

Abstract
Pancreatic adenocarcinoma (PDAC) remains a formidable disease that needs improved therapeutic strategies. Even though immunotherapy has revolutionized treatment for various solid tumor types, it remains largely ineffective in treating individuals with PDAC. This review describes how the application of genome-wide analysis is revitalizing the field of PDAC immunotherapy. Major themes include new insights into the body’s immune response to the cancer, and key immunosuppressive elements that blunt that antitumor immunity. In particular, new evidence indicates that T cell-based antitumor immunity against PDAC is more common, and more easily generated, than previously thought. However, equally common are an array of cellular and molecular defenses employed by the tumor against those T cells. These discoveries have changed how current immunotherapies are deployed and have directed development of novel strategies to better treat this disease. Thus, the impact of genomic analysis has been two-fold: both in demonstrating the heterogeneity of immune targets and defenses in this disease, as well as providing a powerful tool for designing and identifying personalized therapies that exploit each tumor’s unique phenotype. Such personalized treatment combinations may be the key to developing successful immunotherapies for pancreatic adenocarcinoma.
Key words: Pancreatic adenocarcinoma, immunology, genomics, immunotherapy, neoantigen, T cell, CAR T cell, desmoplastic, vaccine, immunosuppression
Introduction
Pancreatic adenocarcinoma (PDAC) is one of the most lethal malignancies and is predicted to become the second leading cause of cancer-related deaths by 2030.1-3 Unfortunately, the vast majority of patients (>80%) present with advanced primary or metastatic disease that is unresectable.4-6 Currently, standard chemotherapy for PDAC remains either nab-paclitaxel plus gemcitabine7 or FOLFIRINOX (leucovorin, 5-fluorouracil, irinotecan and oxaliplatin) which improves the five-year survival rate a mere 8.5 to 11.1 months, respectively.8 Although patients with resectable disease typically have a better prognosis, current surgery and adjuvant chemotherapy regimens only confer a median survival of ~25 months.9 In addition, recent targeted therapy trials aimed at disrupting commonly overactive pathways in pancreatic cancers, such as KRAS and tyrosine kinase receptors, have also failed to improve survival over standard chemotherapy.10,11 While immune checkpoint modulators have demonstrated exciting efficacy in various solid tumors,12-15 these therapies have essentially failed in PDAC patients.16-19 Thus, despite the emergence of new therapies for PDAC, the five-year survival rate for patients with this cancer remains disappointingly low.20 To advance therapeutic options for PDAC patients, a greater understanding of the complex interplay between tumor genomics and the immune landscape of PDAC, including both antitumor immune responses and the suppressive defenses that protect the tumor, is needed. Recent investigations have resulted in the generation of novel immunotherapy strategies and highlight the benefit of using genomic studies to design personalized treatments that effectively address this devastating malignancy. This review highlights how genome-wide analysis is revitalizing the field of PDAC immunotherapy through improved understanding of the complex interaction between the host and PDAC.
Molecular subtypes of pancreatic adenocarcinoma
The vast majority of PDAC harbor a KRAS mutation at codon 1221,22 with the latest data suggesting a 93% prevalence of this mutation among PDAC tumors.23 Significant effort has focused on developing a successful anti-KRAS biologic therapy, yet no KRAS-targeted strategy has impacted clinical practice.24 Further investigation has revealed that this poor response is, in part, due to the tumor’s ability to acquire additional oncogenic mutations.25-27 While the diversity of these mutations initially made defining prognostic patterns difficult,28 application of whole genome and RNA/exome sequencing indicated that these mutations tend to occur in common pathways. Among the most common are RAS/MAPK, Hedgehog, TGFβ, Wnt/Notch, G1/S phase transition, and apoptosis regulation.29,30 Studies have now defined molecular subtypes of PDAC tumors with prognostic and therapeutic indications23,31-34 (Table 1).
Table 1.
