

Abstract
The present study directly tested the hypothesis that the Na+/H+ exchanger 3 (NHE3) in the proximal tubules of the kidney is required for the development of angiotensin II (ANG II)-induced hypertension using proximal tubule-specific NHE3 knockout mice (PT-Nhe3−/−). Specifically, PT-Nhe3−/− mice were generated using the SGLT2-Cre/Nhe3loxlox approach, whereas ANG II-induced hypertension was studied in 12 groups (n=5–12 per group) of adult male and female wild-type (WT) and PT-Nhe3−/− mice. Under basal conditions, systolic (SBP), diastolic (DBP), and mean arterial blood pressure (MAP) were significantly lower in male and female PT-Nhe3−/− than WT mice (P<0.01). A high pressor, 1.5 mg/kg/day, i.p., or a slow pressor dose of ANG II, 0.5 mg/kg/day, i.p., for 2 weeks significantly increased SBP, DBP, and MAP in male and female WT mice (P<0.01), but the hypertensive response to ANG II was markedly attenuated in male and female PT-Nhe3−/− mice (P<0.01). ANG II impaired the pressure-natriuresis response in WT mice, whereas proximal tubule-specific deletion of NHE3 improved the pressure-natriuresis response in ANG II-infused PT-Nhe3−/− mice (P<0.01). AT1 receptor blocker losartan completely blocked ANG II-induced hypertension in both WT and PT-Nhe3−/− mice (P<0.01). However, inhibition of nitric oxide synthase with L-NAME had no effect on ANG II-induced hypertension in WT or PT-Nhe3−/− mice (n.s.). Furthermore, ANG II-induced hypertension was significantly attenuated by an orally absorbable NHE3 inhibitor AVE0657. In conclusion, NHE3 in the proximal tubules of the kidney may be a therapeutical target in hypertension induced by ANG II or with increased NHE3 expression in the proximal tubules.
Keywords: Angiotensin II, hypertension, NHE3, pressure natriuresis, proximal tubule
Introduction
The Na+/H+ exchanger 3 (NHE3) is a ~85 kDa protein encoded by the SLC9A3 gene, and is the most important Na+ transporter in the proximal tubules of the kidney.1–3 NHE3 extrudes a hydrogen ion (H+) into the lumen of the proximal tubules in exchange for the import of a sodium (Na+) ion into the proximal tubule cells, which provides a powerful driving force for directly and indirectly reabsorbing ~50% of filtered Na+ load in the proximal tubules. 1,2 Increased NHE3 expression and activity in the proximal tubules has long been implicated in angiotensin II (ANG II)-dependent hypertension and in spontaneously hypertensive rats (SHR). 4–6 Previous studies have shown that most of prohypertensive factors, including ANG II, 4,7–10 glucocorticoid, 11,12 hyperinsulinemia, 13 hyperglucagonemia, 14 and increased oxidative stress, 10 increase NHE3 expression and increase Na+ reabsorption in the proximal tubules. Increased NHE3 expression or activity has been reported in the proximal tubules of SHR, a rat model of human essential hypertension. 5,15 In vitro, low nanomoles of ANG II induces NHE3 expression in cultured proximal tubule cells. 16–18 In vivo, low subpressor or slow pressor doses of ANG II significantly increase the expression of NHE3 and proximal Na+ reabsorption, which contributes to the development of ANG II-dependent hypertension. 4,10,19 However, a direct cause and effect relationship between ANG II and NHE3 in the proximal tubules of the kidney, impaired pressure-natriuresis response and its resetting in ANG II-induced hypertension has not been previously determined.
We and others have recently shown that the basal blood pressure level is significantly lower in global Nhe3−/−, 1,20 kidney-selective tgNhe3−/− 21,22 or Nhe3loxloxCre mice, 23 as well as proximal tubule-specific PT-Nhe3−/− mice. 24 The reduced basal blood pressure phenotype is associated with a significant decrease in proximal Na+ reabsorption and an augmented pressure-natriuresis response in PT-Nhe3−/− mice. 24 Furthermore, ANG II-induced hypertension is significantly attenuated in global Nhe3−/− 20 and tgNhe3−/− mice with transgenic rescue of the Nhe3 gene in small intestines, 22 thus implicating intestinal and kidney NHE3 in the development of ANG II-dependent hypertension. Yet, whether NHE3 in the proximal tubules of the kidney may be targeted genetically or pharmacologically to attenuate ANG II-induced hypertension has not been investigated previously using genetically modified, proximal tubule-specific PT-Nhe3−/− mice or kidney- or proximal tubule-selective NHE3 inhibitors.
Accordingly, in the present study we first used proximal tubule-specific PT-Nhe3−/− mice 24 to directly test the hypothesis that NHE3 in the proximal tubules of the kidney is required for the full development of ANG II-induced hypertension. Secondly, we performed some proof of the concept experiments to determine whether the pharmacological inhibition of NHE3 in the kidney, primarily in the proximal tubulels and the loop of Henle, with an orally absorbable NHE3 inhibitor can attenuate ANG II-induced hypertension in C57BL/6J mice. Third, since androgen reportedly plays an important role in ANG II-induced hypertension in part by upregulating NHE3 in the proximal tubules and increasing proximal Na+ reabsorption, 25,26 both male and female WT and PT-Nhe3−/− mice were included in the experimental design. The results of the present study provide unequivocal evidence for the 1st time that NHE3 in the proximal tubules of the kidney is required for the full development of ANG II-induced hypertension, and that NHE3 in the proximal tubules may be therapeutically targeted to treat ANG II-dependent hypertension.
Methods
All animal breeding and genotyping procedures, surviving and nonsurviving surgical preparations, experimental protocols, or study materials will be made available to others by contacting Jia L. Zhuo, M.D., Ph.D., at jzhuo@umc.edu The detailed section of Methods and Materials is provided in the ONLINE SUPPLEMENT.
Animals
Adult male and female wild-type (WT), genetically modified, proximal tubule-specific knockout of NHE3 (PT-Nhe3−/−) mice were used in all experimental protocols in the present study, as approved by the Institutional Animal Care and User Committee of the University of Mississippi Medical Center.
Wild-type
As both Nhe3loxlox24,27 and iL1-SGLT2-Cre mouse strains were generated and back-crossed to several generations on the C57BL/6J genetic background, 24,28 both male and female age-matched C57BL/6J mice were used as wildtype controls.
