

Toxicity from the immunosuppressant mycophenolate mofetil is prevented by eliminating gut bacteria expressing β-glucuronidase.
Abstract
Mycophenolate mofetil (MMF) is commonly prescribed and has proven advantages over other immunosuppressive drugs. However, frequent gastrointestinal side effects through an unknown mechanism limit its use. We have found that consumption of MMF alters the composition of the gut microbiota, selecting for bacteria expressing the enzyme β-glucuronidase (GUS) and leading to an up-regulation of GUS activity in the gut of mice and symptomatic humans. In the mouse, vancomycin eliminated GUS-expressing bacteria and prevented MMF-induced weight loss and colonic inflammation. Our work provides a mechanism for the toxicity associated with MMF and a future direction for the development of therapeutics.
INTRODUCTION
Suppression of the immune system is essential after transplantation and is commonly used for the treatment of multiple autoimmune diseases. One of the most widely used immunosuppressive drugs is mycophenolate mofetil (MMF) (1). MMF has important advantages over other drugs (2), but, while effective in enhancing survival, frequent gastrointestinal (GI) side effects can lead to dose reduction or discontinuation, actions linked to increased risk of rejection and patient death after solid organ transplantation (3–5).
The xenobiotic metabolism of MMF within humans has been well characterized (fig. S1) (6). MMF is hydrolyzed to active mycophenolic acid (MPA) and then inactivated by hepatic uridine diphosphate glucuronosyltransferase enzymes to create glucuronidated MPA (MPAG) and a small amount of acyl-MPAG (AcMPAG). Enteric-coated MMF and intravenous MMF are also rapidly converted to MPA and subsequently glucuronidated in the liver. MPAG and AcMPAG are excreted primarily in the urine, but ~10% is transported by the ATP (adenosine triphosphate)–binding cassette subfamily C member 2 (ABCC2) protein into the GI tract where bacteria expressing the enzyme β-glucuronidase (GUS) remove the glucuronic acid (GA) moiety to produce free MPA and GA (7). The resulting GA is available as a carbon source for bacterial metabolism, and the MPA undergoes enterohepatic recirculation.
The gut microbiota, referring to all the microorganisms in this niche, and its associated metagenome (microbiome) influence diverse aspects of human physiology, and changes in its composition affect human health (8). The mechanism(s) contributing to MMF-related GI toxicity have yet to be fully characterized, but we have shown that hematochezia and colonic inflammation in a pediatric heart transplant patient taking MMF was associated with a shift in the composition of the intestinal microbiota and that mice consuming MMF develop weight loss, colonic inflammation, and changes in their gut microbiota (9, 10). In our patient, toxicity resolved only after the discontinuation of MMF, but in our mouse model, MMF-induced GI toxicity was reversed or prevented by the use of broad-spectrum antibiotics and was absent in germ-free animals. These observations suggest that MMF toxicity requires an intact gut microbiota.
In the current study, we sought to further investigate the role of gut bacteria in MMF-induced GI toxicity using our mouse model. We hypothesized that MMF toxicity would be associated with alterations in the composition and function of the microbiome. We identified the ability of a single antibiotic, vancomycin, to both prevent and reverse the pathophysiological effects induced by MMF consumption, and we characterized the impact of vancomycin on the gut microbiome. We then explored the activity of gut bacterial GUS enzymes in the presence of MMF and vancomycin and examined the impact of vancomycin on MPAG catabolism.
Our data suggest that the introduction of MMF alters the composition of the gut microbiota by selecting for GUS-expressing bacteria. Increased GUS expression and activity lead to increased concentrations of MPA in the colon, which is associated with colonic inflammation and weight loss. Human data suggest that increased stool GUS activity can be associated with MMF-related toxicity. Vancomycin eliminated the GUS-expressing bacteria and prevented MMF-induced weight loss and colonic inflammation without reducing therapeutic levels of MPA in the blood. This work demonstrates a mechanism for MMF-induced GI toxicity and suggests a potential clinical intervention. The interaction between MPA and the gut microbiome provides further insight into the substantial problem of drug intolerance and adverse drug reactions driven by microbial metabolism and may also represent a paradigm for how the metabolism of xenobiotics can create local GI pathology.
RESULTS
Vancomycin is sufficient to prevent and reverse MMF-induced weight loss
Using an established mouse model of MMF exposure, we found that vancomycin alone, when administered concurrently with MMF, prevented the weight loss caused by MMF treatment (fig. S2). The weight of vancomycin-treated animals did not differ significantly from that of control, untreated animals after 8 days of MMF exposure (103.2 ± 0.68% versus 98.84 ± 0.82%, P = 0.41). Vancomycin was also able to reverse MMF-induced weight loss even with continued exposure to MMF. As seen previously, MMF caused significant weight loss with animals weighing 75.02 ± 1.5% of their initial weight after 8 days, but in the subsequent 8 days after the introduction of vancomycin, animal weights improved significantly with recovery back to 93.82 ± 1.2% of their initial weight (Fig. 1A). Although animals exposed to MMF and vancomycin were still significantly smaller than control, untreated animals (105.7 ± 1.5%, P = 0.003), they were significantly heavier than animals that continued to be exposed to MMF (69.26 ± 0.93%, P = 0.014). Consistent with the improvement in body weight, vancomycin also improved colon length and restored weight of the cecum, liver, and spleen. However, hematocrit remained significantly reduced (fig. S3). Whole colonic tissue homogenates were used to assess changes in the cytokine and chemokine profile of treated mice. Mice consuming both MMF and vancomycin demonstrated significant decreases (P < 0.05) in the levels of KC (keratinocyte chemoattractant, CXCL1), G-CSF (granulocyte colony-stimulating factor), IL-6 (interleukin-6), MIP-2 (macrophage inflammatory protein 2), LIF (leukemia inhibitory factor), TNFα (tumor necrosis factor–α), and M-CSF (macrophage colony-stimulating factor) and significant increases (P < 0.05) in IL-2, RANTES, MIG, and IP-10 (Fig. 1B and fig. S4). In addition to vancomycin, the effects of individual neomycin, ampicillin, and metronidazole were tested for their ability to reverse MMF-induced weight loss (fig. S2). Metronidazole treatment actually exacerbated the MMF-induced weight loss, and ampicillin and neomycin were less reproducibly effective than vancomycin in preventing MMF-induced weight loss.