Genetic subtypes.
| Collisson et al., 2011 | ||||
|---|---|---|---|---|
| Subtype | Genetic signature | Histology | Clinical implications | |
| Classical | Adhesion-associated and epithelial genes; GATA6hi | Highly differentiated | Sensitive to erlotinib | |
| QM-PDA* | Mesenchymal Genes | Poorly differentiated | Sensitive to gemcitabine | |
| Exocrine-like | Digestive enzyme genes | ELA3A+ and CFTR+ | - | |
| Moffitt et al., 2015 | ||||
| Subtype | Genetic Signature | Histology | Clinical implications | |
| Classical | GATA6hi and SMAD4hi | >10% mucin expression | 1 year survival of 70% | |
| Basal-like | Laminins and Keratins | <10% mucin expression | 1 year survival of 44% | |
| Stromal factors | Collisson's mesenchymal genes and stroma histology likely from CAF° cells not neoplastic cells | Activated stroma = worse prognosis for both classical and basal-like subtypes | ||
| Exocrine factors | Similar to normal exocrine pancreas, not considered a tumor subtype | |||
| Bailey et al., 2016 | ||||
| Subtype | Genetic signature | Histology | Clinical implications | COMPASS Trial |
| Pancreatic progenitor | high PDX1, MUC1 and MUC5AC expression | Includes mucinous non-cystic (colloid) and mucinous IPMN | Responsive to FOLFIRINOX | |
| Squamous | TP53, KDM6A, TP63 N mutations | Includes adeno-squamous carcinomas | Poor prognostic factor | Resistant to FOLFIRINOX |
| ADEX# | Endocrine and exocrine pancreas genes, subclass of pancreatic progenitor | Includes rare acinar cell carcinomas | ||
| Immunogenic | B and T cell genes, upregulation of CTLA4 and PD1 | Includes mucinous non-cystic (colloid) and mucinous IPMN | Potential responsiveness to immune modulators | |
| Cancer Genome Atlas Network, 2017 | ||||
| Subtype | Genetic signature | Histology | Clinical implications | |
| Classical/ pancreatic progenitor | GNAS mutations common; high EVADR, DEANR1, and GATA6-AS1 lncRNAs | |||
| Squamous/ basal-like | TP53 mutations common; high CAV1, low miR-192-5p and miR-194-5p | |||
| ADEX# | Genetic signature may be due to non-neoplastic infiltrate rather than unique neoplasm | Low neoplastic cellularity | ||
| Immunogenic | ||||
*Quasi-mesenchymal-pancreatic ductal adenocarcinoma
°cancer associated fibroblast
#aberrantly differentiated endocrine exocrine.
The first application of PDAC molecular subtypes was to delineate different sensitivities to chemotherapies. One early study found that classical subtype tumors responded more to erlotinib (an EGFR tyrosine kinase inhibitor) while the quasimesenchymal phenotype was more sensitive to gemcitabine.31 The COMPASS clinical trial recently used genome sequencing of PDAC biopsies to categorize tumors as either classical or basal-like prior to FOLFIRINOX therapy, thus highlighting the feasibility and predictive value of pre-therapy genetic analysis.32 This study also demonstrated that classical tumors were more responsive to FOLFIRINOX than basal-like tumors.35 Exploration of PDAC epigenetics has further refined these current subtypes (Table 1) and promises to identify additional therapeutic targets.23 Among these targets are histone acetyltransferases (HATs) and histone deacetylases (HDACs),36,37 and dysregulated/mutated non-coding regions of DNA.38-41
More recently, studies have correlated genetic patterns within the PDAC tumor with its susceptibility to immune attack. Mismatch Repair-Deficient (MMR-D) PDAC, like other MMR-D tumors, arise from mutations in post-replication machinery that allows for a high mutational burden and characteristic microsatellite instability.42,43 MMR-D cancers also demonstrate increased cytotoxic T cell tumor infiltration and higher tumor checkpoint protein expression.34,44 The high mutation rate and T cell infiltration in MMR-D tumors has been correlated with better prognosis45 and response to immunotherapy46 in other cancers. Studies indicate that MMR-D PDAC, particularly those which arise in patients with Lynch Syndrome, will respond to PD-1 blockade similar to other MMR-D solid tumors.47-49 In a recent study, 57% of patients with MMR-D PDAC tumors had objective response rates on PD-1 checkpoint blockade.49 Unfortunately, MMR-D PDAC represents less than 1% of all pancreatic adenocarcinomas.49 Furthermore, genetic screening is currently the only consistent method to detect MMR-D tumors since family history of inherited cancer is not necessarily predictive of risk.50 Of note, global genomic analysis can now identify tumors with high immune infiltrate and checkpoint protein expression (termed immunogenic PDAC) that could be targeted by checkpoint inhibitors or other immunotherapy.33 Further work is required to determine what percentage of tumors have this immunogenic signature and if they differ from tumors that arise from mismatch repair deficiency, particularly in their sensitivity to immunotherapies. Collectively, the identification of PDAC subtypes and their correlation with specific responses to both chemotherapy and immunotherapy is already shaping the development of more targeted and efficacious treatment regimens.