Proximal tubule-specific Nhe3−/− mice (PT-Nhe3−/−) mice
PT-Nhe3−/− mice were generated in this laboratory using the gold standard SGLT2-Cre/ Nhe3loxlox approach, as described previously. 24,27,28
Experimental Protocols
Basal blood pressure and renal functional phenotypes in adult male and female PT-Nhe3−/− mice
Systolic (SBP), diastolic (DBP) and mean arterial blood pressure (MAP) were measured in conscious adult male and female WT and PT-Nhe3−/− mice using both the implanted telemetry (Data Science) and noninvasive tail-cuff approaches using Visitech Blood Pressure Analysis Systems BP-200 Series II (Visitech, NC). 4,20,22 The whole-kidney glomerular filtration rate (GFR) was measured in male and female WT and PT-Nhe3−/− mice using the transdermal GFR monitoring approach with FITC-sinistrin (Figure S1), as described. 29,30, whereas the whole-kidney proximal tubule Na+ reabsorption was measured using the noninvasive lithium clearance technique. 20,22,24,31
Induction of ANG II-induced hypertension in adult male and female WT and PT-Nhe3−/− mice
Six groups (n=5–12 per group) of adult male and female WT and PT-Nhe3−/− mice were studied. Group 1 served as time-control; Group 2 were infused with a high pressor (1.5 mg/kg/day, i.p.) 32 or a slow pressor dose of ANG II via an osmotic minipump (0.5 mg/kg/day, i.p.) supplemented with a 2% NaCl diet for 2 weeks; 4 and Group 3 were treated with ANG II as in Group 2, but concurrently treated with losartan to block AT1 receptors (20 mg/kg/day, p.o.). 4 SBP, DBP, and MAP were measured at basal and weekly after treatments with or without ANG II, 2% high salt diet, or losartan.
The pressure-natriuresis response to a slow pressor dose of ANG II-induced hypertension in male and female WT and PT-Nhe3−/− mice
The pressure-natriuresis response was studied in 4 groups (n=8 per group) of male and female WT and PT-Nhe3−/− mice to determine whether the pressure-natriuresis response was impaired in ANG II-induced hypertension and whether deletion of NHE3 selectively in the proximal tubules attenuates ANG II-induced hypertension by improving the pressure-natriuresis response. 24
Pharmacological inhibition of NHE3 in the kidney attenuates angiotensin II-induced hypertension in C57BL/6J mice
Five groups (n=5–15 per groups) of adult male C57BL/6J mice were studied. Group 1 served as time-controls. Group 2 were treated with an orally absorbable NHE3 inhibitor AVE0657 (a generous gift from Sanofi-Aventis; 20 mg/kg/day, p.o.) to inhibit NHE3 selectively in the kidney, primarily in the proximal tubules and the loop of Henle. Group 3 were infused with a slow pressor dose of ANG II (0.5 mg/kg/day, i.p., via minipump), supplemented with a 2% NaCl diet, to slowly and moderately induce ANG II-dependent hypertension in 2 weeks. 4 Group 4 were infused with ANG II as in Group 3, but treated with AVE0657 for 2 weeks as in Group 2. Group 5 were treated as in Group 4, but adding losartan (20 mg/kg/day, p.o.) to block AT1 receptors. SBP, DBP, and MAP and 24 h fecal and urinary sodium excretion were determined at basal and weekly after treatments.
Induction of L-NAME-induced hypertension in male and female WT and PT-Nhe3−/− mice
To determine whether genetic deletion of NHE3 selectively in the proximal tubules of the kidney prevents L-NAME-induced hypertension, 4 groups (n=10 per group) of adult male and female WT and PT-Nhe3−/− mice were treated without or with the nitric oxide synthase inhibitor L-NAME (50 mg/kg/day, p.o.) for 4 weeks. SBP, DBP, and MAP, and 24 h urinary Na+ and K+ excretion were determined before and weekly after L-NAME treatment.
Statistical analysis
All results are presented as mean ± SEM. The differences between different groups of male and female WT and PT-Nhe3−/− mice in all responses including SBP, DBP and MAP, 24 h urinary and fecal Na+ excretion, the hypertensive responses to ANG II or L-NAME, and the pressure-natriuretic response to ANG II were first analyzed using one-way ANOVA, followed by Student’s unpaired t test if a significant response between groups of WT and PT-Nhe3−/− mice was detected. The significance of statistical differences between responses were set P<0.05.
Results
Proximal tubule-specific deletion of NHE3 attenuates ANG II-induced hypertension in male and female PT-Nhe3−/− mice
As shown in Fig. 1, basal telemetry systolic (WT: 123 ± 5 vs. PT-Nhe3−/−: 107 ± 4 mmHg, P<0.01, n=12 per group), diastolic (WT: 96 ± 6 vs. PT-Nhe3−/−: 82 ± 4 mmHg, P<0.01, n=12 per group), and mean arterial blood pressure (WT: 113 ± 5 vs. PT-Nhe3−/−: 95 ± 5 mmHg, P<0.01, n=12 per group) were significantly lower in conscious male PT-Nhe3−/− mice than in male WT mice (A-C). There were no significant differences in systolic, diastolic and mean arterial pressure between male and female WT, or between male and female PT-Nhe3−/− mice (Fig. 1). In male and female WT mice, a high pressor dose of ANG II markedly increased systolic, diastolic and mean arterial pressure in a time-related manner, reaching a peak pressor response within a week (A-C, P<0.01). While the systolic pressure response to ANG II sustained throughout the experiment (A), the diastolic (B) and mean arterial pressure (C) responses began to decrease 10 days after ANG II infusion (Fig. 1). No significant difference in the pressor response to ANG II was observed between male and female WT mice (Male: 178 ± 8 vs. Female: 179 ± 10 mmHg, n.s.). In male PT-Nhe3−/− mice infused with a similar high pressor dose of ANG II, systolic blood pressure also increased in a time-related manner reaching a peak pressor response within a week of ANG II infusion (Fig. 1A). However, the pressor response to ANG II was markedly attenuated in male PT-Nhe3−/− mice (139 ± 8 mmHg, P<0.01 vs. WT males). Interestingly in female PT-Nhe3−/− mice (Fig. 1D–F), systolic, diastolic and mean arterial pressure responses to ANG II began to decrease 4 days after ANG II infusion (P<0.01). In all male and female WT and PT-Nhe3−/− mice, concurrent pretreatment with the AT1 receptor blocker losartan (20 mg/kg/day, p.o.) effectively blocked all pressor responses to ANG II (P<0.01, Fig. 2F).
Figure 1.

Systolic, diastolic and mean arterial blood pressure responses to a high pressor dose of ANG II infusion, 1.5 mg/kg/day, i.p., via osmotic minipump in conscious, adult male and female wild-type (WT) and PT-Nhe3−/− mice, as measured continuously for 14 days using the direct implanted telemetry technique. Please note the time-dependent increases in systolic, diastolic and mean arterial blood pressure responses in male and female WT mice, and significantly attenuated hypertensive responses to ANG II in male and female PT-Nhe3−/− mice. **P<0.01 vs. WT time-control group; ++P<0.01 vs. PT-Nhe3−/− time-control group, respectively. N=5–12 per group.
Figure 2.

The electron microscopic micrographs showing the ultrastructures of representative glomerulus and proximal tubules in male WT and PT-Nhe3−/− mice (A,B,D,E), and their whole-kidney glomerular filtration rate (C, GFR) and NHE3 proteins in apical membranes of the proximal tubules (F) in response to a slow pressor dose of ANG II infusion (0.5 mg/kg/day, i.p., 2 weeks). Please note that the ultrastructures of glomerular basement membranes (GBM), mesangium (M), capillaries (C), parietal epithelium (PE) of the Bowman’s capsule, and podocytes (PO), and proximal tubular brush border membranes (BBM), microvilli (MV), mitochondria (Mito), and vacuolar apparatus (VA) are comparable between WT and PT-Nhe3−/− mice. GFR was significantly lower in PT-Nhe3−/− mice at basal and in response to ANG II, whereas proximal tubule apical membrane NHE3 proteins were significantly higher in WT mice. **P<0.01 vs. WT or PT-Nhe3−/− control; and ++P<0.01 vs. WT control or in response to ANG II. N=6–10 per group.