Fig. 1. Effect of vancomycin on mouse body weight and colonic inflammation.
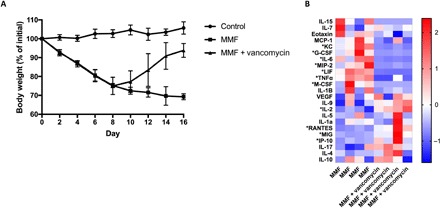
(A) Vancomycin was sufficient to reverse MMF-induced weight loss even in the continued presence of MMF. Data represent two separate experiments with four to nine animals for each group and are expressed as means and SD for each time point. (B) Vancomycin induced significant changes (*P < 0.1) in multiple colonic cytokines and chemokines. Each column represents an individual animal.
Vancomycin abolishes GUS-producing gut bacteria
Using 16S ribosomal RNA (rRNA) amplicon sequencing, we found vancomycin to have broad effects on the composition of the gut microbiota. After 2 days of vancomycin treatment (day 10), the gut microbiome of mice treated with both MMF and vancomycin was significantly (P < 0.05) distinct from the MMF-only group with regard to β-diversity as measured using a nonmetric multidimensional scaling (NMDS) plot of Bray-Curtis dissimilarity and continued to separate from the MMF group over time (fig. S5). After treatment with vancomycin, the α-diversity of the gut microbiome decreased significantly as measured by observed species (277 versus 549, P = 2.8 × 10−4), the Shannon diversity index (1.42 versus 3.83, P < 1.0 × 10−7), and the Simpson’s diversity index (0.59 versus 0.92, P = 8.0 × 10−4) after only 2 days of antibiotic exposure. The Shannon and Simpson’s diversity indices both continued to be significantly lower in the MMF- and vancomycin-treated group for the following 6 days (fig. S5).
At the class level, dominant bacteria included Clostridia, Bacteroidia, and Bacilli during the initial 8 days of MMF treatment (Fig. 2A and fig. S6A). Continued MMF exposure led to an expansion of Gammaproteobacteria, Erysipelotrichia, and, to a lesser extent, Deltaproteobacteria. After the addition of vancomycin on day 8, the Bacteroidia, Clostridia, and Erysipelotrichia taxa were severely diminished, while Bacilli and Gammaproteobacteria flourished. By day 16, after 8 days of combined vancomycin and MMF exposure, the bacterial community was composed almost entirely of class Bacilli, with only a small proportion of Gammaproteobacteria remaining as the next predominant class. The classes Bacilli and Betaproteobacteria were significantly increased (P < 0.05) after the addition of vancomycin (fig. S6A). Considering the differentially depleted classes (Bacteroidia, Clostridia, and Erysipelotrichia present in the MMF-treated animals but eliminated by vancomycin), the genera that were both significantly differentially affected by vancomycin and in relatively high abundance in animals after 8 days of MMF treatment were Bacteroides, Lachnospiraceae NK4A136, Roseburia, and Turicibacter (fig. S6B). We generated species-level resolution for Bacteroides including Bacteroides vulgatus, Bacteroides fragilis, Bacteroides caccae, Bacteroides uniformis, Bacteroides ovatus, and Bacteroides nordii, all of which were detectable in the presence of MMF but rapidly eliminated by vancomycin (Fig. 2B).
Fig. 2. Vancomycin-induced changes in the gut microbiome.
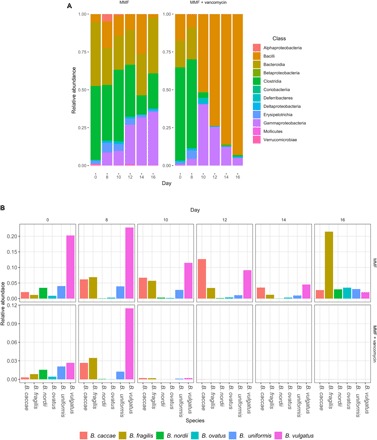
Amplicon sequencing of DNA extracted from mouse fecal pellets was used to determine bacterial composition. (A) Stacked bar plots showing relative abundance of bacterial classes in the presence of MMF with and without vancomycin. Vancomycin decreased overall diversity with a gradual predominance of Bacilli. (B) Bacteroides spp. were abundant in the presence of MMF but were rapidly eliminated by the introduction of vancomycin on day 8.
GUS-predicted gene expression and measured enzyme activity are increased by MMF and abolished by vancomycin
We examined the proportional abundance of the bacterial GUS gene using PICRUSt (Phylogenetic Investigation of Communities using Reconstruction of Unobserved States). We found the predicted proportional abundance of GUS enzyme orthologs [Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology K01195, KEGG Enzyme EC 3.2.1.31] to be comparable between groups before commencing MMF treatment. After 4 days of MMF treatment, the MMF-only group had a significant increase in the predicted proportional abundance of K01195 (P = 0.006), but after 8 days, the abundance in the MMF-only group had decreased slightly (Fig. 3A). Functionally, the activity of GUS enzymes, assessed in fecal pellets of untreated and MMF-treated mice, followed the same pattern, with the treated mice exhibiting a significant increase in enzyme activity after 4 days of exposure (Fig. 3B). During the initial 8 days of MMF-only treatment, prior to the initiation of vancomycin, there were no differences between the two groups with regard to gene expression (P = 0.33) (Fig. 3C). The introduction of vancomycin after 8 days of MMF treatment reduced the predicted proportional abundance of K01195 after only 2 days (day 10). After 8 days of vancomycin treatment (day 16), the abundance of K01195 was reduced to nearly undetectable levels, in comparison to preantibiotic levels (P = 1.2 × 10−7), despite continuation of the MMF exposure. Concordant with reduced gene expression, GUS activity in fecal pellets was abolished by treatment with vancomycin (Fig. 3D).