Beyond subtypes: new insights into pancreatic adenocarcinoma immunology
Unlike MMR-D PDAC, the majority of pancreatic adenocarcinoma tumors are immunologically cold, as defined by the lack of effector T cells in the tumor. Traditionally, this phenomenon is partially attributed to the low mutational load of PDAC that yields few neoantigens for T cells to recognize.51-53 However, new genetic evidence challenges this idea as a recent investigation that profiled tumor antigens from long-term pancreatic cancer survivors revealed that neither abundance of neoantigens nor the levels of CD8+ T cells in PDAC predicted survival.54 Instead, patients with the longest survival tended to have neoantigens which imitated microbial epitopes with stronger likelihood of TCR recognition.
Another study comparing PDAC and melanoma antigen load and T cell responses further challenged the concept that a high number of neoantigens is required for robust antitumor immunity. These investigators discovered that while the number of potential neoantigens in PDAC was an order of magnitude lower than melanoma, almost every PDAC tumor had a mutation that resulted in a predicted neoantigen.55 Yet, despite the lower number of potential neoantigens, T cell infiltration in PDAC was still similar to melanoma. The tumors differed in that the infiltrating T cells in PDAC were less cytotoxic (lower IFNg production) than in melanoma.55 The lower cytotoxicity of PDAC T cells needs to be further elucidated but is consistent with an exhausted phenotype. If true, this would explain why PD-1 blockade is not very effective in PDAC, since it only releases T cells from inhibition but doesn’t reverse their exhausted phenotype.56 A study that sequenced T cell receptors in PDAC tumors offered further corroboration that PDAC can be immunogenic. In this study, PDAC-infiltrating T cells had an elevated level of T cell receptor clonality,57 a marker of an antigen-specific adaptive immune response and favorable prognosis.58,59 Importantly, the clonality and ex vivo expansion rate of PDAC-derived T cells was similar to that found in melanoma biopsies, suggesting that they are equally capable of mounting a disease-specific immune response.57 Additionally, since PDAC tumors have low rates of mutagenesis, the likelihood that common mutations (such as KRAS codon 12 mutations55 or MUC1654) are shared by both the primary and metastatic tumors might improve chances of disease clearance.60
The pro-neoplastic pancreatic adenocarcinoma immune landscape
The dissonance between the seemingly robust T cell infiltration and lack of tumor clearance in PDAC may be explained, at least in part, by to the hostile immune landscape within the tumor. Efforts to understand PDAC at the molecular level have revealed key components to its immunosuppressive environment leading to immune evasion, suppression and exclusion.
PDAC tumors have a variety of protective mechanisms that help them to avoid immune detection (Figure 1A). In addition to yielding few neoantigens,51,52,55 PDAC tumors also downregulate MHC I molecules on their cell surface, thereby rendering them invisible to CD8+ T cells.61,62 Another mechanism to avoid immune recognition is through the release of pancreatic exosomes, which transfer miRNA to neighboring dendritic cells (DCs) decreasing MHC II expression.63 Thus, PDAC tumors conceal their already low antigen signal further by reducing the expression of MHC molecules on both the tumor and antigen presenting cells (APCs). In parallel, the anti-phagocytic molecule CD47 is highly expressed in PDAC tumors, sending a don’t eat me signal that prevents both recognition and phagocytosis by macrophages.64 CD47 blocking antibodies alleviate this inhibition, thus restoring tumor detection and clearance via phagocytosis65 (Figure 1A).
Figure 1.
