

Abstract
Geleophysic dysplasia is a rare, frequently lethal condition characterized by severe short stature with progressive joint contractures, cardiac, pulmonary, and skin anomalies. Geleophysic dysplasia results from dominant fibrillin-1 (FBN1) or recessive ADAMTSL2 mutations, suggesting a functional link between ADAMTSL2 and fibrillin microfibrils. Mice lacking ADAMTSL2 die at birth, which has precluded analysis of postnatal limb development and mechanisms underlying the skeletal anomalies of geleophysic dysplasia. Here, detailed expression analysis of Adamtsl2 using an intragenic lacZ reporter shows strong Adamtsl2 expression in limb tendons. Expression in developing and growing bones is present in regions that are destined to become articular cartilage but is absent in growth plate cartilage. Consistent with strong tendon expression, Adamtsl2 conditional deletion in limb mesenchyme using Prx1-Cre led to tendon anomalies, albeit with normal collagen fibrils, and distal limb shortening, providing a mouse model for geleophysic dysplasia. Unexpectedly, conditional Adamtsl2 deletion using Scx-Cre, a tendon-specific Cre-deleter strain, which does not delete in cartilage, also impaired skeletal growth. Recombinant ADAMTSL2 is shown here to colocalize with fibrillin microfibrils in vitro, and enhanced staining of fibrillin-1 microfibrils was observed in Prx1-Cre Adamtsl2 tendons. The findings show that ADAMTSL2 specifically regulates microfibril assembly in tendons and that proper microfibril composition in tendons is necessary for tendon growth. We speculate that reduced bone growth in geleophysic dysplasia may result from external tethering by short tendons rather than intrinsic growth plate anomalies. Taken together with previous work, we suggest that GD results from abnormal microfibril assembly in tissues, and that ADAMTSL2 may limit the assembly of fibrillin microfibrils.
Keywords: Geleophysic dysplasia, fibrillin microfibrils, ADAMTS-like protein, acromelic dysplasia, skeletal growth, tendon
1. Introduction
Recessive mutations in ADAMTSL2 and dominant mutations in exons 41-42 of FBN1 cause geleophysic dysplasia (GD) (MIM #231050 and MIM #614185, respectively) [1–3]. GD is a rare skeletal disorder that belongs to the group of acromelic dysplasias, defined by short stature with pronounced distal limb shortening (brachydactyly) [4]. Individual acromelic dysplasias are defined by additional, accompanying anomalies in the cardiovascular, pulmonary, or ocular system. GD is characterized by short stature, brachydactyly, and delayed bone age along with joint contractures, tight skin, and hypermuscularity [5–10]. In addition, severe airway and cardiac involvement are common and are lethal in some affected children. An ADAMTSL2 founder mutation in beagle dogs causes Musladin-Lueke syndrome (MLS) [11]. Affected beagles have short stature and distal joint contractures, with severe generalized fibrosis of skin and soft tissues, leading to extremely stiff animals that walk on tiptoes. Since MLS lacks lung or cardiac involvement and other internal organs appear normal, it typically runs a milder, non-lethal course compared to GD, despite being a severely fibrotic condition [11, 12].
ADAMTS-like 2 (ADAMTSL2) is a secreted glycoprotein belonging to the ADAMTS superfamily, which comprises 19 secreted metalloproteases (ADAMTS proteases) with diverse substrates, and 7 ADAMTS-like (ADAMTSL) proteins lacking a protease domain [13–16]. ADAMTSL proteins are therefore thought to have non-proteolytic roles as extracellular matrix (ECM) components or to regulate ADAMTS protease activity as potential co-factors [17, 18]. Recessive mutations in ADAMTS10 (MIM #277600) and ADAMTS17 (MIM #613195) cause disorders of the Weill-Marchesani syndrome (WMS) spectrum, a subgroup of acromelic dysplasias [2, 19–22]. WMS spectrum lacks the severe cardiopulmonary manifestations or juvenile lethality characteristic for GD and, unlike GD, severely affects vision.
Notably, GD and WMS spectrum disorders are each also caused by dominant mutations in FBN1 (MIM #614185 and MIM #608328, respectively) [3, 10, 20, 23]. FBN1 constitutes the major component of fibrillin microfibrils, which are 10-12 nm diameter fibrillar structures in the ECM [24]. They provide structural integrity to tissues and regulate members of the transforming growth factor (TGF)-β superfamily by sequestering bone morphogenetic proteins directly or via binding to latent TGFβ binding proteins [25–28]. The genetics of human acromelic dysplasias thus suggests that a subset of ADAMTS proteases and ADAMTSL proteins operate together with FBN1 in a pathway that regulates musculoskeletal development and growth via the fibrillin microfibril platform [17]. At the molecular level, recombinant ADAMTSL2 binds not only to FBN1, but also to FBN2 [29]. FBN2, together with a third fibrillin isotype, FBN3 (present in humans, but not in mice), is a prominent microfibril component during embryogenesis, whereas FBN1 plays major roles in postnatal growth and tissue integrity [30–32]. Notably, in the Adamtsl2 knockout embryos and newborns, fibrillin-2 (FBN2) microfibrils accumulated at the interface of bronchial smooth muscle cells and the bronchial epithelium [29], suggesting that ADAMTSL2 may modulate the balance between FBN1 and FBN2 during late embryogenesis [17].
Adamtsl2−/− mice die around birth with bronchial occlusion and a cardiac ventricular septal defect [29]. Skeletal patterning and development were unimpaired, suggesting the possibility of defective longitudinal bone growth during the juvenile period. Here, we demonstrate that tissue-specific Adamtsl2 deletion, either in limb mesenchyme or tendons, recapitulates the skeletal phenotype of GD. Together with additional observations from in vitro experiments, mechanisms, the findings provide new insights into the pathophysiology of geleophysic dysplasia and the tissue specific role of ADAMTSL2 with broad relevance to tendon development and skeletal growth regulation.
2. Results
2.1. Adamtsl2 mRNA is expressed in musculoskeletal soft tissues and presumptive articular cartilage, but not in growth plate cartilage.
β-galactosidase staining for an intragenic Adamtsl2-lacZ reporter transgene was used to determine the temporal and spatial pattern of Adamtsl2 mRNA expression during mouse musculoskeletal development and postnatal growth. At embryo age 14.5 (E14.5) and E17.5, whole limb staining showed that Adamtsl2 was most strongly expressed in developing tendons (Figure 1A, B). Strong Adamtsl2 expression was also seen in superficial layers of the presumptive articular cartilage (Figure 1C, D) and in a subset of skeletal muscle cells possibly forming muscle spindles (Figure 1E, F). At postnatal day 12 (P12), Adamtsl2 was strongly expressed in all limb tendons, including the Achilles tendon (Figure 1G, H). In skin, Adamtsl2 was expressed in a thin layer of cells above the panniculus carnosus (Figure 1I, arrows). In skeletal muscle, regions of intense, localized Adamtsl2 expression were observed, possibly representing muscle spindles (Figure 1I, J, arrows in J). In bones and joints, Adamtsl2 was consistently expressed in the superficial layer of the articular cartilage, but not in the growth plate or in bone (Figure 1K–M). In the knee, Adamtsl2 was expressed in superficial cells of the meniscus (Figure 1L arrow heads), and the articular cartilage zones of the ends of the tibia, fibula and femur (Figure 1L, arrows). In the vertebral column, strong Adamtsl2 expression was detected in the inner annulus fibrosus layer of the intervertebral disc, but not in the nucleus pulposus (Figure 1N, O).
Figure 1. Adamtsl2 expression in specific musculoskeletal tissues and conditional deletion in limb mesenchyme and tendon.

(A, B) After whole limb β-gal staining, Adamtsl2 expression (blue) was observed in developing forelimb tendon at E14.5 (A) and E17.5 (B). (C-F) At E17.5, Adamtsl2 was expressed in prospective articular cartilage (C, D; tibiofemoral joint. Panel D represents higher magnification of boxed area in C), and skeletal muscle (E, F). Arrows in panel F indicate focal expression in muscle. (G-O) At PI2, Adamtsl2 was expressed in tendons (G, H), dermis (I; arrows indicate panniculus carnosus), skeletal muscle (I, J), prospective articular cartilage in the hind foot (K) and knee joints (L; arrows indicate joint lining cells, arrowheads indicate outer meniscal cell layer), shoulder joint (M), and the inner annulus fibrosus layer, but not the nucleus pulposus of the intervertebral disc (N, O; N is a tangential section that does not contain the nucleus pulposus). Images A-O are representative of n=2-3. Scale bars in A-O are 50 μm. AT, Achilles tendon, C, calcaneus; F, femur; H, humerus; NP, nucleus pulposus; T, tibia. (P) Gene targeting strategy for limb- and tendon-specific Adamtsl2 deletion. The lacZ-Δneo cassette was removed with FlpE (Adamtsl2fl) and Adamtsl2 was inactivated by excision of exon 5 with Prx1-Cre or Scx-Cre (Adamtsl2-Prx, Adamtsl2-Scx, respectively). (Q) PCR from toe and tail genomic DNA (gDNA). The band shift with primer pair P1-P3 from toe but not tail gDNA indicates excision of exon 5 (red). (R) qRT-PCR of Achilles tendon RNA shows reduction in Adamtsl2 expression after gene deletion (n=3). P-value was calculated with a 2-sided Student t-test.