Fig. 3. MMF–up-regulated GUS expression and activity are prevented by vancomycin.
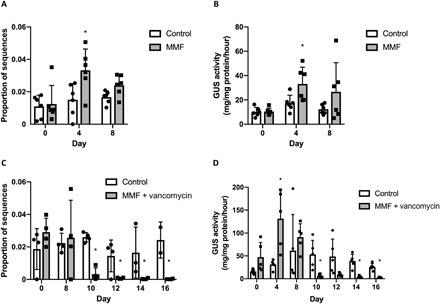
PICRUSt and an enzyme activity assay were used to assess gene expression and GUS activity in mouse fecal pellets. (A) The proportional abundance of KEGG ortholog K01195 increased significantly (*P = 0.006) in the presence of MMF after 4 days. (B) GUS activity in fecal pellets collected from mice consuming MMF also increased significantly (P = 0.03) after 4 days. (C) The introduction of vancomycin on day 8 rapidly eliminated the expression of K01195. (D) Vancomycin, introduced after day 8, abolished GUS activity in fecal pellets (*P < 0.01). All data are plotted as means and SDs with data points for individual samples represented.
Ex vivo imaging of intestinal GUS activity
The effect of MMF and vancomycin on intestinal GUS activity in the intact animal was assessed using the fluorescent substrate fluorescein di-β-d-glucuronide (FDGlcU). Mice gavaged with FDGlcU and exposed to MMF demonstrated an up-regulation of GUS activity in the proximal colon after 8 days of treatment but not in the cecum as compared with controls. Vancomycin significantly reduced GUS activity in both the cecum (day 4, P = 0.05; day 8, P = 0.008) and proximal colon (day 8, P = 0.03) and also caused a qualitative enlargement of the cecum (Fig. 4).
Fig. 4. Intestinal GUS activity is decreased by vancomycin.
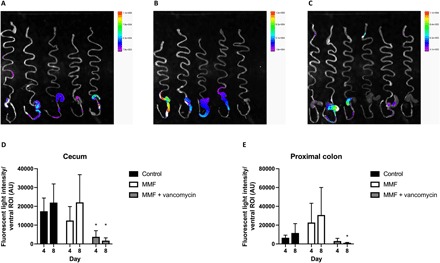
Mice were treated with MMF or MMF and vancomycin for 8 days, and then GUS activity was imaged using the fluorescent substrate FDGlcU. (A) Ex vivo imaging of the GI tract from control mice. (B) Ex vivo imaging of the mouse GI tract after animals consumed MMF for 8 days. (C) Ex vivo imaging of the GI tract of mice exposed to MMF and vancomycin for 8 days. (D) Quantification of GUS activity in the cecum did not demonstrate a significant increase after 4 or 8 days of MMF exposure, but activity decreased significantly after 4 and 8 days of vancomycin treatment. (E) Quantification of GUS activity in the proximal colon did not demonstrate a significant increase with MMF, but activity decreased significantly after 8 days of vancomycin treatment compared with animals receiving MMF only. *P < 0.05.
Vancomycin limits MPAG catabolism
The catabolism of MPAG/AcMPAG (undifferentiated in our fecal assay) to MPA and GA was found to be significantly reduced by the addition of vancomycin in MMF-treated mice, as measured in fecal pellets using ultra-high-performance liquid chromatography (UHPLC) mass spectrometry (MS) (Fig. 5A). During the MMF-only exposure (days 0 to 8), we observed relatively high-average concentrations of MPA in both groups (35.2 ± 4.13 and 33.73 ± 4.50 μM) and relatively low-average concentrations of MPAG (1.85 ± 0.48 and 1.53 ± 0.47 μM). After the addition of vancomycin on day 8, there was a significant increase in the average concentration of MPAG (16.01 ± 0.92 μM, P = 0.01) and a nearly eightfold decrease in the average MPA concentration (4.27 ± 0.73 μM, P < 1 × 10−7). There were no significant changes in average GA concentration in either group over the treatment period. Notably, in serum, there were no significant differences between treatment groups for either MPAG, MPA, or GA on day 16 (Fig. 5B). AcMPAG was not detectable in either group by our assay.
Fig. 5. Changes in MMF metabolites.
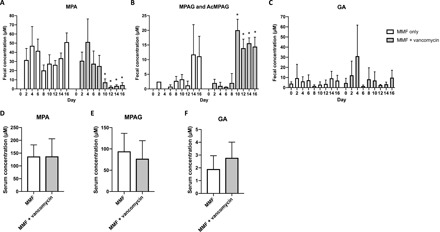
The concentrations of MMF metabolites were measured in mouse fecal pellets and serum. (A to C) Comparison of MPA, MPAG, and GA levels in mice exposed to MMF only for up to 16 days and mice initially exposed to MMF only for 8 days and then with the addition of vancomycin for 8 days. Vancomycin treatment resulted in significant increases in MPAG and significant decreases in MPA levels (P < 0.05). (D to F) In serum from mice consuming MMF, levels of MPA, MPAG, and GA were not affected after 8 days of treatment with vancomycin. AcMPAG was not detected in the serum. Data are means ± SEM. The mean concentration of each metabolite before (days 0 to 8) and after (days 10 to 16) the introduction of vancomycin was compared independently in each treatment group using a two-way analysis of variance (ANOVA), with *P < 0.0005.
Intrarectal administration of MPA promotes weight loss and inflammation
To elucidate the identity of the toxic metabolite, we directly administered MPA and GA into the mouse distal colon. There was a concentration-dependent effect of MPA, which caused significant weight loss (P = 0.002) in treated animals and appeared to promote inflammation as indicated by a trend toward increased activity of colonic myeloperoxidase (MPO) activity (fig. S7).