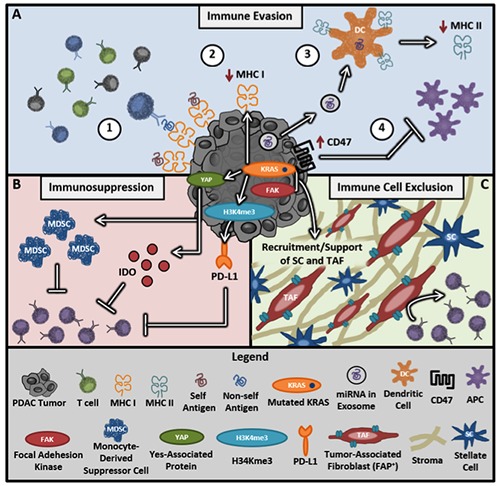
Key immunosuppressive mechanisms of PDAC. A) Immune evasion tactics include 1) low levels of mutation resulting in few non-self antigens, 2) KRAS signaling lowers MHC I expression by tumor cells, 3) exosomes containing miRNA silence dendritic cell (DC) expression of MHC II molecules, and 4) increased expression of anti-phagocytic molecules like CD47 prevent APC processing and tumor clearance. B) The tumor suppresses immune responses through PD-L1 expression, MDSC recruitment, and high IDO expression. C) The highly fibrotic extracellular matrix of the tumor creates a physical barrier preventing T cell infiltration into the tumor.
PDAC also suppresses the T cell anti-tumor response by creating a favorable tumor microenvironment through multiple strategies including the recruitment of suppressive cell subsets, production of enzymes/cytokines, and upregulation of immune checkpoint proteins (Figure 1B). Molecular drivers of immunosuppression include Yes-associated protein (YAP) which signals downstream from KRAS to recruit myeloid-derived suppressor cells (MDSCs) to inhibit adaptive immune responses.66,67 Similarly, cytokine signaling through KRAS and STAT3 drives the production of Indoleamine-2,3-dioxygenase (IDO) at the tumor site.68-70 IDO, an enzyme involved in tryptophan metabolism, drives immunosuppression and correlates with poor clinical outcomes in several malignancies such as breast,71 gastric,72 and liver73 cancer. A recent study reported that IDO is upregulated in 59% of PDAC tissues and that its elevation correlates with poor prognosis, poor tumor differentiation, and higher metastatic burden.74 Targeting of this pathway via the IDO inhibitor indoximod resulted in greater cytotoxic T cell infiltration and tumor reduction in mice,75 identifying IDO as a potential target for future combination therapies.
While monotherapy with PD-1 blockade has proven ineffective in PDAC, evidence suggests that PD-L1 still plays a role in the immunosuppressive landscape of PDAC. Specifically, PD-L1 expression has been reported to be upregulated by the epigenetic modifier H3K4me3 in pancreatic cancer76 and KRAS in lung cancers. 77,78 The fact that both H3K4me3 and KRAS are highly active in the majority of PDAC cancers supports the notion that PD-L1 may indeed be more commonly expressed among PDAC tumors than previously thought.76,79,80 High PD-L1 expression supports the use of checkpoint therapy for PDAC patients in a combinatorial strategy. Furthermore, the multitude of additional immunosuppressive signals in the PDAC microenvironment besides checkpoint proteins, including YAP, IDO and H3K4me3, asserts the benefit of genomic analysis to determine which signals are present in a tumor for optimal therapy design.
Another major barrier to successful anti-PDAC immune responses is the dense extracellular matrix (ECM). Fibrosis in PDAC is directly correlated with aggressiveness of the tumor,81 and this may be due, in part, to passive exclusion of T cells from the tumor.82 Multiple pathways are known to encourage the desmoplastic environment of PDAC and represent possible therapeutic targets. KRAS signaling drives PDAC fibrosis through the recruitment and activation of both tumor-associated fibroblasts (TAF) and stellate cells to further support PDAC progression83-85 (Figure 1C). However, crosstalk between the ECM and the immune microenvironment also plays a role in fibrotic development. For example, inflammatory signaling due to IL-1b, nitrous oxide, and IRAK4 signaling downstream of toll-like receptors are also implicated in fibrosis and resultant chemoresistance.86,87 Further studies that lend a better understanding of the relationship between the desmoplastic microenvironment and the antitumor immune response will be key to unleashing the full potential of PDAC-targeted immunotherapies.