2.2. Conditional Adamtsl2 deletion in limb-mesenchyme leads to acromelic dysplasia.
To define the role of ADAMTSL2 in post-natal limb growth, neonatal lethality of Adamtsl2 null mice was bypassed using Prx1-Cre-mediated limb mesenchyme-specific inactivation (Figure 1P), resulting in Adamtsl2-Prx mice [29, 33]. Adamtsl2-Prx mice were viable and without gross musculoskeletal anomalies. PCR analysis of genomic DNA extracted from toes of Adamtsl2-Prx mice, but not from their tail DNA indicated successful limb-specific excision of the floxed exon-5 of Adamtsl2 by Cre-recombinase (Figure 1Q). In addition, quantitative real-time PCR (qRT-PCR) of Achilles tendon mRNA showed a significant reduction of Adamtsl2 mRNA in Adamtsl2-Prx tendons (Figure 1R).
Alizarin red-Alcian blue stained skeleton preparations from P16 to P20 demonstrated that Adamtsl2-Prx long bone diaphyses were less sculpted, i.e., lacking the characteristic narrowing in the central regions of the diaphysis and that they had a “stubby” appearance (2A, B, F–H, “D” indicates narrowest part of diaphyses). These changes were most visible in metacarpals and metatarsals (Figure 2B, H). All forelimb bones were reduced in length in Adamtsl2-Prx mice (Figure 2C, D). Distal bones were disproportionately shorter, indicating an acromelic limb phenotype. The diaphyseal and metaphyseal regions of Adamtsl2-Prx forelimb bones were significantly wider compared to controls (Figure 2E). Like the forelimbs, Adamtsl2-Prx hind limb bones were shorter and their diaphyseal and metaphyseal regions were significantly wider than controls (Figure 2F–K). At birth (P0), metatarsals and radii of Adamtsl2-Prx mice were significantly shorter, but no significant differences were observed in the length of metacarpals and the ulna (Supplemental Figure 1A). In Adamtsl2-Prx mice older than 9 months, significant bone shortening persisted in metatarsals and the tibia, but was not observed in the forelimb (metacarpals, humerus, radius, ulna) (Supplemental Figure 1B). Neither histological abnormalities in the growth plate of Adamtsl2-Prx limbs nor differences in the height of the growth plate, or its proliferative, pre-hypertrophic and hypertrophic zones were detected (Supplemental Figure 2). No consistent difference in histochemical staining for collagens and proteoglycans, or specifically, for collagen II and collagen X were observed (Supplemental Figure 2). In summary, limb-specific deletion of Adamtsl2 consistently resulted in distal bone shortening in the limbs and in alteration in the shape of the diaphyseal and metaphyseal regions, i.e., recapitulating skeletal anomalies described in GD.
Figure 2. Prx1-Cre mediated Adamtsl2 inactivation results in an acromelic dysplasia (disproportionally shorter distal bones).
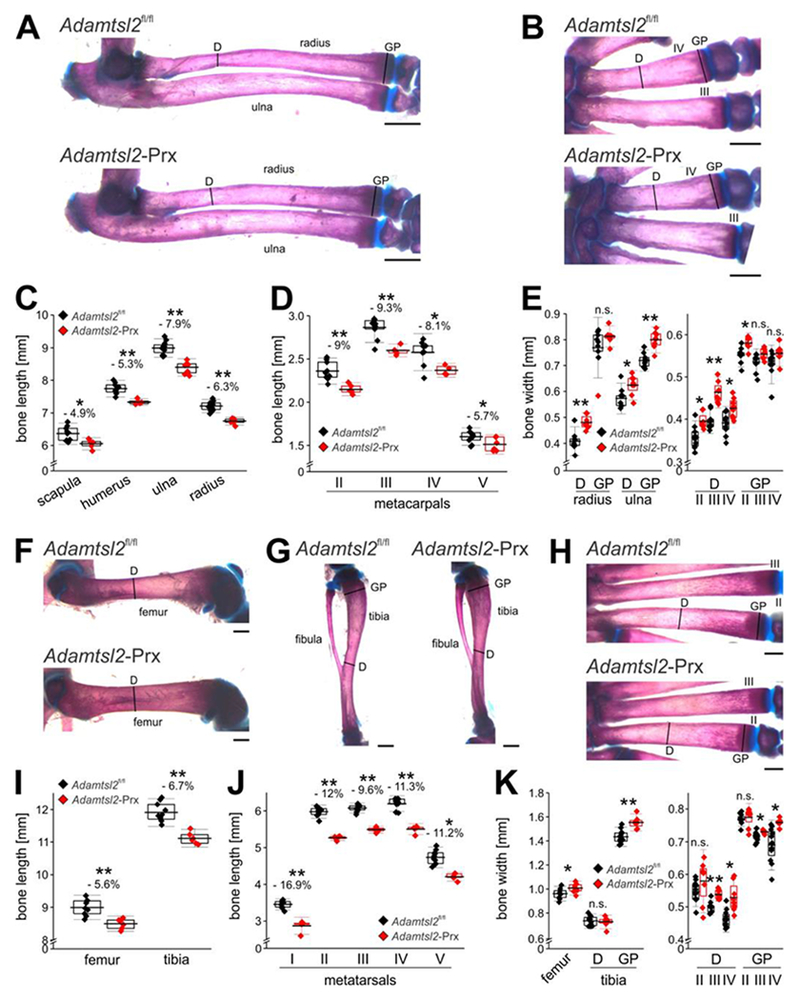
(A, B) Alizarin red-Alcian blue stained radius and ulna (A) and metacarpals (B), showing the sculpting defect in the mid-diaphysis. Lines indicate where bone width was measured. D, diaphysis; GP, growth plate. (C, D) Measured lengths of proximal forelimb long bones (C) and metacarpals (D) indicating disproportionate shortening of the distal bones. (E) Measured widths of diaphysis (D) or growth plate (GP) indicate wider bones in absence of ADAMTSL2. Relative reduction in length or width in C-E is indicated as a percentage. (F-H) Alizarin red-Alcian blue stained femur (F), tibia and fibula (G), and metatarsals (H), showing the sculpting defect in the diaphysis. Lines indicate where bone width was measured. (I, J) Measured lengths of hind limb proximal long bones (I) and metatarsals (J) indicating disproportionately shorter distal bones. (K) Measured widths of diaphysis (D) or growth plate (GP) indicating wider bones in absence of ADAMTSL2. Relative reduction in length or width in I-K is indicated as a percentage. Bones were dissected from littermates at P18 (n=6-10). Relative reduction in length is indicated in %. P-value were calculated with 2-sided Student t-test. * p<0.05; ** p<0.001. The box indicates the 25th-75th percentile, line indicates mean value and whiskers indicate standard deviation. Scale bars in A-C and F-H are 1 mm.
2.3. Abnormal Achilles tendon in Adamtsl2-Prx mice.
In addition to the skeletal anomalies, Achilles tendons from Adamtsl2-Prx mice had altered dimensions and morphology (Figure 3A, B). Specifically, their origins from the gastrocnemius were poorly defined, and they were significantly shorter and wider than controls (Figure 3A, B). During limb dissection, we also noticed that Adamtsl2-Prx Achilles tendons were tethered to surrounding tissue, reminiscent of severe peri-tendon fibrosis reported in Achilles tendon from dogs with Musladin-Lueke syndrome [11]. In contrast, the control Achilles tendons could be readily separated from the surrounding tissue. Because tendons consist predominantly of collagen type I fibrils and genetic loss of several ECM components alters collagen fibril diameter [34–37], we analyzed the cross-sectional diameter of the collagen fibrils by transmission electron microcopy. The profiles of the collagen fibrils were regular and comparable in the two groups. However, based on a two-sample Kolmogorov-Smirnov test and a Kuiper test for variable diameters, the distribution of the collagen fibril diameter was skewed toward larger fibrils in Achilles tendons from Adamtsl2-Prx limbs compared to control tendons (p < 0.001 (Figure 3C, D)).
Figure 3. Altered Achilles tendon morphology and collagen fibril size distribution in the absence of Adanitsl2.