GUS activity in human stool correlates with MMF exposure
Human fecal GUS activity measured in stool samples obtained from adult and pediatric heart transplant recipients was variable but correlated with MMF exposure (Fig. 6). Patient 1, with the highest level of GUS activity, experienced significant neutropenia that was attributed to MMF. Patient 3 is the patient from our case report who had developed hematochezia and lymphonodular hyperplasia related to MMF. Patients 10 and 11 were pediatric patients whose MMF had been discontinued following the recent diagnosis of posttransplant lymphoproliferative disease.
Fig. 6. GUS activity in human stool.
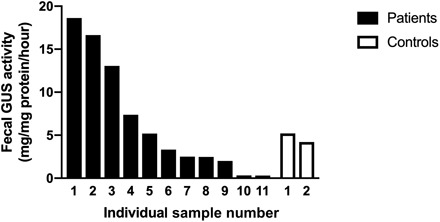
Samples were collected from 11 adult and pediatric heart transplant recipients and 2 healthy, nontransplant controls. The highest level of GUS activity seen in patient 1 correlated with lymphopenia requiring a dose reduction in her MMF. Patient 3 had developed hematochezia and colonic lymphonodular hyperplasia while taking MMF. Patients 10 and 11 had been taken off MMF before sample collection.
DISCUSSION
The use of MMF as an immunosuppressant is frequently limited by GI side effects. Attempts to relieve toxicity by modifying drug delivery, either through use of an enteric coating or intravenous administration, have not been successful (11, 12). The metabolism of MMF and MPA has been well described, but the mechanism of toxicity has remained elusive. Previously, we have demonstrated the requirement for an intact gut microbiome for MMF to cause GI toxicity in the mouse (10). Here, we now demonstrate the ability of a single antibiotic, vancomycin, to both prevent and reverse MMF-induced GI toxicity through the reduction of GUS expression, activity, and GUS-producing bacteria, and, consequently, decreasing catabolism of the MMF metabolite MPAG to free MPA. In our mouse model, reduced fecal concentrations of MPA were associated with reduced GI toxicity.
Complete eradication of the gut microbiome with a cocktail of broad-spectrum antibiotics was able to prevent and reverse MMF-induced GI toxicity (10). From those four antibiotics, it appears that only vancomycin is necessary and sufficient for the prevention and reversal of MMF-induced weight loss. Vancomycin is used clinically primarily for its ability to treat infections caused by methicillin-resistant Staphylococcus aureus and colitis caused by Clostridium difficile. Its impact in reversing MMF toxicity was unexpected. Neomycin and ampicillin have broader activity against both Gram-negative and Gram-positive organisms, and metronidazole targets obligate anaerobes and specific protozoans. Vancomycin was also used to reverse MMF-induced colonic inflammation as demonstrated by improvements in cecum, liver, and spleen weights and colon length. Colonic tissue homogenates also showed changes in multiple cytokines and chemokines consistent with a reduction in the proinflammatory state induced by MMF exposure. This inflammatory stimulus was previously postulated to be a pathogenic bacterium (i.e., Escherichia/Shigella) that expanded due to MMF-induced dysregulation of the microbiome (10, 13). However, our present data suggest that the factor initiating the inflammatory response may be free MPA, released after bacterial GUS metabolism in the lumen, which interacts with the epithelium in the distal GI tract.
Using 16S rRNA amplicon sequencing, we determined which bacteria thrived during MMF exposure, presumably due to their consumption of free GA as a nutrient source (7, 14), and were eliminated by the addition of vancomycin. After 8 days of MMF-only treatment, there was a consistent abundance of the bacterial classes Betaproteobacteria, Bacteroidia, Bacilli, Gammaproteobacteria, Erysipelotrichia, and Alphaproteobacteria. After initiating vancomycin treatment, the classes Bacteroidia, Clostridia, and Erysipelotrichia were severely depleted, allowing for expansion of the classes Gammaproteobacteria and Bacilli. Given the reportedly narrow spectrum of activity of vancomycin primarily against Gram-positive organisms, this response was surprising. However, a similar response was also observed in humans treated with oral vancomycin (15, 16). In the study by Isaac et al. (15), the authors found that humans consuming vancomycin for 21 days demonstrated a loss of gut microbe diversity and alterations in most bacterial taxa, including reductions in the Bacteroidetes (which includes the class Bacteroidia) and Firmicutes phyla. This study and our data suggest that vancomycin has broad effects on the gut microbiome, including Gram-negative organisms, or, more specifically, is affecting the gut microbe ecosystem and removing key (as yet unidentified) microbes, causing the collapse of important populations such as the Bacteroidia. The potential pathogenic bacteria Escherichia/Shigella (which cannot be distinguished based on 16S rRNA sequencing) of class Gammaproteobacteria, previously thought to be a primary cause of inflammation following MMF-induced dysbiosis of the microbiota (10, 13), expanded during vancomycin treatment, while inflammation and toxicity improved. The gut bacteria Bacteroides spp. and Roseburia spp., both enriched in the presence of MMF and eliminated by vancomycin, encode for GUS enzymes and have demonstrated GUS activity in vitro (17, 18). B. fragilis, B. uniformis, B. caccae, and B. vulgatus all increased in abundance during MMF treatment, and these bacterial species were eliminated by vancomycin, suggesting the capacity for bacterial GUS activity was reduced. This hypothesis was supported by our PICRUSt analysis and in vivo imaging of GUS activity.