Collectively, genotyping and histopathologic characterization of PDAC tumors has deepened our understanding of three key immunoresistant mechanisms: immune evasion, immunosuppression and immune cell exclusion (Figure 1). These findings lay the groundwork for novel therapies focused on either driving T cell responses to PDAC or subduing its immunosuppressive microenvironment.
Novel immunotherapies for generating T cell responses to pancreatic adenocarcinoma
The majority of immunotherapies for PDAC have focused primarily on inducing or enhancing tumor-specific T cell responses. These therapies fall into three major categories, including: increasing antigen-specific responses (via vaccination), improving T cell recognition of tumor-associated proteins (via T cell receptor selection or design), and targeting the immunosuppressive tumor microenvironment.
Vaccines produced early evidence that an antitumor immune response could be generated against common, previously nonimmunogenic PDAC tumor antigens. As a proof of concept, an early study using the GVAX vaccine (irradiated allogeneic PDAC cells that express the immunostimulatory cytokine GM-CSF88) revealed a correlation between clinical response and an increase in antigen-specific CD8+ T cells.89 In subsequent preclinical and clinical trials, the GVAX vaccine has been reported to induce cytotoxic T cell infiltration and increased PD-L1 expression in the tumor.79,80,90 In mouse models, the addition of PD-1 checkpoint blockade following GVAX vaccination increased survival by 38% and curative responses by 25.5%.80 These data suggest that combination therapy of vaccination and PD-L1 inhibition could have a synergistic effect to enhance clinical responses. Several trials have been or are currently ongoing based on a combinatorial strategy with GVAX (NCT02243371, NCT03161379, NCT03190265, NCT02451982, NCT03767582). Additionally, vaccination using peptide-pulsed, patient-derived DCs given concurrently with the (TLR)-3 agonist poly-ICLC in a phase 1 trial was well tolerated with a median overall survival of 7.7 months.91
A better understanding of specific genetic alterations in PDAC such as unique post-translational modifications has expanded vaccine strategies. A classic example is MUC1, which is aberrantly glycosylated in a wide number of cancers including PDAC.92 A phase 1 clinical trial in which advanced pancreatic cancer patients were vaccinated with dendritic cells carrying the abnormal MUC1 antigen demonstrated an immunological response in two of seven patients. In these patients, there was significantly increased IFNγ and granzyme B production, but the treatment did not slow disease progression in any of the seven.93 Another protein that is uniquely modified by tumors (including PDAC) is ɑ-enolase, which is often citrullinated during overactive autophagy. In preclinical work, vaccinating with citrullinated ɑ-enolase peptides induced a strong proinflammatory Th1 response with survival benefits in mice with melanoma, lung, and pancreatic cancer.94
Antigenicity can also be induced through synthetic modification of peptides. As an example, a recent study synthetically modified pancreatic lysates with the ɑ-gal epitope from non-primate mammals to take advantage of naturally occurring anti-Gal antibodies specific for that epitope. This vaccination strategy increased not only anti-Gal antibody responses, but also expanded lymphocytes specific for MUC1 and mesothelin antigens.95 Overall, the expanding experience with vaccination therapies has demonstrated that this approach may boost T cell responses against PDAC tumors and serve as a compelling partner in immunotherapy combination strategies (Figure 2A).
Figure 2.
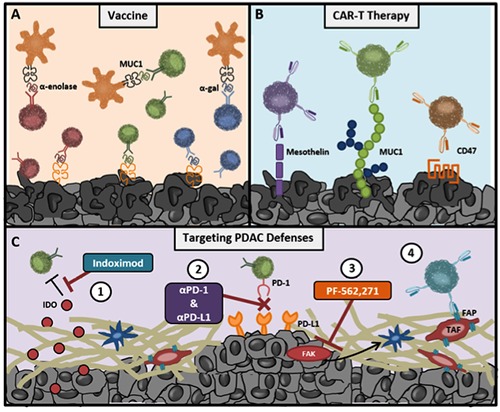
Advances in immunotherapy informed by genomic analysis of PDAC tumors. A) Vaccine strategies can take advantage of either natural (Muc1 and ɑ-enolase) or man-made (ɑ-gal) post-translational modifications of self-proteins. B) PDAC tumors express high levels of aberrant surface proteins, which can be targeted by T cells with chimeric antigen receptors (CARs). C) Both immunosuppressive (1&2) and desmoplastic (3&4) features of the PDAC immune environment are being targeted by novel immunotherapies.