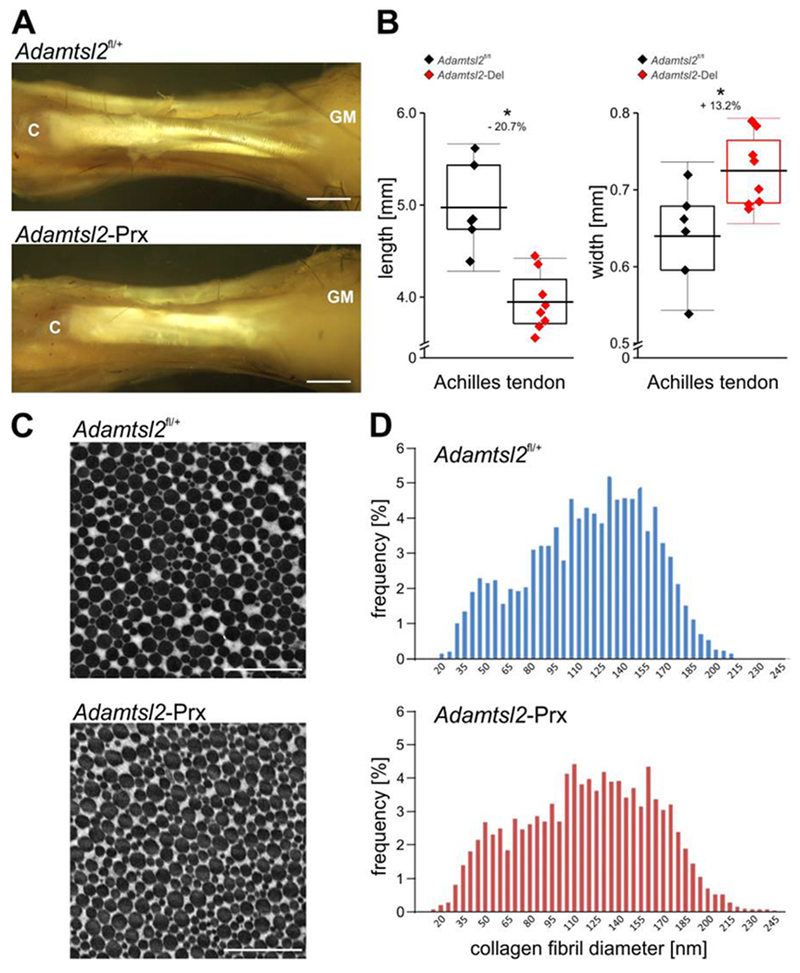
(A) A dissection of Achilles tendon at P27 shows reduced length and altered morphology of ADAMTSL2-deficient tendon (increased width, poorer definition of sub-tendons at their origin from the gastrocnemius muscle (GM); C, calcaneus). Images shown are representative of n=6-8 and represent littermates. Scale bars are 1 mm. (B) Adamtsl2-deficient Achilles tendons are significantly shorter and wider than wild-type (n=6-8). P-values, calculated with a two-sided Student t-test and relative reduction in length or increase in width in % are indicated. * p<0.05. The box indicates the 25th-75th percentile, line indicates mean value and whiskers indicate standard deviation. (C) Electron micrograph of cross-section through the Achilles tendon mid-substance showing that mutant and wild-type collagen fibrils have comparable cross-sectional morphology. Images shown are representative of n=10-12 images and n=3 littermates per genotype. Scale bars represent 1 μm. (D) Histograms of the distribution of the collagen fibril diameters indicated comparable distribution profile between the genotypes (10-12 digital images were quantified from n=3 mice per genotype). Collagen fibrils in the Adamtsl2-Prx tendons were slightly larger compared to wild-type control tendons (p<0.001).
Given intense Adamtsl2 expression in tendons and pronounced morphological alterations in Adamtsl2-Prx Achilles tendons, we undertook tendon-specific Adamtsl2 deletion using Scx-Cre (Adamtsl2-Scx). The mT/mG reporter mouse, which switches cellular fluorescence from red to green in the presence of Cre-recombinase demonstrated deletion specifically in tendons, including the Achilles tendon but not in the distal region of long bones nor in skeletal muscle (Figure 4A–D). Although Achilles tendons from Adamtsl2-Scx limbs were significantly shorter, their visually increased width did not reach statistical significance over control tendons (Figure 4E, F).
Figure 4. Scx-Cre mediated Adamtsl2 inactivation in tendon results in altered tendon morphology and non-autonomous bone shortening.

(A, B) Fluorescence imaging of the ankle from newborn Adamtsl2-Scx mice shows green fluorescence in tendons indicating specific Scx-Cre recombinase activity (A, arrows indicating Achilles tendon) compared to red fluorescence in mG/mT mice without Scx-Cre (B, arrows). Scale bars in A and B are 1 mm. Images shown are representative of n=2 littermates. (C, D) Longitudinal section through distal tibia (T) showing Scx-Cre activity in Achilles tendon tissue (AT, green), but neither gastrocnemius muscle (GM), tibial bone (red) (C) nor the calcaneal insertion (C) of the Achilles tendon showed Scx-Cre activity. (D). Scale bars in C and D represent 100 μm. (E) Gross view of Achilles tendon shows altered dimensions from the dorsal (left-hand panels) or lateral aspects (right-hand panels). Images shown are representative of n=8-10 from n=4-5 littermates. Scale bars in E are 1 mm. (F) Adamtsl2-deficient Achilles tendons are significantly shorter and tended to be wider (n=8-10). Relative reduction in length is indicated in %. (G) Alizarin red-Alcian blue stained tibiae showing normal morphology. Scale bars represent 1 mm. (H) Bone length measurements of forelimb and hind limb long bones and metatarsals demonstrates bone shortening in Adamtsl2-Scx limbs (n=8-10). Relative reduction in length is indicated in %. P-values were calculated with 2-sided Student t-test. * p<0.05; ** p<0.01, n.s. not significant. The box indicates the 25th-75th percentile, the line indicates mean value and whiskers indicate standard deviation.
2.4. Adamtsl2-Scx limbs have reduced bone length.
As in Adamtsl2-Prx limbs, several Adamtsl2-Scx forelimb and hind limb bones were shorter, with a ~6% reduction in length (Figure 4G, H), but disproportionate shortening of distal bones found in Adamtsl2-Prx limbs was not seen in Adamtsl2-Scx limbs. The length of metacarpals from Adamtsl2-Scx forelimbs was not different compared to control limbs and a diaphyseal modeling defect was not evident (data not shown). Thus, deletion of Adamtsl2 in tendons led to milder changes in bone length than those arising from Adamtsl2 deletion in total limb mesenchyme, suggesting contributions of other tissues in Adamtsl2-Prx limbs.
2.5. ADAMTSL2 deficiency alters tenocyte organization and shape.
Longitudinal sections through the tendon mid-substance revealed a disorganization of tenocytes in Adamtsl2-Prx tendons (Figure 5A, B). In control tendons, tenocytes form regular linear arrays oriented along the longitudinal axis of the tendon. However, in Adamtsl2-Prx tendon, the shape of the tenocytes was more irregular with loss of register in tenocyte arrays. Quantification of nuclear shape parameters as a surrogate for cell shape revealed a significant increase in the nuclear aspect ratio and in the nuclear irregularity index based on DAPI-stained sections (Figure 5C). Multiphoton imaging (second harmonic generation) demonstrated a disarray of collagen fiber orientation and higher fluorescence intensity (Figure 5D, E). Quantification of collagen fiber alignment after Fourier transformation revealed no changes in the directionality, but a significant increase in the dispersion, indicating a wider range of directional angles in Adamtsl2-Prx tendons (Figure 5F). Adamtsl2 deficient Achilles tendon ECM had more intense FBN1 staining in the in the vicinity of the tenocyte arrays than control tendons (Figure 5G, H). FBN2 staining intensity was low and demonstrated no obvious difference between Adamtsl2 deficient and control tendons (data not shown). Fbn1, Fbn2, Fn1, Col1a1, Col3a1, Scx, and Tnmd mRNA levels were not altered in Adamtsl2-Prx tendons (Supplemental Figure 3).
Figure 5. Disorganization of tenocyte arrays and extracellular matrix in Adamtsl2-Prx tendon.