PICRUSt predicts the functional content of a metagenome from 16S rRNA amplicon sequencing data (19). Functional orthologs are annotated in the KEGG Orthology database (20). In bacterial DNA extracted from mouse fecal pellets, PICRUSt predicted an increase in the abundance of GUS orthologs in response to MMF after 4 days. This suggests a rapid increase in bacterial metabolism in response to the introduction of MMF, but this increase does not appear to be sustained. This increase in predicted gene expression was supported by our in vitro assay for GUS activity, which showed a similar, transient increase in activity. In humans, levels of GUS activity were quite variable but did correlate with MMF-related side effects for two of the patients with the highest level of GUS activity and with the lowest levels seen in patients who had been recently taken off MMF as part of a reduction in their immunosuppression related to the diagnosis of malignancy. Levels of stool GUS activity in humans on MMF may therefore represent a potential biomarker to assess for drug-related toxicity, although this remains to be tested in a larger cohort.
Assessment of bacterial GUS activity in vivo based on activation of a fluorescent substrate after GUS hydrolysis (21) demonstrated an increase in overall bacterial GUS activity in the proximal colon, but not the cecum, of MMF-treated mice as compared with controls that was abolished by treatment with vancomycin. In fecal pellets, relatively higher levels of MPA and lower levels of MPAG were measured during MMF treatment. The introduction of vancomycin induced a sharp increase in MPAG concentration, in tandem with a drastic reduction in the concentration of MPA, demonstrating that vancomycin does alter bacterial metabolism of MPAG in the colon. Reduced colonic concentrations of MPA in our model coincided with reduced GI and systemic toxicity, suggesting that MPA is the major contributor to MMF-induced GI inflammation and toxicity. The serum concentration of MPA was not significantly reduced in the vancomycin-treated animals. However, our assay did not specifically distinguish between MPAG and AcMPAG. In our previous work, MMF resulted in a modest increase in hepatic gene expression of the MPAG transporter Abcc2, but levels did not decrease with antibiotic exposure (10).
Multiple human clinical reports have indirectly implicated GUS involvement in MMF metabolism (22, 23), and recent work by others further supports our hypothesis that MPA is responsible for the GI toxicity experienced by patients. MPA depleted the intracellular guanosine pool in human intestinal LS180 cells, potentially leading to reduced membrane integrity (24). Qasim et al. demonstrated that MPA directly modulated intestinal epithelial tight junctions in Caco-2 cell monolayers by up-regulating myosin light-chain kinase, leading to increased phosphorylation of myosin II regulatory light chain and increased tight junction permeability (25, 26). This may occur via MPA-stimulated activation of the midkine-dependent PI3K (phosphatidylinositol 3-kinase) pathway (27). Our previous work supports these observations of MPA-induced epithelial barrier dysfunction as we found that MMF exposure led to an increase in serum lipopolysaccharide, an indirect measure of intestinal epithelial barrier function (10, 28). Together, these data suggest that MPA may trigger local and systemic toxicity by disrupting GI epithelial barrier function, potentially allowing toxic metabolites to cross into the tissue and circulation.
In summary, our data suggest that bacterial GUS metabolism of MPAG to free MPA in the colonic lumen is responsible for the GI toxicity associated with MMF use. The GUS pathway responsible for MMF metabolism affects multiple other commonly prescribed drugs, including nonsteroidal anti-inflammatory drugs, acetaminophen, irinotecan, and cannabinoids (29). The ability of vancomycin to reduce toxicity suggests a possible therapeutic approach to treat or prevent MMF-induced GI toxicity.
MATERIALS AND METHODS
Animals
All animal procedures were approved by the University of Calgary Animal Care Committee (protocol AC14-0068) and were in accordance with the Canadian Council on Animal Care policies. Female C57BL/6 mice aged 7 to 10 weeks (Charles River) were used in all experiments. Mice were housed in sterilized, filter-top cages under specific pathogen–free conditions, separated by treatment group, and fed control chow (Teklad Global 16% protein rodent diet TD.00217) for a minimum of 1 week before commencing any experiment. MMF-containing chow was administered ad libitum at a concentration of 0.563% (w/w) (30). Vancomycin (500 mg/liter) was administered in the drinking water ad libitum. Consumption of both chow and water was monitored daily.
Sample collection and assays
Mice were individually placed in clean, ethanol-sterilized cages, and immediately upon defecation, each fecal pellet was placed in a sterile collection tube, snap frozen, and stored at −80°C. Whole blood was collected from anesthetized animals by cardiac puncture. Serum was isolated by centrifugation at 1500g × 15 min and stored at −80°C. Liver, spleen, and intestine were collected immediately after euthanasia. Each tissue was weighed, and colon length was measured before being placed in microcentrifuge tubes, snap frozen, and then stored at −80°C. Hematocrit was determined using a microhematocrit capillary tube reader. Whole colonic tissue was homogenized in cell lysis buffer, and protein concentration was determined using the Precision Red Protein Assay (Cytoskeleton). Samples were sent to Eve Technologies for analysis using their 32-multiplex Mouse Cytokine/Chemokine Array.
Bacterial community analysis
Bacterial genomic DNA was isolated from mouse fecal pellets (31), and concentration was determined using a Qubit 2.0 fluorometer (Invitrogen). Variable regions 3 and 4 of the 16S rRNA gene were amplified by polymerase chain reaction using barcoded primers (32) before sequencing on an Illumina MiSeq in the Nicole Perkins Microbial Communities Core Laboratory at the University of Calgary. Primers and poor-quality reads were removed using Cutadapt v1.16 and then processed using DADA2 v1.6.0 (33). Taxonomy was assigned using the Silva Taxonomy Training Set v128 and the Silva Species Assignment Set v128 (34). Visual and statistical analyses of amplicon sequencing data were performed using the R packages Phyloseq (35) and Vegan v2.5-2 with support from dplyr 0.7.5 and ggplot2 v3.0.0. Reads of nonbacterial origin and samples with insufficient depth (<10,000 reads) were removed. Observed species, Shannon diversity index, and Simpson’s index were determined. Multiple indices were used to ensure that a potential bias did not arise from unequal numbers of reads between samples. β-Diversity was assessed using an NMDS plot of Bray-Curtis dissimilarity. The R package DESeq2 v1.18.1 and microbiomeSeq v0.1 were used to calculate significantly differentially abundant taxa in the vancomycin-treated group over time. Prior to further analysis and visualization of taxa-specific differences between samples or treatment groups, reads in each sample were normalized to relative abundance.