Adoptive cell therapy with either naturally arising tumor-infiltrating lymphocytes (TILs) or with T cells genetically redirected with antigen receptors (TCRs or CARs, respectively) produce some of the highest response rates and odds of complete responses in patients with various solid tumors and leukemias.96-103 Although TIL/TCR therapy can target any non-self antigen from either surface or intracellular peptides, this strategy is dependent on MHC presentation, which, as previously discussed (Figure 1A), is often downregulated in PDAC tumors. CAR T cells were designed, in part, to address these limitations. CARs are comprised of an antigen- specific scFv antibody linked to the CD3ζ TCR signaling domain as well as costimulatory domains (such as CD28 or 4-1BB). Because CAR T cells are antibody based, this strategy does not depend on antigen presentation by MHC molecules, but rather recognizes the unprocessed protein on the cell surface. An additional benefit of CAR T cells is the potential to turn a wide array of non-immunogenic, or even immunosuppressive, proteins into reliable targets for an antitumor response (Figure 2B).
CAR T cell therapy was recently FDA approved to treat patients with B cell malignancies based on initial evidence that up to ~89% of these patients have a complete response from their disease. 104,105 CAR T cell therapy has also been developed for PDAC with early notable success. In a recent preclinical study, investigators isolated two highly specific TCRs against KRAS codon 12 mutations G12V and G12D from mice immunized with human PDAC cells. Infusion of human T cells redirected with these TCRs resulted in slowed tumor growth in xenograft mouse models.106 Multiple additional CAR T cells have been developed against highly expressed molecules on the surface of PDAC cells, including: mesothelin,107 MUC1,108 and the prostate cancer antigen PSCA.109A phase 1 clinical trial reported that infusion of mesothelin- targeted CAR T cells led to tumor debulking in three of six PDAC patients as noted by decreased metabolically active tumor on PET-CT110 (NCT01897415). Additional phase 1 clinical trials targeting mesothelin (lentiviral transduced; NCT03323944) and PSCA (NCT02744287) are also currently underway. CAR T cells targeting CD47 have also shown promise in early preclinical studies. Interestingly, this strategy transforms a basic cloaking defense of the tumor into a reliably expressed target antigen. In a xenograft mouse model, CD47-specific CAR T cells limited growth of PDAC tumors by greater than 50%.111 These novel therapies underscore that our growing understanding of tumor genomics can not only guide molecular therapies, but can also suggest targets for future cellular therapies.
Novel immunotherapies for disabling the immunosuppressive pancreatic adenocarcinoma microenvironment
Growing evidence suggests that in order for patients to fully benefit from T cell enhancing immunotherapies, the immunosuppressive microenvironment of their tumors will also need to be targeted. Major advancements have been made in therapies that target the immunosuppressive defenses of PDAC. For example, intratumoral delivery of nanoparticles loaded with the immune-activating chemotherapy oxaliplatin and the IDO inhibitor indoximod resulted in the decrease of FoxP3+ regulatory T cells, increased recruitment and expansion of effector T cells, and enhanced tumor regression. 75 Another study combined GVAX with IDO inhibition, which resulted in enhanced T cell infiltration and function and increased survival from ~40% to ~90%.112
In addition to IDO and checkpoint inhibitors, alternative methods of breaking down the immunosuppressive defenses of PDAC using chemotherapeutic agents have been reported. Combination of gemcitabine with a CD40 agonist results in a T cell-dependent regression of subcutaneous murine PDAC tumors.113 The depletion of extratumoral macrophages in these mice increased T cell infiltration into spontaneous tumors, demonstrating a potential future target for enhancing T cell-mediated PDAC regression. CD40 agonists have also improved penetration of chemotherapy into the tumor through macrophage-dependent depletion of tumor stroma.114 A recent phase 1 study determined that the combination of CD40 agonists and gemcitabine was well tolerated and capable of producing a therapeutic response in four out of 22 PDAC patients115 (NCT00711191). A new phase 1 trial testing the combination of CD40 agonist and gemcitabine/Nab-Paclitaxel with or without Nivolumab is currently ongoing (NCT03214250).