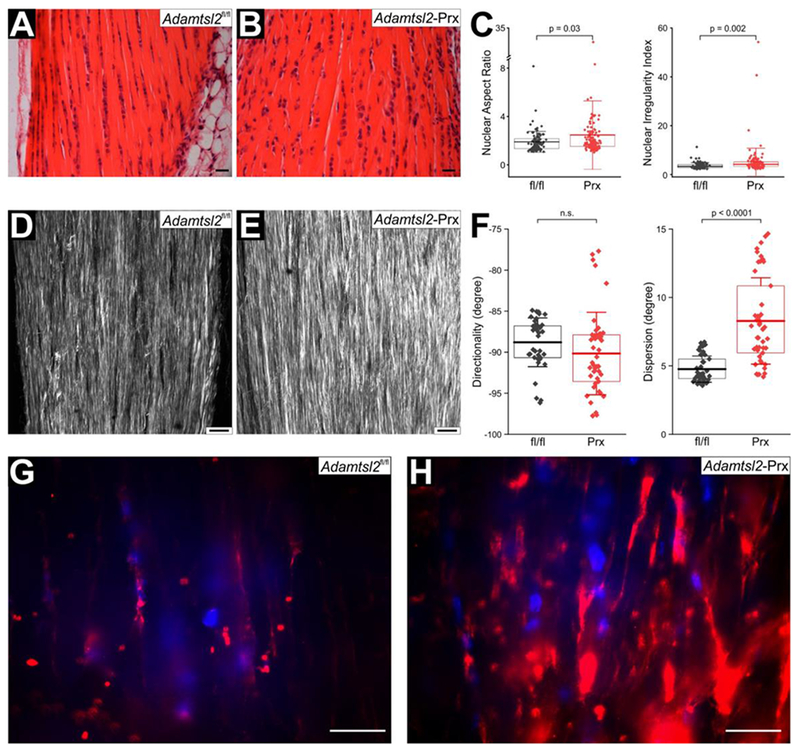
(A, B) Hematoxylin & eosin staining of longitudinal sections through Achilles tendon show disturbance of linear tenocyte arrays in Adamtsl2-Prx tendon. Images shown are representative of n=3 littermates per genotype. (C) Quantification of nuclear aspect ratio and nuclear irregularity index from DAPI-stained sections from Adamtsl2fl/fl (n= 104) and Adamtsl2-Prx (n=135) tendons show significant differences suggesting altered cell shape. P-values were calculated with 2-sided Student t-test. The box indicates the 25th-75th percentile, the line indicates mean value and whiskers indicate standard deviation. (D, E) Second harmonic generation imaging of collagen fibers shows more intense collagen signal and disorganized collagen fibers in the absence of ADAMTSL2. Images shown are representative of n=3 littermates. (F) Quantification of directionality and dispersion of the collagen fibers in Adamtsl2fl/fl (n= 42 optical sections) and Adamtsl2-Prx (n=47 optical sections) after Fourier transformation show no changes in overall direction, but a significantly higher dispersion of directionality angles. P-values were calculated with 2-sided Student t-test. n.s.; not significant. The box indicates the 25th-75th percentile, the line indicates mean value and whiskers indicate standard deviation. (G, H) Fibrillin-1 (FBN1) staining is cell-associated, and stronger in Adamtsl2-Prx tendons. Images shown are representative of n=3 littermates. Scale bars represent 20 μm.
2.6. ADAMTSL2 binds fibrillin microfibrils.
We showed previously that recombinant ADAMTSL2 directly bound to the N- and C-terminal halves of recombinant FBN1 and FBN2 [3, 29]. To discern whether ADAMTSL2 also localized to the corresponding supramolecular complexes, i.e., to fibrillin microfibrils, we added purified recombinant ADAMTSL2 to human dermal fibroblasts and asked whether it colocalized with endogenous fibrillin microfibrils. Recombinant ADAMTSL2 co-localized with both FBN1 and FBN2 microfibrils, but the colocalization with FBN2 was weaker because fewer FBN2-stained microfibrils were formed by these cells (Figure 6A). To capture early time points of microfibril formation and to analyze the potential co-localization of ADAMTSL2 with fibronectin fibrils, whose assembly precedes that of fibrillin microfibrils, we performed co-immunostaining experiments 24h after cell seeding and recombinant ADAMTSL2 supplementation (Figure 6B). We found that ADAMTSL2 colocalized with nascent FBN1 microfibrils, but not with FBN2 at the 24h time point. ADAMTSL2 also colocalized to some, but not all, fibronectin-positive fibrils. Given the previously published co-localization of FBN1 or FBN2 with fibronectin [38] and co-localization of FBN1 and FBN2 [39, 40], our data suggest that ADAMTSL2 predominantly localizes to FBN1 microfibrils in the ECM of human dermal fibroblasts. Taken together with increased FBN1 immunostaining in Adamtsl2-Prx tendons, the data suggest a specific regulatory role for ADAMTSL2 in limiting FBN1 levels in tendon microfibrils after birth.
Figure 6. ADAMTSL2 co-localizes with fibrillin microfibrils in cultured fibroblasts.

(A) ADAMTSL2 co-localizes with fibrillin-1 (FBN1) and to a lesser extent with fibrillin-2 (FBN2) containing microfibrils assembled by human dermal fibroblasts 48h after seeding cells and addition of recombinant ADAMTSL2. (B) 24h after seeding cells and ADAMTSL2 addition, ADAMTSL2 localized to FBN1 microfibrils and fibronectin fibers. No localization to FBN2 microfibrils was observed. Nuclei were counterstained with DAPI. Scale bars are 20 μm.
3. Discussion
Here, limb-specific and tendon-specific conditional Adamtsl2 deletion are shown to provide mouse models for GD, overcoming the roadblock imposed by death of Adamtsl2−/− newborn mice [29]. Intense Adamtsl2 expression in tendons, tenocyte disarray, ADAMTSL2 binding to fibrillin microfibrils and excess FBN1 microfibrils in tendons upon ADAMTSL2 deletion strongly suggests that ADAMTSL2 limits formation of fibrillin microfibrils in the ECM close to the tenocytes and such has a tissue-autonomous function in tendon, altering tendon dimensions and growth. In contrast, the impact on bone length appears to be non-autonomous since Adamtsl2 expression was not observed in growth plate cartilage, and tendon-specific anomalies affected bone length. Bone growth results from chondrocyte proliferation, hypertrophy and ECM synthesis in the cartilage growth plates, located at both ends of long bones. The major mediators of growth are well–characterized and include Indian hedgehog and parathyroid hormone-related protein acting locally in the growth plate, and endocrine factors such as growth hormone, and vitamin D [41–43]. On the other hand, limb shortening is known to occur after soft tissue contractures, juvenile paralysis or experimental manipulation of soft tissues [44–46]. The skeletal impact of soft tissue anomalies is thought to arise from mechanical constraints imposed by reduced soft tissue extensibility, consistent with retardation of growth plate activity by exogenous tethering or compressive force [47, 48]. Notably, all tendons cross at least one joint, allowing the muscle-tendon unit to exert compressive force on one or more growth plates. Moreover, tendon-specific deletion with ricx-driven Cre-recombinase, which, unlike Prx-Cre, spares recombination in cartilage, specifically led to bone shortening. Thus, the bone shortening observed after Adamtsl2 deletion could arise secondary to defective tendon growth.
Previous work showed that linear tenocyte arrays in maturing tendons were specifically associated with fibrillin microfibrils, which were postulated to represent fiducial elements essential for tenogenesis [49]. Although collagen fibrillogenesis was grossly unimpaired, the findings suggest that accumulation of FBN1 in tendon microfibrils affects the overall growth and morphogenesis of the Achilles tendon, including alignment of cells and collagen fibers, which are higher-order assemblies of collagen fibrils. The observed minor differences in skeletal growth following Prx1-Cre and Scx-Cre mutagenesis may arise from ADAMTSL2 deletion in muscle and other mesenchymal cells by Prx1-Cre, whereas it was not deleted by Scx-Cre in these tissues. Interestingly, Fbn1 deletion using Scx-Cre results in bone lengthening i.e., a mouse model for skeletal manifestations of Marfan syndrome (Dr. Francesco Ramirez, Icahn School of Medicine at Mount Sinai, personal communication). The genesis of the cortical diaphyseal and metaphyseal bone sculpting defect is unclear since ADAMTSL2 is not expressed in bone or periosteum. We suggest that this could also arise from the influence of contracted tendons and soft tissue in the juvenile period, where bone remodeling is sensitive to mechanical forces [50]. Recently, Delhon et al conditionally inactivated Adamtsl2 in cartilage using an independent Adamtsl2 floxed allele and a Col2a1Cre deleter strain, and observed a significant reduction of overall growth and limb length [51]. Col2a1Cre-mediated deletion also occurs in the axial skeleton, whereas Prx1-Cre is limb-specific, explaining the overall reduction of body length in the Col2a1Cre experiments. However, their experiments suggested a significant impact on growth plate morphology and expression of collagen II and collagen X genes, which is inconsistent with our observation of normal collagen II and collagen X immunostaining. Furthermore, they detected Adamtsl2 mRNA in growth plate chondrocytes using in situ hybridization, which the intragenic lacZ reporter did not disclose in our experiments. It is possible that low levels of Adamtsl2 mRNA, undetectable in our analysis, may have a significant impact on the growth plate, but it is unclear why this impact was not evident in the Prx1-Cre-mediated Adamtsl2 deletion we report here, since deletion would also occur in growth plate chondrocytes. Taken together, the two studies suggest that ADAMTSL2 may regulate limb-growth both autonomously in growth plate cartilage, and non-autonomously via tendon and soft tissue effects, the latter being the major conclusion of our analysis. Both of these mouse models will facilitate understanding of GD mechanisms at the molecular level.