Glomerular and proximal tubular structural responses to ANG II in male and female WT and PT-Nhe3−/− mice
Glomerular and proximal tubular ultrastructural responses to a slow pressor dose of ANG II infusion were compared using high resolution transmission electron microscopic imaging (Fig. 2 & Fig. S3). A moderate degree of glomerular mesangial expansion was observed in both male and female WT and PT-Nhe3−/− mice. No significant differences in the ultrastructure of glomerular basement membranes (GBM), glomerular mesangium (M), the endothelium and the glomerular capillaries (C), the parietal epithelium of the Bowman’s capsule (PE), or podocytes (PO), were identified between male or female (not shown) WT and PT-Nhe3−/− mice (Fig. 2A & Fig. 2B). Basal GFR, as determined by the transdermal GFR monitoring with FITC-sinistrin, was significantly lower in male and female PT-Nhe3−/− mice than WT controls (Fig. 2C). However, ANG II significantly decreased GFR by ~15% to similar extents in male and female WT and PT-Nhe3−/− mice (Fig. 2C). In ANG II-treated male and female WT and PT-Nhe3−/− mice, the size of the proximal tubules, the orientation and length of brush border membranes (BBM) and microvilli (MV), mitochondria, as well as vacuolar apparatus were largely similar (Fig. 2D & Fig. 2E & Fig. S3). However, the level of NHE3 proteins in freshly isolated apical membranes of the proximal tubules of the kidney was significantly increased in WT male mice in response to a slow pressor dose of ANG II infusion (Fig. 2F & Fig. S2).
Proximal tubule-specific deletion of NHE3 in the kidney improves the pressure-natriuresis response to ANG II in PT-Nhe3−/− mice
The pressure-natriuresis response was studied in anesthetized male WT and PT-Nhe3−/− mice infused with a slow pressor dose of ANG II (0.5 mg/kg/day, i.p.) and a 2% NaCl diet for 7 days (Fig. 3). Renal perfusion pressure (RPF) was raised equally by ~30 mmHg with occluding mesenteric and celiac arteries and/or the abdominal aorta below the renal arteries. Urinary Na+ excretion (UNaV) increased from basal 6.34 ± 0.30 μmol/h to 26.54 ± 5.85 μmol/h in male WT mice (P<0.01). Thus, the pressure-natriuresis response increased ~4.0-fold in WT mice (P<0.01; Fig. 3). In male PT-Nhe3−/− mice, UNaV increased from basal 8.10 ± 0.6 μmol/h to 52.50 ± 5.10 μmol/h (P<0.01; Fig. 3). The pressure-natriuresis response increased ~6.5-fold in PT-Nhe3−/− mice (P<0.01, Fig. 3). The difference in the pressure-natriuresis response between male WT and PT-Nhe3−/− mice was statistically significant (P<0.01). Furthermore, the accumulative 24 h urinary Na+ excretion was significantly higher in ANG II-infused PT-Nhe3−/− than ANG II-infused WT mice at basal, 3, 7, and 14 days after ANG II infusion (Fig. S4, P<0.01).
Figure 3.

The hypertensive (A) and pressure-natriuresis (B,C), heart rate (D), and heart (E) or kidney wt. (F) to body wt. ratio responses to a slow and low pressor dose of ANG II infusion, 0.5 mg/kg/day, i.p., and 2% NaCl diet for 2 weeks in male WT and PT-Nhe3−/− mice. Please note that the hypertensive response to ANG II was significantly attenuated in PT-Nhe3−/− mice, compared with WT mice (P<0.01). In response to an increase of ~30 mmHg in renal perfusion pressure (RPP), the pressure-natriureiss response increased ~4-fold in WT mice, whereas the response increased ~6.5-fold in PT-Nhe3−/− mice (**P<0.01). Both net urinary Na+ excretion (not shown) and as a fraction to a 10 mmHg increase in RPP were significantly higher in PT-Nhe3−/− mice (p<0.01; n=8) than in WT mice (p<0.01; n=8). There were no significant differences in heart rate, heart wt. or kidney wt. to body wt. ratio responses between WT and PT-Nhe3−/− mice. **P<0.01 vs. control, basal or WT mice; ++P<0.01 vs. ANG II; ##P<0.01 vs. WT corresponding treatment groups.
An orally absorbable NHE3 inhibitor AVE0657 attenuates ANG II-induced hypertension in WT mice by selectively inhibiting NHE3 in the kidney
An orally absorbable NHE3 inhibitor AVE0757 was used in the present study to determine the scientific premise that the pharmacological inhibition of NHE3 selectively in the kidney, primarily in the proximal tubules and the loop of Henle, attenuates ANG II-induced hypertension. As expected, a slow pressor dose of ANG II (0.5 mg/kg/day, i.p.) and 2% NaCl diet significantly increased systolic blood pressure from 116 ± 2 mmHg to 147 ± 4 mmHg (Fig. 4, P<0.01). Concurrent treatment with AVE0657 (20 mg/kg/day, p.o.) significantly attenuated systolic blood pressure to 125 ± 4 mmHg in ANG II-infused mice (Fig. 4, P<0.01). Concurrent treatment with AVE0657 and losartan (20 mg/kg/day, p.o.), completely normalize blood pressure in ANG II-infused mice to control (115 ± 5 mmHg, P<0.01). Unlike global Nhe3−/−20 or tgNhe3−/− mice, 22 AVE0657 had no effect on 24 h fecal Na+ excretion, which was similar to WT time-control or PT-Nhe3−/− mice (Fig. S5). However, AVE0657 significantly increased 24 h urinary Na+ excretion (P<0.01) without significantly altering urinary K+ excretion in control or ANG II-infused mice (Fig. 4). AVE0657 treatment alone had no significant effects on systolic blood pressure (−5 ± 2 mmHg, n.s.; Fig. S6), 24 h water drinking (Fig. S7) or 24 h urine output in WT mice (Fig. S8).
Figure 4.

Effects of the NHE3 inhibitor AVE0657 (20 mg/kg/day, p.o., Sanofi-Aventis) on ANG II-induced hypertension in male C57BL/6J mice infused with a slow pressor dose of ANG II at 0.5 mg/kg/day, i.p., for 2 weeks. AVE0657 is an orally absorbable NHE3 inhibitor which is filtered by the glomerulus into the proximal tubules to inhibit NHE3 in the apical membranes (A). AVE0657 significantly attenuated ANG II-induced hypertension (B), and the response was associated with a significant natriuretic response after 7 days’ administration (D), without affecting 24 h urinary K+ (E) or fecal Na+ excretion (F). **P<0.01 vs. time-control group; ++P<0.01 vs. ANG II group.