For input into PICRUSt v1.1.3, reads were reprocessed into operational taxonomic units (OTUs) (36). Read quality was assessed using FastQC v0.11.5, and paired-end reads were merged and filtered using PEAR v0.9.10. Chimeric sequences were removed using VSEARCH v1.11. Sequences were clustered into OTUs using a closed reference OTU picking pipeline using QIIME v1.91 (37) and mapped to the Greengenes Database v13_8. Within PICRUSt, the OTU table was 16S rRNA copy number normalized, and then functional predictions of KEGG orthologs were predicted using PICRUSt’s Hidden State Prediction algorithm.
Ex vivo imaging of GUS activity
We performed ex vivo tissue imaging of intestinal bacterial GUS activity through quantification of fluorescence following hydrolysis of FDGlcU (Sigma-Aldrich) to fluorescein. Three hours prior to imaging, each animal was gavaged with FDGlcU (7.3 μmol/kg). Immediately before imaging, mice were euthanized, and abdominal organs were excised and imaged using an In Vivo Xtreme 4MP imaging platform (Bruker). Reflectance imaging (2-s exposure time) was followed by fluorescence imaging (5-s exposure time, 470-nm excitation and 535-nm emission filters) and x-ray imaging (10-s exposure time). Pixel binning was kept constant at 4 × 4. Images were acquired and analyzed using the Bruker molecular imaging software MI SE v7.1.3.20550. GUS activity was quantified by measuring the mean fluorescence after background subtraction in a given region of interest, which was kept constant over the time period of imaging.
Metabolite extraction and MS
Fecal metabolites were extracted from fecal pellets (50 mg) using 500 μl of 4°C 50% methanol followed by homogenization with ceramic beads using a Bead Ruptor 4 (Omni). Insoluble debris was separated by centrifugation at 10,000g × 15 min at 4°C, and 300 μl was collected. The remaining material was reextracted in an additional 300 μl of 4°C 50% methanol following the same procedure, and supernatants from the two-stage extraction were combined. Metabolites present in whole blood were extracted by diluting samples 1:20 (v/v) in 4°C 50% methanol. Insoluble material was removed by centrifugation at 14,600g × 5 min at 4°C, and the supernatant was harvested. All metabolite extracts were stored at −80°C until quantitative analyses could be completed. All MS analyses were conducted at the Calgary Metabolomics Research Facility (CMRF). Metabolites in the extracts were quantified using UHPLC MS. Fecal metabolites were separated by hydrophilic interaction liquid chromatography (Syncronis HILIC column, Thermo Fisher Scientific), and serum metabolites were separated using reversed-phase chromatography (Accucore Vanquish C18+, Thermo Fisher Scientific). MS analyses were conducted using a Q-Exactive HF Mass Spectrometer (Thermo Fisher Scientific) in negative ion full-scan mode. Metabolites were assigned by high-resolution MS and coelution of the metabolite with a commercial metabolite standard (MPA and GA from Thermo Fisher Scientific, and MPAG and AcMPAG from Toronto Research Chemicals). Metabolite concentrations were measured by fitting peak intensities of extracted ion chromatograms to calibration reference curves prepared using standards. Data were analyzed using the MAVEN software package (38).
Enzymatic assay for GUS activity
Fecal pellets were homogenized in phosphate-buffered saline (0.01 M, pH 7.0) using a Bead Ruptor 4 (Omni) and then sonicated using a Branson 3510 Water Bath Ultrasonicator. Activity of extracted bacterial GUS was measured by hydrolysis of phenolphthalein glucuronide (Sigma-Aldrich) to free phenolphthalein and quantified by spectrophotometry (39). GUS activity was calculated as mg of free phenolphthalein liberated/mg protein/hour of incubation.
Intrarectal administration of MPA and GA
Injections were performed daily for 8 days using a 1-ml syringe and a polyethylene catheter (PE-50). With the mouse immobilized, the catheter was inserted 3 to 4 cm intrarectally, and 200 μl of either vehicle (50% dimethyl sulfoxide), GA (17.5 mM), or MPA (1.75 or 17.5 mM) was slowly injected by hand. After removal of the catheter, the rectum was held closed for a minimum of 2 min. Body weight of each animal was recorded daily. After 8 days, animals were sacrificed. Colonic tissue was homogenized in hexadecyltrimethylammonium buffer using a Polytron homogenizer (Kinematica). O-dianisidine (Sigma-Aldrich) was added to the cell lysate, and its oxidation was measured by absorbance at 450 nm using a SpectraMax Plus 384 microplate reader. MPO activity (U/mg tissue) was calculated from absorbance based on a standard curve.
Statistical analysis
All statistical analysis was conducted using R and Prism 7 (GraphPad). Data are expressed as means ± SD. For each set of data, normality was determined using the Shapiro-Wilk Normality Test. For data with two treatment groups, each experimental group mean was compared pairwise against the control group mean using either Welch’s t test or Wilcoxon rank sum test with a correction for multiple comparisons using the Benjamini-Hochberg procedure. Means of parametric data with >2 treatment variables compared at a single time point were analyzed using a one-way ANOVA. Significant ANOVA results were followed by Tukey’s honestly significant difference post hoc test. Means of nonparametric data with >2 treatment variables compared at a single time were analyzed using pairwise Wilcoxon rank sum tests with P value correction for multiple testing using the Benjamini-Hochberg procedure. To compare means of two treatment groups over time, a two-way ANOVA was used, followed by Tukey’s honestly significant difference post hoc test. If the two-way ANOVA residuals were nonparametric, the Kruskal-Wallis rank sum test, followed by Dunn’s Kruskal-Wallis multiple comparison post hoc test, was used. Intergroup significance in β-diversity was computed using the Adonis test applied pairwise, using the Benjamini-Hochberg procedure for P value correction. A value of P < 0.05 was considered significant, except for the assessment of colonic tissue cytokines and chemokines, the pairwise group comparisons of observed species for α-diversity, and β-diversity comparisons between MMF- and vancomycin-treated animals, where a value P < 0.1 was considered significant.