Previously, embryos and newborn mice with global loss of ADAMTSL2 were noted to have an excess of FBN2 microfibrils in bronchial ECM associated with bronchial epithelial dysplasia and occlusion of bronchial lumena [29]. FBN2 is highly expressed during embryogenesis, with reduced expression post-natally, whereas FBN1 is expressed pre- and well as post-natally [30, 31]. ADAMTSL2 has at least two binding sites on both fibrillin isotypes [1, 52]. A major transition in fibrillin gene expression in mice thus occurs at birth, when FBN2 expression wanes and FBN1 becomes the dominant fibrillin isotype. Previously observed FBN2 accumulation in embryonic Adamtsl2−/− bronchi, taken together with intense FBN1 staining in post-natal Adamtsl2–deficient Achilles tendon suggest a general function for ADAMTSL2 in limiting microfibril assembly independent of fibrillin isotype [3, 29]. A prior demonstration that ADAMTSL2 binds to both FBN1 and FBN2 was extended here to demonstrate their binding to microfibrils, which in addition to fibrillins, contain numerous other molecules [1, 29]. However, when recombinant ADAMTSL2 was added to human dermal fibroblasts, no consistent change in the fibrillin microfibril staining was observed. One possibility is that dermal fibroblasts do not recapitulate the precise expression of ADAMTS proteins occurring in tenocytes, such that other family members may blunt the impact of exogenous ADAMTSL2. Alternatively, ADAMTSL2 may modulate protease activity in the ECM, facilitating microfibril turnover in tissues. In this regard, mutation of two ADAMTS proteases, ADAMTS10 and ADAMTS17 result in acromelic dysplasias, and ADAMTS 10 has been shown to have proteolytic activity against FBN2 and fibrillin-1 [53, 54].
The enduring paradox of FBN1 mutations is that the majority of these mutations cause MFS, but a subset of mutations, located in the fifth transforming growth factor-β–like / 8-cysteine domain of FBN1 (encoded by exons 41 and 42), also known as TB5, lead to acromelic dysplasias [10]. Jensen et al demonstrated that FBN1 TB5 mutants causing GD were secreted and assembled into microfibrils, contrasting with MFS mutations affecting this region, which were not [55]. TB5 mutants leading to GD block the binding of this domain to heparan-sulfate, which regulates microfibril assembly [56, 57]. Taken together, with the present findings, we postulate that abnormal fibrillin microfibril assembly is the general molecular basis of GD, and the specific role of ADAMTSL2 may be to inhibit microfibril assembly, whether it be assembly of FBN2 fibrils in the embryonic period or FBN1 fibrils in tendons during the postnatal period.
4. Methods
All reagents were purchased from Sigma-Aldrich or ThermoFisher Scientific unless specified.
4.1. Mouse strains
Adamtsl2-Prx mice were generated by deleting the Frt-site-flanked lacZ-Δneo cassette from Adamtsl2+− mice (KOMP allele: Adamtsl2tm1a(KOMP)Wtsi, NIH, Bethesda, MD, USA) using B6.Cg-Tg(ACTB-FLPe)9205Dym/J (Jackson Laboratory, Bar Harbor, ME USA) [29]. FLPe (FLP recombinase)-mediated deletion generated the Adamtsl2fl allele, where exon 5 is flanked by loxP sites and is amenable to Cre-mediated excision (Figure 1D). Mice were backcrossed >10 generations into C57BL/6 and both females and males were used. Mutant mice were compared to littermate controls. A B6.Cg-Tg(Prrx1-cre)1Cjt/J (Prx1-Cre) male mouse was obtained from Jackson Laboratory and the Scx-Cre strain was previously described [58]. Mice were used under a protocol approved by the animal use committee of the Cleveland Clinic (IACUC protocols 2012-0818 and 2015-1458).
4.2. Genotyping
Genomic DNA from toe and tail tissue was isolated using DirectPCR (Viagen, Los Angeles, CA, USA). PCR products were amplified using Taq polymerase (New England BioLabs Inc., Ipswich, MA, USA) and a forward primer located in exon 4 {Adamtsl2 wild-type allele, P1: 5′-gtaccagctctgcagagtgc-3′) in combination with reverse primers located in the En2-splice acceptor site in the lacZ-Δneo {Adamtsl2 knock-out allele, P2: 5′-cactgagtctctggcatctc-3′) or in exon 6 (Adamtsl2 wild-type or conditional allele, respectively, P3: 5′-ctctcaggtcggtgagcttg-3′). PCR products were separated by agarose gel electrophoresis and visualized with ethidium bromide.
4.3. β-Galactosidase (β-gal) staining
Tissue was fixed in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA, USA) overnight and stained with potassium ferrocyanide/potassium ferricyanide/5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal) (Denville Scientific Inc., Holliston, MA, USA) as described previously [59].
4.4. Bone and tendon morphometry
Limbs were dissected, and the Achilles tendon exposed and photographed in situ. The soft tissue was removed, and bones were fixed in 80% ethanol and dehydrated in 96% ethanol and acetone over several days for Alizarin red / Alcian blue staining. Staining was performed with a solution of 30 mg Alcian blue and 5 mg Alizarin red in 20 ml acetic acid / 80 ml 95% ethanol for several days at room temperature. Tissues were rinsed with 95% ethanol and cleared with 1% aqueous potassium hydroxide followed by serial transfer in 20%, 50% and 80% glycerin prepared in 1% aqueous potassium hydroxide. Cleared and stained bones were photographed, and bone length was measured using Image J (NIH, Bethesda, MD). Mutants were compared to littermate controls (Adamtsl2fl/fl, Prx1-Cre, Scx-Cre) in all analyses and statistical significance was determined with a two-sided Student t-test using the Origin 2017 software package (OriginLab, Northampton, MA, USA).
4.5. Gene expression analysis
Achilles tendons were homogenized with an Ultra Turrax homogenizer (IKA-Works Inc., Wilmington, NC, USA) in Trizol and total RNA was extracted according to the manufacturer’s protocol. 1 μg of total RNA was reverse transcribed using the High Efficiency cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA). Quantitative real-time PCR (qPCR) was performed with EvaGreen (VWR, Radnor, PA, USA) in a total volume of 10 μl using 0.125 μl cDNA template. qPCR reactions were performed in triplicate with a CFX96 real-time system (Bio-Rad, Hercules, CA, USA).
4.6. Electron microscopy
All chemicals for electron microscopy were purchased from Electron Microscopy Sciences (Hatfield, PA, USA). Achilles tendons were fixed immediately upon dissection and then overnight in cold 4% paraformaldehyde, 2.5% glutaraldehyde, 0.1 M sodium cacodylate and 8 mM CaCl2, pH 7.4, rinsed in cacodylate buffer and post-fixed for 1 h with 1% osmium tetroxide. Fixed tendons were dehydrated in a graded ethanol series followed by 100% propylene oxide, infiltrated and embedded over a 3-day period in a mixture of Embed 812, acidic methyl anhydride, dodecenylsuccinic anhydride and 2,4,6-Tris(dimethylaminomethyl)phenol (DMP-30) and polymerized overnight at 60 °C. Cross-sections of tendons (90 nm) were prepared using a Leica ultramicrotome (Leica Microsystems Inc., Buffalo Grove, IL, USA) and post-stained with 2% aqueous uranyl acetate and 1% phosphotungstic acid, pH 3.2. The sections were examined and imaged at 80 kV using a JEOL 1400 transmission electron microscope (JEOL Ltd., Tokyo, Japan) equipped with a Gatan Orius widefield side mount CC Digital camera (Gatan Inc., Pleasanton, CA, USA). Tendon diameter analysis was obtained from pooled data from one tendon from each of three littermates of each genotype. Digital images from each tendon were taken from nonoverlapping areas at 60,000X. 10-12 images were randomized and masked before fibril diameters were measured using a RM Biometrics-Bioquant Image Analysis System (Nashville, TN, USA). 3586 and 3797 fibril diameters for the control and the Adamtsl2-Prx genotype, respectively, were measured. Fibril diameters were measured along the minor axis of the fibril cross-section. Tendon diameter measurements were pooled into groups by genotype and represented as histograms. A Kolmogorow-Smirnow and a Kuiper test was performed to determine if the two datasets were different.