Proximal tubule-specific deletion of NHE3 in the kidney doesn’t alter L-NAME-induced hypertension in PT-Nhe3−/− mice
In the present study, separate groups of male and female WT and PT-Nhe3−/− mice were treated without or with L-NAME (50 mg/kg/day, p.o.) for 4 weeks to slowly increase blood pressure. Figure 5 shows that L-NAME-treatment induced time-related, moderate but significant increases in systolic blood pressure, which peaked at 2 weeks and sustained for 4 weeks in both WT (Basal: 115 ± 3 vs. L-NAME: 126 ± mmHg, P<0.05), and PT-Nhe3−/− mice (Basal: 103 ± 3 mmHg vs. L-NAME: 119 ± 3 mmHg, P<0.05) (Fig. 5A). Similar L-NAME-induced blood pressure responses were observed in female WT and PT-Nhe3−/− mice (n.s.) (Fig. 5A). L-NAME-induced hypertension was associated with antinatriuresis in WT mice (P<0.05 vs. basal) (Fig. 5D), but it was conversely associated with natriuresis in female PT-Nhe3−/− mice (P<0.01) (Fig. 5D). There were no significant differences in heart rate (Fig. 5B), heart wt. (Fig. 5C) to or kidney wt. to body wt. ratio (Fig. 5F), and urinary K+ excretion (Fig. 5E) between male and female WT and PT-Nhe3−/− mice in response to L-NAME treatment.
Figure 5.

Proximal tubule-specific deletion of NHE3 in the kidney doesn’t alter the hypertensive response to L-NAME treatment in male and female PT-Nhe3−/− mice (A), compared with male and female WT mice. However, L-NAME had an antinatriuretic effect in both WT and PT-Nhe3−/− mice (D). **P<0.01 vs. basal of the same strain of mice; ++P<0.01 vs. male WT or PT-Nhe3−/− mice. N=9–10 per group.
Discussion
An important role of NHE3 in the proximal tubules of the kidney in ANG II-dependent hypertension is deduced primarily from previous studies showing the stimulatory effects of ANG II on NHE3 expression and activity in cultured proximal tubule cells. 12,16–18,33,34 In rats or mice, subpressor or slow pressor doses of ANG II were found to significantly increase the expression of NHE3 in the kidney, stimulate proximal Na+ reabsorption, and elevate blood pressure. 4,10,19,35 Since NHE3 expression or activity was found in the proximal tubules of SHR, a well-recognized animal model of human essential hypertension, 5,15,36 NHE3 in the proximal tubules has been implicated in the pathogenesis of hypertension. However, the evidence supporting an important role of proximal tubule NHE3 in ANG II-induced hypertension or SHR from previous studies remain indirect, because a cause and effect relationship has not been established previously. Against this background, the present study directly tested the hypothesis that NHE3 in the proximal tubules of the kidney plays a significant role in the pathogenesis of ANG II-dependent hypertension, and that deletion of NHE3 selectively in the proximal tubules attenuates ANG II-induced hypertension. Using mutant mice with proximal tubule-specific deletion of NHE3 in the kidney, the present study provides unequivocal evidence that NHE3 in the proximal tubules of the kidney plays a key role in the development of ANG II-induced hypertension. Furthermore, the results from the present study using an orally absorbable, kidney-selective NHE3 inhibitor AVE0657, strongly support a proof of the concept that deletion or pharmacological inhibition of NHE3 selectively in the proximal tubules of the kidney will significantly lower blood pressure in animal models of hypertension associated with the activation of the intratubular renin-angiotensin system in the kidney.
The key strength of the present study is that the hypothesis was tested using a proximal tubule-specific genetic approach for the 1st time to directly determine the cause and effect relationship between NHE3 in the proximal tubules and ANG II-induced hypertension. This proximal tubule-specific approach is clearly advantageous compared with the global or whole-body, and the kidney-specific approaches, because the former approach can theoretically exclude the confounding effects or contributions of NHE3 expression and actions from other organs or other kidney tubular segments in ANG II-induced hypertension. In recent studies, we have determined the roles of intestinal and kidney NHE3 in the development of ANG II-induced hypertension using global Nhe3−/− and kidney-selective tgNhe3−/− mouse models. 20,22,37 In those studies, Nhe3−/− and tgNhe3−/− mice were infused with a high pressor does of ANG II (1.5 mg/kg/day, i.p., via osmotic minipump) for 2 weeks or acutely infused intravenously under anesthesia. The hypertensive response to ANG II was significantly attenuated in Nhe3−/− and tgNhe3−/− mice, suggesting that systemic and tissue (intestinal and kidney) effects of NHE3 were involved. 20,22 However, a significant limitation for using these two mutant NHE3 mouse models is that the relative contribution of intestinal versus kidney or proximal tubule NHE3 to the development of ANG II-induced hypertension cannot be determined. Indeed, NHE3 is abundantly expressed in apical membranes of small intestines in the digestive system, where ANG II stimulates Na+ absorption, contributing to increases in body Na+ intake and blood pressure homeostasis. 1,20–22,38 Even in the kidney, a low level of NHE3 is also expressed in the ascending limb (TAL) of the loop of Henle, 39,40 where ANG II may induce NHE3 expression and alter Na+ transport. Thus the present study is both conceptually and technically innovative to use the proximal tubule-specific NHE3 knockout model to determine the role of NHE3 in ANG II-induced hypertension. Using the state of art SGLT2-Cre/Nhe3fl/fl approach, NHE3 is genetically deleted only in the proximal tubules, while leaving those NHE3 intact in the TAL of the loop of Henle and in small intestines. In the present study, hypertension induced by a high pressor (1.5 mg/kg/day, i.p.) and a slow pressor dose of ANG II (0.5 mg/kg/day, i.p.) was attenuated in male and female PT-Nhe3−/− mice, confirming a key role of NHE3 in the proximal tubules.
The present study has reported the potential mechanisms by which deletion of NHE3 selectively in the proximal tubules of the kidney attenuates hypertension induced by a slow pressor dose of ANG II. Our current results support the scientific premise that in ANG II-induced hypertension, ANG II activates AT1 receptors to increase NHE3 expression and activity in the proximal tubules, which stimulates proximal tubule Na+ reabsorption and impairs the pressure-natriuresis response. Thus, deletion of NHE3 from the proximal tubules removes a major target of ANG II in the renal proximal tubules, inhibits proximal tubule Na+ reabsorption, and significantly improves the pressure-natriuresis response in PT-Nhe3−/− mice. This interpretation is consistent with our recent study in which proximal tubule-specific NHE3 deletion significantly lowers basal blood pressure by inhibiting proximal tubule Na+ reabsorption and promoting the pressure-natriuresis response. 24 Our conclusion is also supported by previous studies showing that impaired pressure-natriuresis response occurs in all animal models of ANG II-dependent hypertension, including 2K1C, 41 TGR(mRen-2)27, 42 and ANG II-infused hypertension. 43,44 However, a careful examination and comparison of electron micrographs of glomerular and proximal tubular ultrastructures between ANG II-infused WT and PT-Nhe3−/− mice found no significant differences in glomerular capillaries, mesangium, and podocytes, or the microvilli of the brush border membranes and mitochondria of the proximal tubules. We further found that deletion of NHE3 selectively in the proximal tubules doesn’t significantly alter L-NAME-induced hypertension in PT-Nhe3−/− mice. Taken together, the results of the present study provide direct evidence supporting a key role of proximal tubule NHE3 in the development of ANG II-induced, but not L-NAME-induced hypertension.