Supplementary Material
Acknowledgments
We acknowledge the assistance of B. Petri and the Mouse Phenomics Resource Laboratory in the Snyder Institute for Chronic Diseases at the University of Calgary with the in vivo mouse imaging experiments. Funding: This project was supported by a research grant from the Canadian National Transplant Research Program (to S.C.G.). M.R.T. was a recipient of a Cumming School of Medicine graduate scholarship. Metabolomics data were acquired at the CMRF, which is supported by the International Microbiome Centre and the Canada Foundation for Innovation (CFI-JELF 34986). I.A.L. was supported by an Alberta Innovates Translational Health Chair. Author contributions: M.R.T., K.L.F., S.A.H., and S.C.G. designed the study. M.R.T., K.L.F., H.R., and A.M. performed the experiments. M.R.T. and S.C.G. analyzed the data. M.R.T. and I.A.L. performed the MS assays and analysis. M.R.T. and S.C.G. wrote the manuscript. All authors reviewed and approved the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaax2358/DC1
Fig. S1. MMF metabolism.
Fig. S2. Effect of individual antibiotics (ampicillin, neomycin, metronidazole, or vancomycin) on mouse body weight in the presence of MMF.
Fig. S3. Tissue weights.
Fig. S4. Effect of 8 days of exposure to MMF alone or MMF with vancomycin on individual cytokines and chemokines.
Fig. S5. Changes in bacterial diversity.
Fig. S6. Changes in bacterial abundance.
Fig. S7. Intrarectal metabolite effects.
REFERENCES AND NOTES
- 1.Staatz C. E., Tett S. E., Pharmacology and toxicology of mycophenolate in organ transplant recipients: An update. Arch. Toxicol. 88, 1351–1389 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Kobashigawa J. A., Meiser B. M., Review of major clinical trials with mycophenolate mofetil in cardiac transplantation. Transplantation 80, S235–S243 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Panarelli N. C., Drug-induced injury in the gastrointestinal tract. Semin. Diagn. Pathol. 31, 165–175 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Calmet F. H., Yarur A. J., Pukazhendhi G., Ahmad J., Bhamidimarri K. R., Endoscopic and histological features of mycophenolate mofetil colitis in patients after solid organ transplantation. Ann. Gastroenterol. 28, 366–373 (2015). [PMC free article] [PubMed] [Google Scholar]
- 5.Knoll G. A., MacDonald I., Khan A., Van Walraven C., Mycophenolate mofetil dose reduction and the risk of acute rejection after renal transplantation. J. Am. Soc. Nephrol. 14, 2381–2386 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Lamba V., Sangkuhl K., Sanghavi K., Fish A., Altman R. B., Klein T. E., PharmGKB summary: Mycophenolic acid pathway. Pharmacogenet. Genomics 24, 73–79 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pellock S. J., Redinbo M. R., Glucuronides in the gut: Sugar-driven symbioses between microbe and host. J. Biol. Chem. 292, 8569–8576 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clemente J. C., Ursell L. K., Parfrey L. W., Knight R., The impact of the gut microbiota on human health: An integrative view. Cell 148, 1258–1270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flannigan K. L., Rajbar T., Moffat A., McKenzie L. S., Dicke F., Rioux K., Workentine M. L., Louie T. J., Hirota S. A., Greenway S. C., Changes in composition of the gut bacterial microbiome after fecal microbiota transplantation for recurrent Clostridium difficile infection in a pediatric heart transplant patient. Front. Cardiovasc. Med. 4, 17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flannigan K. L., Taylor M. R., Pereira S. K., Rodriguez-Arguello J., Moffat A. W., Alston L., Wang X., Poon K. K., Beck P. L., Rioux K. P., Jonnalagadda M., Chelikani P. K., Galipeau H. J., Lewis I. A., Workentine M. L., Greenway S. C., Hirota S. A., An intact microbiota is required for the gastrointestinal toxicity of the immunosuppressant mycophenolate mofetil. J. Heart Lung Transplant. 37, 1047–1059 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Pescovitz M. D., Conti D., Dunn J., Gonwa T., Halloran P., Sollinger H., Tomlanovich S., Weinstein S., Inokuchi S., Kiberd B., Kittur D., Merion R. M., Norman D., Shoker A., Wilburn R., Nicholls A. J., Arterburn S., Dumont E., Intravenous mycophenolate mofetil: Safety, tolerability, and pharmacokinetics. Clin. Transplant. 14, 179–188 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Gardiner K. M., Tett S. E., Staatz C. E., Is conversion from mycophenolate mofetil to enteric-coated mycophenolate sodium justifiable for gastrointestinal quality of life? Drugs R D 18, 271–282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tourret J., Willing B. P., Dion S., MacPherson J., Denamur E., Finlay B. B., Immunosuppressive treatment alters secretion of ileal antimicrobial peptides and gut microbiota, and favors subsequent colonization by uropathogenic Escherichia coli. Transplantation 101, 74–82 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Peekhaus N., Conway T., What’s for dinner?: Entner-Doudoroff metabolism in Escherichia coli. J. Bacteriol. 180, 3495–3502 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Isaac S., Scher J. U., Djukovic A., Jiménez N., Littman D. R., Abramson S. B., Pamer E. G., Ubeda C., Short- and long-term effects of oral vancomycin on the human intestinal microbiota. J. Antimicrob. Chemother. 72, 128–136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Louie T. J., Byrne B., Emery J., Ward L., Krulicki W., Nguyen D., Wu K., Cannon K., Differences of the fecal microflora With Clostridium difficile therapies. Clin. Infect. Dis. 60 (Suppl. 2), S91–S97 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Pollet R. M., D’Agostino E. H., Walton W. G., Xu Y., Little M. S., Biernat K. A., Pellock S. J., Patterson L. M., Creekmore B. C., Isenberg H. N., Bahethi R. R., Bhatt A. P., Liu J., Gharaibeh R. Z., Redinbo M. R., An atlas of β-glucuronidases in the human intestinal microbiome. Structure 25, 967–977.