4.7. Histology and immunostaining
Limbs were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) in PBS for 24h. Those containing mineralized bone were subsequently decalcified in 14% EDTA solution, changed every 3 days for 3-4 weeks and paraffin-embedded. 8-10 μm sections were used for hematoxylin and eosin staining, Masson trichrome, and Safranin-O stain according to standard protocols. For immunostaining of collagen II, antigen retrieval with Histo/Zyme, pH 7.2 was performed after rehydration of paraffin-embedded tissue sections. Sections were blocked with 10% normal goat serum (NGS) (Jackson ImmunoResearch, West Grove, PA, USA) in PBS for 30 minutes at room temperature and incubated with a polyclonal anti-collagen II antibody (GeneTex, #20300) diluted 1:200 in blocking buffer at 4°C overnight. The collagen II antibody was visualized with rhodamine-red goat anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA, USA) diluted 1:200 in blocking buffer. Sections were mounted in ProLong Diamond Antifade Mountant with DAPI. Staining for collagen X was performed using the same protocol except for heat-mediated antigen retrieval by incubating the slides in 10 mM citrate, 2 mM EDTA, 0.05% Tween-20, pH 6.2 overnight in a water bath at 60°C, instead of enzyme-mediated antigen retrieval. The collagen X antibody was diluted 1:20 in 10% normal goat serum and incubated for two hours at room temperature [60]. For immunostaining with FBN1 and FBN2 antibodies, tissues were rehydrated, and antigen retrieval was performed with 10 mM citrate, 2 mM EDTA, 0.05% Tween-20, pH 6.2 for 4 × 1.5 min in a microwave oven. Sections were cooled in antigen-retrieval buffer rinsed with water and 2 × PBS and blocked for 1h in 5% normal goat serum in PBS. Sections were incubated with FBN1 antibody [57] diluted in blocking buffer overnight at 4 °C, rinsed 3 × 10 min in PBS, and incubated with secondary antibody (goat anti-rabbit-Alexa-564) (Jackson ImmunoResearch, West Grove, PA, USA) diluted in blocking buffer for 1h at RT. Sections were rinsed 3 × 10 min in PBS and mounted using VectaShield Gold with DAPI. Sections were photographed on an Olympus BX51 upright microscope (Olympus, Center Valley, PA, USA) using a Leica DFC7000T camera and Leica Application Suite v4.6 imaging software (Leica Microsystems, Wetzlar, Germany). Nuclear morphology was analyzed with the ImageJ NII plug-in [61]. Statistical analysis was performed using Origin 2017 software package (OriginLab, Northampton, MA, USA).
4.8. Second-harmonic generation / Two-photon imaging
Paraformaldehyde-fixed, paraffin-embedded tendon sections were rehydrated and coverslipped in 50% glycerol in PBS. Images were acquired using a Leica TCS SP5 II Confocal/Multi-Photon high-speed upright microscope with a 25X water immersion lens in forward scattering mode (Leica Microsystems). The excitation wavelength was set to 880 nm and the non-descanned detector (NDD) was set to collect signals between 430 and 450 nm. Z-stacks were acquired at a scan speed of 600 Hz in bidirectional mode with a line average of 5. Images were processed using ImageJ-Fiji software (NIH, Bethesda, MD). Directionality was quantified with the ImageJ “Directionality” plug-in on Fourier-transformed images. Statistical analysis was performed using the Origin 2017 software package (OriginLab, Northampton, MA, USA).
4.9. Cell culture and co-localization studies
Human dermal fibroblasts derived from explant cultures of circumcised foreskin were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, and 5 mM L-glutamine (complete DMEM) in a 5% CO2 atmosphere in a humidified incubator at 37 °C. 50,000 cells / chamber were seeded in 8-well chamber slides (BD Bioscience, San Jose, CA, USA) and after 16 h 50 μg of recombinant mouse ADAMTSL2 [14], was added in complete DMEM [29]. After 24 h, 48 h, or 72 h cells were fixed in ice-cold 70% acetone / 30% methanol (v/v) and co-immunostained for recombinant ADAMTSL2 using monoclonal mouse α-myc antibody (clone 9E10, Invitrogen) or the polyclonal rabbit antibodies anti-FBN1, anti-FBN2, or anti-fibronectin (pAB 2033, MilliporeSigma, Burlington, MA) [40, 57, 62, 63].
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Highlights:
Adamtsl2 is expressed in several musculoskeletal tissues, such as tendon and muscle, but not in the growth plate.
Deletion of ADAMTSL2 in the limb resulted in a geleophysic dysplasia-like phenotype, e.g. shorter bones with a more severe shortening of the distal skeletal elements.
ADAMTSL2-deficient Achilles tendons are shorter than wild-type tendons.
Conditional deletion of ADAMTSL2 in tendon resulted in tendon shortening and short limb phenotype, suggesting an autonomous role for ADAMTSL2 in tendon growth and a non-autonomous role in regulating bone growth.
ADAMTSL2 deficiency in tendon results in a disarray of tenocytes and an accumulation of fibrillin-1 in the vicnity of tenocytes.
ADAMTSL2 colocalizes with fibrillin microfibrils assembled by cultured fibroblasts.
6. Acknowledgements
This work was supported by the National Institutes of Health (grant numbers AR070748 to D.H., AR53890 to S. A, and AR44745 to D. B.). We thank Dr. Dieter Reinhardt (McGill University, Montreal, Canada) for providing the fibrillin-1 antibody and Dr. Robert Mecham (Washington University, St. Louis, USA) for providing the fibrillin-2 antibody. The anti-collagen type X antibody (X53) was a gift from Ryan Coghlan and William Horton (Shriner’s Hospital, Portland, OR, USA). We thank Dr. Judith Drazba from the Digital Imaging Core (Cleveland Clinic Lerner Research Institute) for help with two-photon imaging. This work utilized a Leica SP5 confocal/multi-photon microscope that was purchased with partial funding from National Institutes of Health SIG (grant number 1S10RR026820-01). We thank Thomas Adams (University of Southern Florida) for help with the statistical analysis of the collagen fibril distribution.
Non-standard Abbreviations:
- ADAMTSL
a disintegrin-like and metalloproteinase domain with thrombospondin-type 1 motiflike
- DMEM
Dulbecco’s Modified Eagle Medium
- ECM
extracellular matrix
- FBN1
fibrillin-1
- GD
geleophysic dysplasia
- KOMP
Knockout Mouse Project
- MFS
Marfan syndrome
- PBS
phosphate-buffered saline
- TGFβ
ransforming growth factor-β
- WMS
Weill Marchesani syndrome
- RT-qPCR
real-time quantitative PCR
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
7. References
- [1].Le Goff C, Morice-Picard F, Dagoneau N, Wang LW, Perrot C, Crow YJ, Bauer F, Flori E, Prost-Squarcioni C, Krakow D, Ge G, Greenspan DS, Bonnet D, Le Merrer M, Munnich A, Apte SS, and Cormier-Daire V, ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation, Nat. Genet 40 (2008) 1119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Allali S, Le Goff C, Pressac-Diebold I, Pfennig G, Mahaut C, Dagoneau N, Alanay Y, Brady AF, Crow YJ, Devriendt K, Drouin-Garraud V, Flori E, Genevieve D, Hennekam RC, Hurst J, Krakow D, Le Merrer M, Lichtenbelt KD, Lynch SA, Lyonnet S, MacDermot K, Mansour S, Megarbane A, Santos HG, Splitt M, Superti-Furga A, Unger S, Williams D, Munnich A, and Cormier-Daire V, Molecular screening of ADAMTSL2 gene in 33 patients reveals the genetic heterogeneity of geleophysic dysplasia, J. Med. Genet 48 (2011) 417–21. 10.1136/img.2010.087544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Goff C. Le, Mahaut C, Wang LW, Allali S, Abhyankar A, Jensen S, Zylberberg L, Collod-Beroud G, Bonnet D, Alanay Y, Brady AF, Cordier MP, Devriendt K, Genevieve D, Kiper PO, Kitoh H, Krakow D, Lynch SA, Merrer M. Le, Megarbane A, Mortier G, Odent S, Polak M, Rohrbach M, Sillence D, Stolte-Dijkstra I, Superti-Furga A, Rimoin DL, Topouchian V, Unger S, Zabel B, Bole-Feysot C, Nitschke P, Handford P, Casanova JL, Boileau C, Apte SS, Munnich A, and Cormier-Daire V, Mutations in the TGFbeta binding-proteinlike domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias, Am. J. Hum. Genet 89 (2011) 7–14. 10.1016/i.aihg.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Le Goff C, and Cormier-Daire V, Genetic and molecular aspects of acromelic dysplasia, Pediatr Endocrinol Rev 6 (2009) 418–23. [PubMed] [Google Scholar]