The results of the present study using an orally absorbable NHE3 inhibitor AVE0657 in ANG II-induced hypertension strongly suggest that NHE3 in the proximal tubules of the kidney may be a potential pharmacological target for treating hypertension associated with the activation of intratubular ANG II or upregulation of NHE3 expression in the proximal tubules. The present study is highly significant and translationally relevant, because no diuretic or antihypertensive drug has been developed to target the proximal tubules, with the exception of carbonic anhydrase inhibitors that are mild diuretic. Currently, three classes of diuretics are used as the 1st line of anti-hypertensive drugs; a) thiazides, which inhibit the thiazide-sensitive Na+/Cl− cotransporter; b) loop diuretics, which inhibit the NKCC2 cotransporter in the TAL; and c) K+-sparing diuretics, which block aldosterone-sensitive ENaC. 45,46 However, long-term use of distal nephron-targeting diuretics is also associated with increased ANG II formation and Na+ reabsorption in the proximal tubules. No study has been reported to determine whether NHE3 in the proximal tubules of the kidney should be targeted to treat hypertension. Nevertheless, a recent proof of the concept study has shown that a non-absorbable NHE3 inhibitor, SAR218034 (Sanofi-Aventis), significantly decreased blood pressure in old SHR lean rats by inhibiting intestinal Na+ absorption or dietary Na+ intake without severe salt wasting. 47 Sanofi-Aventis has recently developed a different NHE3 inhibitor, i.e., AVE0657, which is orally absorbable so it doesn’t inhibit NHE3 in small intestines, but it enters the circulation and is filtered into the proximal tubules to inhibit NHE3 in the kidney (Sanofi-Aventis, personal communications). In the present study, we demonstrated that at 20 mg/kg/day, AVE0657 significantly attenuated ANG II-induced hypertension in C57BL/6J mice to a similar level seen in PT-Nhe3−/− mice. The antihypertensive effect of AVE0657 in ANG II-infused mice was associated with significant increased 24 h urinary Na+ excretion without altering 24 h fecal Na+ excretion and urinary K+ excretion. We recently reported that 24 h fecal Na+ excretion increased 10-fold in global Nhe3−/− mice compared with wild-type mice, due to a marked Na+ absorptive defect as a result of NHE3 deletion from small intestines. 20 In the present study, we found no differences in 24 h fecal Na+ excretion in C57BL/6J mice treated with or without AVE0657, or between C57BL/6J and PT-Nhe3−/− mice. These results support the scientific premise that unlike the non-absorbable NHE3 inhibitor SAR218034, AVE0657 doesn’t inhibit Na+ absorption by small intestines, so it doesn’t cause salt wasting from the gut as does SAR218034 47 or global Nhe3−/− mice. 1,20,22,38 However, we should recognize that in addition to targeting NHE3 in the proximal tubules, AVE0657 may also inhibit NHE3 in the TAL to a much smaller extent due to a low level of NHE3 expression in this tubular segment. Nevertheless, the development of orally absorbable NHE3 inhibitors that selectively target NHE3 in the proximal tubules and the TAL may have a significant impact on human hypertension associated with activation of intratubular ANG II, increases in NHE3 expression and proximal tubule Na+ reabsorption, and impairment of pressure-natriuresis.
Finally, as mandated by the NIH’s Policies on Consideration of Sex as Biological Variables in the experimental design, 48,49 the present study has studied and compared the sex differences in the hypertensive responses to ANG II or L-NAME treatment in female WT and PT-Nhe3−/− mice. We did not find significant sex differences in the hypertensive response to ANG II or to L-NAME in male and female WT mice. However, we found a sex difference in the hypertensive response to ANG II between female WT and PT-Nhe3−/− mice. In female WT mice, ANG II induced a largely sustained hypertension throughout ANG II infusion, but the hypertensive effect to ANG II was markedly attenuated in female PT-Nhe3−/− mice 3–4 days after ANG II infusion. The reasons underlying this sex difference in ANG II-induced hypertension in male and female PT-Nhe3−/− mice are unknown, but they are likely due to the effects of female hormones on the systemic vascular response to ANG II, 50,51 or to the compensatory responses of other Na+ transporters or cotransporters along the renal tubules in the absence of NHE3 in the proximal tubules. 52–55
Perspectives
In summary, the present study was the first to directly determine the cause and effect relationship between ANG II and NHE3 in the proximal tubules of the kidney in the development of ANG II-induced hypertension using a genetically modified mouse model with proximal tubule-specific deletion of NHE3 and an orally absorbable NHE3 inhibitor AVE0657 (Fig. 6). We demonstrated that deletion of NHE3 selectively in the proximal tubules of the kidney significantly attenuated hypertension induced by a high pressor and a slow pressor dose of ANG II infusion. Since no differences were found in the glomerular and proximal tubular ultrastructures in response to ANG II between WT and PT-Nhe3−/− mice, our results suggest that the antihypertensive effect of proximal tubule-specific NHE3 deletion was directly due to the removal of NHE3 proteins and its Na+-retaining effect from the proximal tubules, which leads to a significant improvement of the pressure-natriuresis response in ANG II-infused PT-Nhe3−/− mice. Furthermore, our study demonstrates for the 1st time that an orally absorbable NHE3 inhibitor AVE0657 significantly attenuated ANG II-induced hypertension in WT mice. These findings are highly significant and translationally relevant, because a) ~90% of NHE3 is expressed in the proximal tubules alone, 40,56 b) NHE3 is the most important Na+ transporter in the proximal tubules, 2,3,57 and c) NHE3 is a major target for ANG II. 37 Thus the results of the present study support the scientific premise that NHE3 in the proximal tubules of the kidney may be therapeutically targeted in treating hypertension associated with activation of intratubular ANG II and increased NHE3 expression or activity in the kidney.
Figure 6.