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pellock S. J., Walton W. G., Biernat K. A., Torres-Rivera D., Creekmore B. C., Xu Y., Liu J., Tripathy A., Stewart L. J., Redinbo M. R., Three structurally and functionally distinct β-glucuronidases from the human gut microbe Bacteroides uniformis. J. Biol. Chem. 293, 18559–18573 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langille M. G. I., Zaneveld J., Caporaso J. G., McDonald D., Knights D., Reyes J. A., Clemente J. C., Burkepile D. E., Vega Thurber R. L., Knight R., Beiko R. G., Huttenhower C., Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanehisa M., Goto S., KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen M., Cheng K.-W., Chen Y.-J., Wang C.-H., Cheng T.-C., Chang K.-C., Kao A.-P., Chuang K.-H., Real-time imaging of intestinal bacterial β-glucuronidase activity by hydrolysis of a fluorescent probe. Sci. Rep. 7, 3142 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ratna P., Mathew B. S., Annapandian V. M., Saravanakumar K., Basu G., Tamilarasi V., Fleming D. H., Pharmacokinetic drug interaction of mycophenolate with co-amoxiclav in renal transplant patients. Transplantation 91, e36–e38 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Kodawara T., Masuda S., Yano Y., Matsubara K., Nakamura T., Masada M., Inhibitory effect of ciprofloxacin on β-glucuronidase-mediated deconjugation of mycophenolic acid glucuronide. Biopharm. Drug Dispos. 35, 275–283 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Heischmann S., Dzieciatkowska M., Hansen K., Leibfritz D., Christians U., The immunosuppressant mycophenolic acid alters nucleotide and lipid metabolism in an intestinal cell model. Sci. Rep. 7, 45088 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qasim M., Rahman H., Ahmed R., Oellerich M., Asif A. R., Mycophenolic acid mediated disruption of the intestinal epithelial tight junctions. Exp. Cell Res. 322, 277–289 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Turner J. R., Rill B. K., Carlson S. L., Carnes D., Kerner R., Mrsny R. J., Madara J. L., Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am. J. Physiol. 273, C1378–C1385 (1997). [DOI] [PubMed] [Google Scholar]
- 27.Khan N., Binder L., Pantakani D. V. K., Asif A. R., MPA modulates tight junctions’ permeability via Midkine/PI3K pathway in Caco-2 cells: a possible mechanism of leak-flux diarrhea in organ transplanted patients. Front. Physiol. 8, 438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Derikx J. P. M., Luyer M. D. P., Heineman E., Buurman W. A., Non-invasive markers of gut wall integrity in health and disease. World J. Gastroenterol. 16, 5272–5279 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wallace B. D., Wang H., Lane K. T., Scott J. E., Orans J., Koo J. S., Venkatesh M., Jobin C., Yeh L. A., Mani S., Redinbo M. R., Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 330, 831–835 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jonnalagadda M., Brown C. E., Chang W.-C., Ostberg J. R., Forman S. J., Jensen M. C., Engineering human T cells for resistance to methotrexate and mycophenolate mofetil as an in vivo cell selection strategy. PLOS ONE 8, e65519 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whelan F. J., Verschoor C. P., Stearns J. C., Rossi L., Luinstra K., Loeb M., Smieja M., Johnstone J., Surette M. G., Bowdish D. M. E., The loss of topography in the microbial communities of the upper respiratory tract in the elderly. Ann. Am. Thorac. Soc. 11, 513–521 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Bartram A. K., Lynch M. D. J., Stearns J. C., Moreno-Hagelsieb G., Neufeld J. D., Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end illumina reads. Appl. Environ. Microbiol. 77, 3846–3852 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., Holmes S. P., DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pruesse E., Quast C., Knittel K., Fuchs B. M., Ludwig W., Peplies J., Glöckner F. O., SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McMurdie P. J., Holmes S., phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLOS ONE 8, e61217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Comeau A. M., Douglas G. M., Langille M. G. I., Microbiome Helper: A custom and streamlined workflow for microbiome research. mSystems 2, e00127-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caporaso J. G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F. D., Costello E. K., Fierer N., Peña A. G., Goodrich J. K., Gordon J. I., Huttley G. A., Kelley S. T., Knights D., Koenig J. E., Ley R. E., Lozupone C. A., McDonald D., Muegge B. D., Pirrung M., Reeder J., Sevinsky J. R., Turnbaugh P. J., Walters W. A., Widmann J., Yatsunenko T., Zaneveld J., Knight R., QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melamud E., Vastag L., Rabinowitz J. D., Metabolomic analysis and visualization engine for LC−MS data. Anal. Chem. 82, 9818–9826 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Asano Y., Hiramoto T., Nishino R., Aiba Y., Kimura T., Yoshihara K., Koga Y., Sudo N., Critical role of gut microbiota in the production of biologically active, free catecholamines in the gut lumen of mice. Am. J. Physiol. Gastrointest. Liver Physiol. 303, G1288–G1295 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaax2358/DC1
Fig. S1. MMF metabolism.
Fig. S2. Effect of individual antibiotics (ampicillin, neomycin, metronidazole, or vancomycin) on mouse body weight in the presence of MMF.
Fig. S3. Tissue weights.
Fig. S4. Effect of 8 days of exposure to MMF alone or MMF with vancomycin on individual cytokines and chemokines.
Fig. S5. Changes in bacterial diversity.
Fig. S6. Changes in bacterial abundance.
Fig. S7. Intrarectal metabolite effects.