- [5].Giray O, Kyr M, Bora E, Saylam G, Ugurlu B, and Gurel D, Clinical and morphological phenotype of geleophysic dysplasia, Ann. Trop. Paediatr 28 (2008)161–4. 10.1179/146532808X302206. [DOI] [PubMed] [Google Scholar]
- [6].Panagopoulos P, Fryssira H, Koutras I, Daskalakis G, Economou A, Benetou V, and Antsaklis A, Geleophysic dysplasia: a patient with a severe form of the disorder, J. Obstet. Gynaecol 25 (2005) 818–20. 10.1080/01443610500336058. [DOI] [PubMed] [Google Scholar]
- [7].Santolaya JM, Groninga LC, Delgado A, Monasterio JL, Camarero C, and Bilbao FJ, Patients with geleophysic dysplasia are not always geleophysic, Am. J. Med. Genet 72 (1997) 85–90. [PubMed] [Google Scholar]
- [8].Pontz BF, Stoss H, Henschke F, Freisinger P, Karbowski A, and Spranger J, Clinical and ultrastructural findings in three patients with geleophysic dysplasia, Am. J. Med. Genet 63 (1996) 50–4. [pii] 10.1002/(SICI)1096-8628(19960503)63:1<50::AID-AJMG11>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- [9].Shohat M, Gruber HE, Pagon RA, Witcoff LJ, Lachman R, Ferry D, Flaum E, and Rimoin DL, Geleophysic dysplasia: a storage disorder affecting the skin, bone, liver, heart, and trachea, J. Pediatr 117 (1990) 227–32. [DOI] [PubMed] [Google Scholar]
- [10].Sakai LY, and Keene DR, Fibrillin protein pleiotropy: Acromelic dysplasias, Matrix Biol. (2018) 10.1016/i.matbio.2018.09.005. [DOI] [PubMed] [Google Scholar]
- [11].Bader HL, Ruhe AL, Wang LW, Wong AK, Walsh KF, Packer RA, Mitelman J, Robertson KR, O’Brien DP, Broman KW, Shelton GD, Apte SS, and Neff MW, An ADAMTSL2 founder mutation causes Musladin-Lueke Syndrome, a heritable disorder of beagle dogs, featuring stiff skin and joint contractures, PloS one 5(2010)e12817 10.1371/iournal.pone.0012817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Packer RA, Logan MA, Guo LT, Apte SS, Bader H, O’Brien DP, Johnson G, and Shelton GD, Clinical Phenotype of Musladin-Lueke Syndrome in 2 Beagles, J. Vet. Intern. Med 31 (2017) 532–538. 10.1111/jvim.14654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Apte SS, A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms, J. Biol. Chem 284 (2009) 31493–7. 10.1074/ibc.R109.052340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Koo BH, Le Goff C, Jungers KA, Vasanji A, O’Flaherty J, Weyman CM, and Apte SS, ADAMTS-like 2 (ADAMTSL2) is a secreted glycoprotein that is widely expressed during mouse embryogenesis and is regulated during skeletal myogenesis, Matrix Biol. 26 (2007) 431–41. https://doi.org/S0945-053X(07)00042-X [pii] 10.1016/j.matbio.2007.03.003. [DOI] [PubMed] [Google Scholar]
- [15].Mead TJ, and Apte SS, ADAMTS proteins in human disorders, Matrix Biol. 71-72 (2018) 225–239. 10.1016/i.matbio.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dubail J, and Apte SS , Insights on ADAMTS proteases and ADAMTS-like proteins from mammalian genetics, Matrix Biol. 44-46 (2015) 24–37. 10.1016/i.matbio.2015.03.001. [DOI] [PubMed] [Google Scholar]
- [17].Hubmacher D, and Apte SS, ADAMTS proteins as modulators of microfibril formation and function, Matrix Biol. 47 (2015) 34–43. 10.1016/i.matbio.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fessler JH, Kramerova I, Kramerov A, Chen Y, and Fessler LI, Papilin, a novel component of basement membranes, in relation to ADAMTS metalloproteases and ECM development, Int. J. Biochem. Cell Biol 36(2004)1079–84. 10.1016/i.biocel.2003.12.010. [DOI] [PubMed] [Google Scholar]
- [19].Morales J, Al-Sharif L, Khalil DS, Shinwari JM, Bavi P, Al-Mahrouqi RA, Al-Raihi A, Alkuraya S, Meyer BF, and Al Tassan N, Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature, Am. J. Hum. Genet 85 (2009) 558–68. https://doi.org/S0002-9297(09)00407-8 [pii] 10.1016/j.ajhg.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kutz WE, Wang LW, Dagoneau N, Odrcic KJ, Cormier-Daire V, Traboulsi EI, and Apte SS, Functional analysis of an ADAMTS10 signal peptide mutation in Weill-Marchesani syndrome demonstrates a long-range effect on secretion of the full-length enzyme, Hum. Mutat 29 (2008) 1425–34. 10.1002/humu.20797. [DOI] [PubMed] [Google Scholar]
- [21].Faivre L, Gorlin RJ, Wirtz MK, Godfrey M, Dagoneau N, Samples JR, Le Merrer M, Collod-Beroud C Boileau, A. Munnich, and V. Cormier-Daire, In frame fibrillin-1 gene deletion in autosomal dominant Weill-Marchesani syndrome, J. Med. Genet 40 (2003) 34–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dagoneau N, Benoist-Lasselin C, Huber C, Faivre L, Megarbane A, Alswaid A, Dollfus H, Alembik Y, Munnich A, Legeai-Mallet L, and Cormier-Daire V, ADAMTS10 Mutations in Autosomal Recessive Weill-Marchesani Syndrome, Am. J. Hum. Genet 75 (2004) 801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cecchi A, Ogawa N, Martinez HR, Carlson A, Fan Y, Penny DJ, Guo DC, Eisenberg S, Safi H, Estrera A, Lewis RA, Meyers D, and Milewicz DM, Missense mutations in FBN1 exons 41 and 42 cause Weill-Marchesani syndrome with thoracic aortic disease and Marfan syndrome, American iournal of medical genetics. Part A 161A (2013) 2305–10. 10.1002/aimg.a.36044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hubmacher D, and Reinhardt D. In: Mecham RP ed. The Extracellular Matrix; an Overview. Berlin Heidelberg: Springer-Verlag; 2011:233–265. [Google Scholar]
- [25].Sengle G, Charbonneau NL, Ono RN, Sasaki T, Alvarez J, Keene DR, Bachinger HP, and Sakai LY, Targeting of bone morphogenetic protein growth factor complexes to fibrillin, J. Biol. Chem 283 (2008) 13874–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sengle G, Ono RN, Sasaki T, and Sakai LY, Prodomains of transforming growth factor beta (TGFbeta) superfamily members specify different functions: extracellular matrix interactions and growth factor bioavailability, J. Biol. Chem 286 (2011) 5087–99. 10.1074/ibc.M110.188615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Todorovic V, and Rifkin DB, LTBPs, more than just an escort service, J. Cell. Biochem 113 (2012) 410–8. 10.1002/icb.23385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ramirez F, Caescu C, Wondimu E, and Galatioto J, Marfan syndrome; A connective tissue disease at the crossroads of mechanotransduction, TGFbeta signaling and cell stemness, Matrix Biol. 71–72 (2018) 82–89. 10.1016/j.matbio.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hubmacher D, Wang LW, Mecham RP, Reinhardt DP, and Apte SS, Adamtsl2 deletion results in bronchial fibrillin microfibril accumulation and bronchial epithelial dysplasia-a novel mouse model providing insights into geleophysic dysplasia, Dis Model Mech 8 (2015) 487–99. 10.1242/dmm.017046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mariencheck MC, Davis EC, Zhang H, Ramirez F, Rosenbloom J, Gibson MA, Parks WC, and Mecham RP, Fibrillin-1 and fibrillin-2 show temporal and tissue-specific regulation of expression in developing elastic tissues, Connect. Tissue Res 31 (1995) 87–97. [DOI] [PubMed] [Google Scholar]
- [31].Zhang H, Hu W, and Ramirez F, Developmental expression of fibrillin genes suggests heterogeneity of extracellular microfibrils, J. Cell Biol 129 (1995) 1165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sabatier L, Miosge N, Hubmacher D, Lin G, Davis EC, and Reinhardt DP, Fibrillin-3 expression in human development, Matrix Biol. 30(2011) 43–52. 10.1016/i.matbio.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Logan M, Martin JF, Nagy A, Lobe C, Olson EN, and Tabin CJ, Expression of Cre Recombinase in the developing mouse limb bud driven by a Prx1 enhancer, Genesis (New York, N.Y. : 2000) 33 (2002) 77–80. [DOI] [PubMed] [Google Scholar]
- [34].Chen S, and Birk DE, The regulatory roles of small leucine-rich proteoglycans in extracellular matrix assembly, The FEBS journal 280(2013)2120–37. 10.1111/febs.12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Svensson L, Aszodi A, Reinholt FP, Fassler R, Heinegard D, and Oldberg A, Fibromodulin-null mice have abnormal collagen fibrils, tissue organization, and altered lumican deposition in tendon, J. Biol. Chem 274 (1999) 9636–47. [DOI] [PubMed] [Google Scholar]