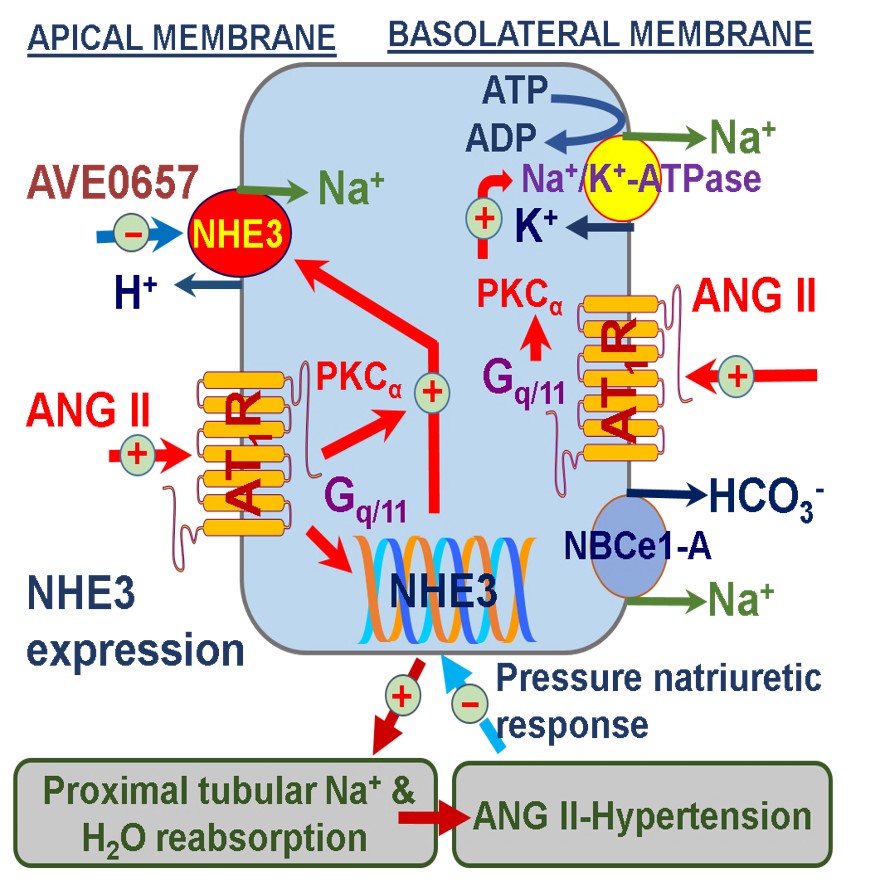
The important role of NHE3 in the proximal tubules of the kidney in the development of ANG II-dependent hypertension. We hypothesize that the circulating and intratubular ANG II binds to high affinity and high density of AT1 receptors on both apical and basolateral membranes of the proximal tubules and activates downstream signaling pathways such as protein kinase Cα (PKCα) to increase the expression and activity of NHE3 and Na+/K+-ATPase. The sustained upregulation of NHE3 and Na+/K+-ATPase by ANG II stimulates proximal tubular Na+ reabsorption, increases body Na+ and fluid retention, impairs the pressure-natriuresis response, and ultimately contributes to the development of ANG II-dependent hypertension.37 At the early stage of hypertension, the rise in arterial pressure induces the pressure-natriuresis response to prevent further increase in blood pressure. However, the pressure-natriuresis response resets to higher pressures during established hypertension. Modified from Reference 37 with permission.
Supplementary Material
{kind=link}
Novelty and Significance.
What Is New?
The Na+/H+ exchanger 3 (NHE3) in the proximal tubules of the kidney is directly and indirectly responsible for mediating the reabsorption of over 50% of filtered Na+ load, whereas ANG II has a powerful Na+-retaining effect in the proximal tubules. The direct cause and effect relationship between ANG II and NHE3 in ANG II-dependent hypertension has never been investigated previously.
This is the 1st study to use the mutant mice with deletion of NHE3 selectively in the proximal tubules of the kidney and an orally absorbable NHE3 inhibitor AVE0657 to test and confirm the hypothesis that the deletion or pharmacological inhibition of NHE3 selectively in the proximal tubules attenuates ANG II-induced hypertension by improving the pressure-natriuresis response.
What Is Relevant?
The impairment of and resetting of the pressure-natriuresis response in the kidney is a key mechanism in most, if not all, forms of human hypertension, especially in ANG II-dependent hypertension.
Only about 50% of hypertensive patients have their blood pressure under adequate control despite treated with different classes of antihypertensive drugs. The present study provides unequivocal evidence that NHE3 in the proximal tubules of the kidney may be therapeutically targeted to treat hypertension induced by ANG II or due to increased expression and activity of NHE3 in the proximal tubules of the kidney.
Summary.
This study used a novel tissue-specific, genetically modified mouse model to demonstrate for the 1st time that deletion, or pharmacological inhibition, of NHE3 selectively in the proximal tubules of the kidney effectively attenuates ANG II-induced hypertension by improving the pressure-natriuresis response. The results support the hypothesis that NHE3 in the proximal tubules may be therapeutically targeted to treat hypertension induced by activation of intratubular ANG II and increased expression and activity of NHE3 in the proximal tubules of the kidney.
Acknowledgements
We greatly thank Dr. Manoocher Soleimani of the University of Cincinnati College of Medicine for generously providing us with breeding pairs of Nhe3flox/flox mice, Drs. Isabelle Rubera and Michel Tauc of LP2M UMR-CNRS 7370, Université Côte d’Azur, Nice Cedex 2, France, as well as Dr. Rong Li of Johns Hopkin University for generously providing us with breeding pairs of iL1-sglt2-Cre mice for our mutant mouse breeding programs. We also particularly thank Sanofi-Aventis, Frankfurt, Germany for generously providing the invaluable, orally absorbable NHE3 inhibitor AVE0657, their confidential advices on the pharmacodynamics, pharmacokinetics, and doses of AVE0657 for mouse studies, and for approving the publication of results associated with AVE0657 in the present study.
Sources of Funding
This work was supported in part by grants from National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK; 2RO1DK067299-06A2, 2R01DK067299-10A1), and NIDDK/National Institute of General Medical Sciences (1R01DK102429-01, 2R01DK102429-03A1), and National Heart, Lung, And Blood Institute (1R56HL130988-01) to Dr. Jia L. Zhuo.
Footnotes
Disclosures
None.
