity 31, 609–620 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Zheng Y., Josefowicz S., Chaudhry A., Peng X. P., Forbush K., Rudensky A. Y., Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 463, 808–812 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaudhry A., Rudra D., Treuting P., Samstein R. M., Liang Y., Kas A., Rudensky A. Y., CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 326, 986–991 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koch M. A., Tucker-Heard G., Perdue N. R., Killebrew J. R., Urdahl K. B., Campbell D. J., The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 10, 595–602 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campbell D. J., Koch M. A., Treg cells: Patrolling a dangerous neighborhood. Nat. Med. 17, 929–930 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Cipolletta D., Feuerer M., Li A., Kamei N., Lee J., Shoelson S. E., Benoist C., Mathis D., PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 486, 549–553 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu F., Sharma S., Edwards J., Feigenbaum L., Zhu J., Dynamic expression of transcription factors T-bet and GATA-3 by regulatory T cells maintains immunotolerance. Nat. Immunol. 16, 197–206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Isoda T., Moore A. J., He Z., Chandra V., Aida M., Denholtz M., van Hamburg J. P., Fisch K. M., Chang A. N., Fahl S. P., Wiest D. L., Murre C., Non-coding transcription instructs chromatin folding and compartmentalization to dictate enhancer-promoter communication and T cell fate. Cell 171, 103–119.e18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cismasiu V. B., Adamo K., Gecewicz J., Duque J., Lin Q., Avram D., BCL11B functionally associates with the NuRD complex in T lymphocytes to repress targeted promoter. Oncogene 24, 6753–6764 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Hosokawa H., Ungerbäck J., Wang X., Matsumoto M., Nakayama K. I., Cohen S. M., Tanaka T., Rothenberg E. V., Transcription factor PU.1 represses and activates gene expression in early T cells by redirecting partner transcription factor binding. Immunity 48, 1119–1134.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li P., Burke S., Wang J., Chen X., Ortiz M., Lee S.-C., Lu D., Campos L., Goulding D., Ng B. L., Dougan G., Huntly B., Gottgens B., Jenkins N. A., Copeland N. G., Colucci F., Liu P., Reprogramming of T cells to natural killer-like cells upon Bcl11b deletion. Science 329, 85–89 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trapnell C., Hendrickson D. G., Sauvageau M., Goff L., Rinn J. L., Pachter L., Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 31, 46–53 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng Y., Josefowicz S. Z., Kas A., Chu T.-T., Gavin M. A., Rudensky A. Y., Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature 445, 936–940 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Buenrostro J. D., Giresi P. G., Zaba L. C., Chang H. Y., Greenleaf W. J., Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim D., Langmead B., Salzberg S. L., HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tarasov A., Vilella A. J., Cuppen E., Nijman I. J., Prins P., Sambamba: Fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K., Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramírez F., Dündar F., Diehl S., Grüning B. A., Manke T., deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaaw0706/DC1
Fig. S1. Treg-specific deletion of Bcl11b results in systemic autoimmunity comparable to that observed in mouse lacking functional Foxp3 expression.
Fig. S2. Marked immune activation in KO mice.
Fig. S3. Functional annotation and GSEA of Bcl11b-dependent up-regulated (Up) or down-regulated (Down) genes.
Fig. S4. Motif analysis to identify potential transcription factor binding sites.
Fig. S5. Characterization of genes harboring Lost or Gained Foxp3 peaks in Bcl11b-deficient Treg cells.
Table S1. Data tables related to Fig. 3.
Table S2. Data tables related to Fig. 4 (A to E).
Table S3. Data tables related to Fig. 4F to I.
Table S4. Data tables related to Fig. 5.
Table S5. Data tables related to Fig. 6.