- [36].Danielson KG, Baribault H, Holmes DF, Graham H, Kadler KE, and Iozzo RV, Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility, J. Cell Biol 136 (1997) 729–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ameye L, Aria D, Jepsen K, Oldberg A, Xu T, and Young MF, Abnormal collagen fibrils in tendons of biglycan/fibromodulin-deficient mice lead to gait impairment, ectopic ossification, and osteoarthritis, FASEB J. 16 (2002) 673–80. 10.1096/fi.01-0848com. [DOI] [PubMed] [Google Scholar]
- [38].Sabatier L, Chen D, Fagotto-Kaufmann C, Hubmacher D, McKee MD, Annis DS, Mosher DF, and Reinhardt DP, Fibrillin assembly requires fibronectin, Mol. Biol. Cell 20 (2009) 846–58. https://doi.org/E08-08-0830 [pii] 10.1091/mbc.E08-08-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Beene LC, Wang LW, Hubmacher D, Keene DR, Reinhardt DP, Annis DS, Mosher DF, Mecham RP, Traboulsi EI, and Apte SS, Nonselective assembly of fibrillin 1 and fibrillin 2 in the rodent ocular zonule and in cultured cells: implications for Marfan syndrome, Invest. Ophthalmol. Vis. Sci 54 (2013) 8337–44. 10.1167/iovs.13-13121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lin G, Tiedemann K, Vollbrandt T, Peters H, Batge B, Brinckmann J, and Reinhardt DP, Homo- and heterotypic fibrillin-1 and −2 interactions constitute the basis for the assembly of microfibrils, J. Biol. Chem 277 (2002) 50795–804. 10.1074/ibc.M210611200 M210611200 [pii]. [DOI] [PubMed] [Google Scholar]
- [41].Shim KS, Pubertal growth and epiphyseal fusion, Annals of pediatric endocrinology & metabolism 20 (2015) 8–12. 10.6065/apem.2015.20.L8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lui JC, Nilsson O, and Baron J, Recent research on the growth plate: Recent insights into the regulation of the growth plate, J. Mol. Endocrinol 53(2014)T1–9. 10.1530/JME-14-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Maes C, Signaling pathways effecting crosstalk between cartilage and adjacent tissues: Seminars in cell and developmental biology: The biology and pathology of cartilage, Seminars in cell & developmental biology 62 (2017) 16–33. 10.1016/i.semcdb.2016.05.007. [DOI] [PubMed] [Google Scholar]
- [44].Callewaert BL, Loeys BL, Ficcadenti A, Vermeer S, Landgren M, Kroes HY, Yaron Y, Pope M, Foulds N, Boute O, Galan F, Kingston H, Van der Aa N, Salcedo I, Swinkels ME, Wallgren-Pettersson C, Gabrielli O, De Backer J, Coucke PJ, and De Paepe AM, Comprehensive clinical and molecular assessment of 32 probands with congenital contractural arachnodactyly: report of 14 novel mutations and review of the literature, Hum. Mutat 30 (2009) 334–41. 10.1002/humu.20854. [DOI] [PubMed] [Google Scholar]
- [45].Hoyer J, Kraus C, Hammersen G, Geppert JP, and Rauch A, Lethal cutis laxa with contractural arachnodactyly, overgrowth and soft tissue bleeding due to a novel homozygous fibulin-4 gene mutation, Clin. Genet 76 (2009) 276–81. 10.1111/i.1399-0004.2009.01204.x. [DOI] [PubMed] [Google Scholar]
- [46].Putnam EA, Zhang H, Ramirez F, and Milewicz DM, Fibrillin-2 (FBN2) mutations result in the Marfan-like disorder, congenital contractural arachnodactyly, Nat. Genet 11 (1995) 456–8. 10.1038/ng1295-456. [DOI] [PubMed] [Google Scholar]
- [47].Di Nino DL, Crochiere ML, and Linsenmayer TF, Multiple mechanisms of perichondrial regulation of cartilage growth, Dev. Dyn 225 (2002) 250–9. 10.1002/dvdy.10160. [DOI] [PubMed] [Google Scholar]
- [48].Long F, and Linsenmayer TF, Regulation of growth region cartilage proliferation and differentiation by perichondrium, Development 125 (1998) 1067–73. [DOI] [PubMed] [Google Scholar]
- [49].Ritty TM, Roth R, and Heuser JE, Tendon cell array isolation reveals a previously unknown fibrillin-2-containing macromolecular assembly, Structure 11 (2003) 1179–88. [DOI] [PubMed] [Google Scholar]
- [50].Pearson OM, and Lieberman DE, The aging of Wolff’s “law”: ontogeny and responses to mechanical loading in cortical bone, Am J Phys Anthropol Suppl 39 (2004) 63–99. 10.1002/aipa.20155. [DOI] [PubMed] [Google Scholar]
- [51].Delhon L, Mahaut C, Goudin N, Gaudas E, Piquand K, Le Goff W, Cormier-Daire V, and Le Goff C, Impairment of chondrogenesis and microfibrillar network in Adamtsl2 deficiency, FASEB J. 33 (2019) 2707–2718. 10.1096/fi.201800753RR. [DOI] [PubMed] [Google Scholar]
- [52].Hubmacher D, Wang LW, Mecham RP, Reinhardt DP, and Apte SS, Adamtsl2 deletion results in bronchial fibrillin microfibril accumulation and bronchial epithelial dysplasia--a novel mouse model providing insights into geleophysic dysplasia, Dis Model Mech 8 (2015) 487–99. 10.1242/dmm.017046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kutz WE, Wang LW, Bader HL, Majors AK, Iwata K, Traboulsi EI, Sakai LY, Keene DR, and Apte SS, ADAMTS10 protein interacts with fibrillin-1 and promotes its deposition in extracellular matrix of cultured fibroblasts, J. Biol. Chem 286 (2011) 17156–67. 10.1074/ibc.M111.231571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wang LW, Kutz WE, Mead TJ, Beene LC, Singh S, Jenkins MW, Reinhardt DP, and Apte SS, Adamts10 inactivation in mice leads to persistence of ocular microfibrils subsequent to reduced fibrillin-2 cleavage, Matrix Biol. (2018) 10.1016/i.matbio.2018.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jensen SA, Iqbal S, Bulsiewicz A, and Handford PA, A microfibril assembly assay identifies different mechanisms of dominance underlying Marfan syndrome, stiff skin syndrome and acromelic dysplasias, Hum. Mol. Genet 24 (2015) 4454–63. 10.1093/hmg/ddv181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Cain SA, McGovern A, Baldwin AK, Baldock C, and Kielty CM, Fibrillin-1 mutations causing Weill-Marchesani syndrome and acromicric and geleophysic dysplasias disrupt heparan sulfate interactions, PloS one 7 (2012) e48634 10.1371/journal.pone.0048634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tiedemann K, Batge B, Muller PK, and Reinhardt DP, Interactions of fibrillin-1 with heparin/heparan sulfate, implications for microfibrillar assembly, J. Biol. Chem 276 (2001) 36035–42. 10.1074/ibc.M104985200 M104985200 [pii]. [DOI] [PubMed] [Google Scholar]
- [58].Blitz E, Viukov S, Sharir A, Shwartz Y, Galloway JL, Pryce BA, Johnson RL, Tabin CJ, Schweitzer R, and Zelzer E, Bone ridge patterning during musculoskeletal assembly is mediated through SCX regulation of Bmp4 at the tendon-skeleton junction, Dev. Cell 17 (2009) 861–73. 10.1016/j.devcel.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].McCulloch DR, Nelson CM, Dixon LJ, Silver DL, Wylie JD, Lindner V, Sasaki T, Cooley MA, Argraves WS, and Apte SS, ADAMTS metalloproteases generate active versican fragments that regulate interdigital web regression, Dev. Cell 17 (2009) 687–98. https://doi.org/S1534-5807(09)00390-6 [pii] 10.1016/j.devcel.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Girkontaite I, Frischholz S, Lammi P, Wagner K, Swoboda B, Aigner T, and Von der Mark K, Immunolocalization of type X collagen in normal fetal and adult osteoarthritic cartilage with monoclonal antibodies, Matrix Biol. 15 (1996) 231–8. [DOI] [PubMed] [Google Scholar]
- [61].Filippi-Chiela EC, Oliveira MM, Jurkovski B, Callegari-Jacques SM, da Silva VD, and Lenz G, Nuclear morphometric analysis (NMA): screening of senescence, apoptosis and nuclear irregularities, PloS one 7 (2012) e42522 10.1371/journal.pone.0042522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hubmacher D, Schneider M, Berardinelli SJ, Takeuchi H, Willard B, Reinhardt DP, Haltiwanger RS, and Apte SS , Unusual life cycle and impact on microfibril assembly of ADAMTS17, a secreted metalloprotease mutated in genetic eye disease, Sci Rep 7 (2017) 41871 10.1038/srep41871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Weinbaum JS, Broekelmann TJ, Pierce RA, Werneck CC, Segade F, Craft CS, Knutsen RH, and Mecham RP, Deficiency in microfibril-associated glycoprotein-1 leads to complex phenotypes in multiple organ systems, J. Biol. Chem 283(2008) 25533–43. https://doi.org/M709962200 [pii] 10.1074/jbc.M709962200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.