References
- 1.Schultheis PJ, Clarke LL, Meneton P, et al. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat Genet 1998; 19(3):282–285. [DOI] [PubMed] [Google Scholar]
- 2.Wang T, Yang CL, Abbiati T, Schultheis PJ, Shull GE, Giebisch G, Aronson PS. Mechanism of proximal tubule bicarbonate absorption in NHE3 null mice. Am J Physiol 1999; 277(2 Pt 2):F298–F302. [DOI] [PubMed] [Google Scholar]
- 3.Zhuo JL, Li XC. Proximal nephron. Compr Physiol. 2013; [3(3)]:1079–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li XC, Zhuo JL. Phosphoproteomic analysis of AT1 receptor-mediated signaling responses in proximal tubules of angiotensin II-induced hypertensive rats. Kidney Int. 2011; 80:620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.LaPointe MS, Sodhi C, Sahai A, Batlle D. Na+/H+ exchange activity and NHE-3 expression in renal tubules from the spontaneously hypertensive rat. Kidney Int. 2002; 62(1):157–165. [DOI] [PubMed] [Google Scholar]
- 6.Kelly MP, Quinn PA, Davies JE, Ng LL. Activity and expression of Na+-H+ exchanger isoforms 1 and 3 in kidney proximal tubules of hypertensive rats. Circ Res. 1997; 80(6):853–860. [DOI] [PubMed] [Google Scholar]
- 7.He P, Klein J, Yun CC. Activation of Na+/H+ exchanger NHE3 by angiotensin II is mediated by inositol 1,4,5-triphosphate (IP3) receptor-binding protein released with IP3 (IRBIT) and Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 2010; 285(36):27869–27878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riquier-Brison AD, Leong PK, Pihakaski-Maunsbach K, McDonough AA. Angiotensin II stimulates trafficking of NHE3, NaPi2, and associated proteins into the proximal tubule microvilli. Am J Physiol Renal Physiol. 2010; 298(1):F177–F186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du Z, Wan L, Yan Q, Weinbaum S, Weinstein AM, Wang T. Regulation of glomerulotubular balance II: impact of angiotensin II on flow-dependent transport. Am J Physiol Renal Physiol. 2012; 303(11):F1507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banday AA, Lokhandwala MF. Angiotensin II-mediated biphasic regulation of proximal tubular Na+/H+ exchanger 3 is impaired during oxidative stress. Am J Physiol Renal Physiol. 2011; 301(2):F364–F370. [DOI] [PubMed] [Google Scholar]
- 11.Pao AC, Bhargava A, Di SF, Quigley R, Shao X, Wang J, Thomas S, Zhang J, Shi M, Funder JW, Moe OW, Pearce D. Expression and role of serum and glucocorticoid-regulated kinase 2 in the regulation of Na+/H+ exchanger 3 in the mammalian kidney. Am J Physiol Renal Physiol. 2010; 299(6):F1496–F1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang D, Zhang H, Lang F, Yun CC. Acute activation of NHE3 by dexamethasone correlates with activation of SGK1 and requires a functional glucocorticoid receptor. Am J Physiol Cell Physiol. 2007; 292(1):C396–C404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuster DG, Bobulescu IA, Zhang J, Wade J, Moe OW. Characterization of the regulation of renal Na+/H+ exchanger NHE3 by insulin. Am J Physiol Renal Physiol. 2007; 292(2):F577–F585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amemiya M, Kusano E, Muto S, Tabei K, Ando Y, Alpern RJ, Asano Y. Glucagon acutely inhibits but chronically activates Na+/H+ antiporter 3 activity in OKP cells. Exp Nephrol. 2002; 10(1):26–33. [DOI] [PubMed] [Google Scholar]
- 15.Hayashi M, Yoshida T, Monkawa T, Yamaji Y, Sato S, Saruta T. Na+/H+-exchanger 3 activity and its gene in the spontaneously hypertensive rat kidney. J Hypertens. 1997; 15(1):43–48. [PubMed] [Google Scholar]
- 16.Li XC, Hopfer U, Zhuo JL. AT1 receptor-mediated uptake of angiotensin II and NHE-3 expression in proximal tubule cells through the microtubule-dependent endocytic pathway. Am J Physiol Renal Physiol. 2009; 297(5):F1342–F1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu L, Dixit MP, Nullmeyer KD, Xu H, Kiela PR, Lynch RM, Ghishan FK. Regulation of Na+/H+ exchanger-NHE3 by angiotensin-II in OKP cells. Biochim Biophys Acta. 2006; 1758(4):519–526. [DOI] [PubMed] [Google Scholar]
- 18.Li XC, Hopfer U, Zhuo JL. Novel signaling mechanisms of intracellular angiotensin II-induced NHE3 expression and activation in mouse proximal tubule cells. Am J Physiol Renal Physiol. 2012; 303(12):F1617–F1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reilly AM, Harris PJ, Williams DA. Biphasic effect of angiotensin II on intracellular sodium concentration in rat proximal tubules. Am J Physiol. 1995; 269(3 Pt 2):F374–F380. [DOI] [PubMed] [Google Scholar]
- 20.Li XC, Shull GE, Miguel-Qin E, Zhuo JL. Role of the Na+/H+ exchanger 3 in angiotensin II-induced hypertension. Physiol Genomics. 47(10), 479–487. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noonan WT, Woo AL, Nieman ML, Prasad V, Schultheis PJ, Shull GE, Lorenz JN. Blood pressure maintenance in NHE3-deficient mice with transgenic expression of NHE3 in small intestine. Am J Physiol Regul Integr Comp Physiol. 2005; 288(3):R685–R691. [DOI] [PubMed] [Google Scholar]
- 22.Li XC, Shull GE, Miguel-Qin E, Chen F, Zhuo JL. Role of the Na+/H+ exchanger 3 in angiotensin II-induced hypertension in NHE3-deficient mice with transgenic rescue of NHE3 in small intestines. Physiol Rep. 2015; 3(11):e12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fenton RA, Poulsen SB, de la Mora CS, Soleimani M, Dominguez Rieg JA, Rieg T. Renal tubular NHE3 is required in the maintenance of water and sodium chloride homeostasis. Kidney Int. 2017; 92:397–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li XC, Soleimani M, Zhu D, Rubera I, Tauc M, Zheng X, Zhang J, Chen X, Zhuo JL. Proximal tubule-specific deletion of the NHE3 (Na+/H+ exchanger 3) promotes the pressure-natriuresis response and lowers blood pressure in mice. Hypertension. 2018; 72(6):1328–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quan A, Chakravarty S, Chen JK, Chen JC, Loleh S, Saini N, Harris RC, Capdevila J, Quigley R. Androgens augment proximal tubule transport. Am J Physiol Renal Physiol. 2004; 287(3):F452–F459. [DOI] [PubMed] [Google Scholar]
- 26.Reckelhoff JF, Zhang H, Granger JP. Testosterone exacerbates hypertension and reduces pressure-natriuresis in male spontaneously hypertensive rats. Hypertension. 1998; 31(1 Pt 2):435–439. [DOI] [PubMed] [Google Scholar]
- 27.Li HC, Du Z, Barone S, Rubera I, McDonough AA, Tauc M, Zahedi K, Wang T, Soleimani M. Proximal tubule specific knockout of the Na⁺/H⁺ exchanger NHE3: effects on bicarbonate absorption and ammonium excretion. J Mol Med (Berl) 2013; 91:951–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rubera I, Poujeol C, Bertin G, Hasseine L, Counillon L, Poujeol P, Tauc M. Specific Cre/Lox recombination in the mouse proximal tubule. J Am Soc Nephrol. 2004; 15(8):2050–2056. [DOI] [PubMed] [Google Scholar]
- 29.Schreiber A, Shulhevich Y, Geraci S, Hesser J, Stsepankou D, Neudecker S, Koenig S, Heinrich R, Hoecklin F, Pill J, Friedemann J, Schweda F, Gretz N, Schock-Kusch D. Transcutaneous measurement of renal function in conscious mice. Am J Physiol Renal Physiol. 2012; 303(5):F783–F788. [DOI] [PubMed] [Google Scholar]
- 30.Schock-Kusch D, Xie Q, Shulhevich Y, Hesser J, Stsepankou D, Sadick M, Koenig S, Hoecklin F, Pill J, Gretz N. Transcutaneous assessment of renal function in conscious rats with a device for measuring FITC-sinistrin disappearance curves. Kidney Int. 2011; 79(11):1254–1258. [DOI] [PubMed] [Google Scholar]
- 31.Li XC, Cook JL, Rubera I, Tauc M, Zhang F, Zhuo JL. Intrarenal transfer of an intracellular cyan fluorescent fusion of angiotensin II selectively in proximal tubules increases blood pressure in rats and mice. Am J Physiol Renal Physiol. 2011; 300:F1076–F1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li XC, Navar LG, Shao Y, Zhuo JL. Genetic deletion of AT1a receptors attenuates intracellular accumulation of angiotensin II in the kidney of AT1a receptor-deficient mice. Am J Physiol Renal Physiol. 2007; 293:F586–F593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li XC, Zhuo JL. Selective knockdown of AT1 receptors by RNA interference inhibits Val5-Ang II endocytosis and NHE-3 expression in immortalized rabbit proximal tubule cells. Am J Physiol Cell Physiol. 2007; 293:C367–C378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li XC, Zhuo JL. Intracellular ANG II directly induces in vitro transcription of TGF-β1, MCP-1, and NHE-3 mRNAs in isolated rat renal cortical nuclei via activation of nuclear AT1a receptors. Am J Physiol Cell Physiol. 2008; 294(4):C1034–C1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li XC, Zhuo JL. Proximal tubule-dominant transfer of AT1a receptors induces blood pressure responses to intracellular angiotensin II in AT1a receptor-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2013; 304:R588–R598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu H, Sagnella GA, Dong Y, Miller MA, Onipinla A, Markandu ND, MacGregor GA. Molecular variants of the sodium/hydrogen exchanger type 3 gene and essential hypertension. J Hypertens. 2004; 22(7):1269–1275. [DOI] [PubMed] [Google Scholar]
- 37.Li XC, Zheng X, Chen X, Zhao C, Zhu D, Zhang J, Zhuo JL. The genetic and genomic evidence for an important role of the Na+/H+ exchanger 3 in the blood pressure regulation and angiotensin II-induced hypertension. Physiol Genomics 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woo AL, Noonan WT, Schultheis PJ, Neumann JC, Manning PA, Lorenz JN, Shull GE. Renal function in NHE3-deficient mice with transgenic rescue of small intestinal absorptive defect. Am J Physiol Renal Physiol. 2003; 284(6):F1190–F1198. [DOI] [PubMed] [Google Scholar]
- 39.Biemesderfer D, Rutherford PA, Nagy T, Pizzonia JH, bu-Alfa AK, Aronson PS. Monoclonal antibodies for high-resolution localization of NHE3 in adult and neonatal rat kidney. Am J Physiol. 1997; 273(2 Pt 2):F289–F299. [DOI] [PubMed] [Google Scholar]
- 40.Biemesderfer D, Pizzonia J, Abu-Alfa A, Exner M, Reilly R, Igarashi P, Aronson PS. NHE3: a Na+/H+ exchanger isoform of renal brush border. Am J Physiol. 1993; 265(5 Pt 2):F736–F742. [DOI] [PubMed] [Google Scholar]
- 41.Rostand SG, Lewis D, Watkins JB, Huang WC, Navar LG. Attenuated pressure natriuresis in hypertensive rats. Kidney Int. 1982; 21(2):331–338. [DOI] [PubMed] [Google Scholar]
- 42.Gross V, Roman RJ, Cowley AW Jr. Abnormal pressure-natriuresis in transgenic renin gene rats. J Hypertens. 1994; 12(9):1029–1034. [PubMed] [Google Scholar]
- 43.Mattson DL, Raff H, Roman RJ. Influence of angiotensin II on pressure natriuresis and renal hemodynamics in volume-expanded rats. Am J Physiol. 1991; 260(6 Pt 2):R1200–R1209. [DOI] [PubMed] [Google Scholar]
- 44.Wang CT, Chin SY, Navar LG. Impairment of pressure-natriuresis and renal autoregulation in ANG II-infused hypertensive rats. Am J Physiol Renal Physiol. 2000; 279(2):F319–F325. [DOI] [PubMed] [Google Scholar]
- 45.Mancia G, De BG, Dominiczak A, et al. 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens. 2007; 25(6):1105–1187. [DOI] [PubMed] [Google Scholar]
- 46.Ernst ME, Moser M. Use of diuretics in patients with hypertension. N Engl J Med. 2009; 361(22):2153–2164. [DOI] [PubMed] [Google Scholar]
- 47.Linz D, Wirth K, Linz W, Heuer HO, Frick W, Hofmeister A, Heinelt U, Arndt P, Schwahn U, Bohm M, Ruetten H. Antihypertensive and laxative effects by pharmacological inhibition of sodium-proton-exchanger subtype 3-mediated sodium absorption in the gut. Hypertension. 2012; 60(6):1560–1567. [DOI] [PubMed] [Google Scholar]
- 48.Sandberg K, Umans JG. Recommendations concerning the new U.S. National Institutes of Health initiative to balance the sex of cells and animals in preclinical research. FASEB J. 2015; 29(5):1646–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maric-Bilkan C, Arnold AP, Taylor DA, Dwinell M, Howlett SE, Wenger N, Reckelhoff JF, Sandberg K, Churchill G, Levin E, Lundberg MS. Report of the National Heart, Lung, and Blood Institute Working Group on Sex Differences Research in Cardiovascular Disease: Scientific Questions and Challenges. Hypertension. 2016; 67(5):802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ji H, Zheng W, Wu X, Liu J, Ecelbarger CM, Watkins R, Arnold AP, Sandberg K. Sex chromosome effects unmasked in angiotensin II-induced hypertension. Hypertension. 2010; 55(5):1275–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ji H, Zheng W, Li X, Liu J, Wu X, Zhang MA, Umans JG, Hay M, Speth RC, Dunn SE, Sandberg K. Sex-specific T-cell regulation of angiotensin II-dependent hypertension. Hypertension. 2014; 64(3):573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nguyen MT, Lee DH, Delpire E, McDonough AA. Differential regulation of Na+ transporters along nephron during ANG II-dependent hypertension: distal stimulation counteracted by proximal inhibition. Am J Physiol Renal Physiol. 2013; 305(4):F510–F519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nguyen MT, Han J, Ralph DL, Veiras LC, McDonough AA. Short-term nonpressor angiotensin II infusion stimulates sodium transporters in proximal tubule and distal nephron. Physiol Rep. 2015; 3(9):e12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mirabito KM, Hilliard LM, Kett MM, Brown RD, Booth SC, Widdop RE, Moritz KM, Evans RG, Denton KM. Sex- and age-related differences in the chronic pressure-natriuresis relationship: role of the angiotensin type 2 receptor. Am J Physiol Renal Physiol. 2014; 307(8):F901–F907. [DOI] [PubMed] [Google Scholar]
- 55.Hilliard LM, Nematbakhsh M, Kett MM, Teichman E, Sampson AK, Widdop RE, Evans RG, Denton KM. Gender differences in pressure-natriuresis and renal autoregulation: role of the Angiotensin type 2 receptor. Hypertension. 2011; 57(2):275–282. [DOI] [PubMed] [Google Scholar]
- 56.Tse CM, Brant SR, Walker MS, Pouyssegur J, Donowitz M. Cloning and sequencing of a rabbit cDNA encoding an intestinal and kidney-specific Na+/H+ exchanger isoform (NHE-3). J Biol Chem. 1992; 267(13):9340–9346. [PubMed] [Google Scholar]
- 57.Lorenz JN, Schultheis PJ, Traynor T, Shull GE, Schnermann J. Micropuncture analysis of single-nephron function in NHE3-deficient mice. Am J Physiol. 1999; 277(3 Pt 2):F447–F453. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.