Reducing tumor fibrosis is another powerful strategy to induce tumor sensitivity to immunotherapy. For example, blocking focal adhesion kinase (FAK) decreased stromal activity and fibrosis, halting overall growth and metastasis of the tumor.116 Combining checkpoint inhibition with FAK inhibition doubled survival time in mice whose tumors were previously unresponsive to checkpoint blockade.117 In another study, depletion of fibroblast activation protein (FAP)-expressing fibroblasts in the tumor rendered pancreatic tumors sensitive to checkpoint therapy.118 To this end, CAR T cells have also been developed against the FAP+ fibroblasts, which are a key component of PDAC tumors.119 Blockade of cholecystokinin receptor signaling also reduces tumor growth and fibrosis. Similar to FAK inhibition, it enhances survival outcomes of mice treated with checkpoint inhibition.120 These examples indicate that inhibition of the tumor stroma may represent a critical strategy for sensitizing PDAC tumors to immunotherapies (Figure 2C).
While preclinical work has identified a multitude of single therapies that successfully generate immunity to PDAC, these agents in isolation are largely ineffective when translated to patients. The efficacy of the combinatorial approach vs. monotherapy has borne out in a couple of recent clinical trials121,122 (UMIN000005248, UMIN000000769). Many of the other studies discussed in this review have also indicated the power of multimodal immunotherapies, and the majority of current NIH clinical trials in PDAC immunotherapy are examining a combinatorial approach to treating this disease (Table 2). Furthermore, the success of combinations of single agents that on their own were not effective in PDAC also advocates for revisiting therapies previously determined obsolete. For example, as single agents, KRAS inhibitors have failed to make a marked improvement in patient survival despite the near universality of KRAS mutation in PDAC. However, KRAS inhibitors may prove powerful additions to T cell activating immunotherapies as KRAS is now known to be central to driving the immunosuppressive microenvironment (Figure 1).
Table 2.
Clinical trials.
| Category | Interventions | Clinical trial ID |
|---|---|---|
| anti-CTLA-4 | Ipilimumab (anti-CTLA-4) + Gemcitabine | NCT01473940 |
| anti-CTLA-4/ anti-PD- | Tremelimumab (anti-CTLA-4) + Durvalumab (anti-PD-L1) | NCT02527434 |
| Ipilimumab (anti-CTLA-4) or Nivolumab (anti-PD-1) + VX15/2503 (anti-SEMA4D antibody) | NCT03373188 | |
| Niraparib (PARP inhibitor) + Nivolumab (anti-PD-1) or Ipilimumab (anti-CTLA-4) | NCT03404960 | |
| anti-PD-1/ PD-L1 | Pembrolizumab (anti-PD-1) + ACP-196 | NCT02362048 |
| Pembrolizumab (anti-PD-1) + Vaccinia virus (p53 vector) | NCT02432963 | |
| Nivolumab (anti-PD-1) + Cabiralizumab (anti-CSF-1R) | NCT02526017 | |
| Pembrolizumab (anti-PD-1) + AMG820 (anti-CSF-1R) | NCT02713529 | |
| Durvalumab (anti-PD-L1) + Galunisertib (TGFbeta inhibitor) | NCT02734160 | |
| Nivolumab (anti-PD-1) + Paricalcitol (Vitamin D analog) + Chemotherapies | NCT02754726 | |
| Pembrolizumab (anti-PD-1) + BL-8040 (CXCR4 antagonist) | NCT02826486 | |
| Pembrolizumab (anti-PD-1) + BL-8040 (CXCR4 antagonist) | NCT02907099 | |
| Nivolumab (anti-PD-1) + IRE | NCT03080974 | |
| Pembrolizumab (anti-PD-1) + XL888 (hsp90 inhibitor) | NCT03095781 | |
| Nivolumab (anti-PD-1) + Daratumumab (anti-CD38) | NCT03098550 | |
| Pembrolizumab (anti-PD-1) + Olaptesed (anti-CXCL12) | NCT03168139 | |
| Atezalizumab (anti-PD-L1) + BL-8040 (CXCR4 antagonist) + RO6874281 (anti-FAP) + Chemotherapies | NCT03193190 | |
| Nivolumab (anti-PD-1)+ APX005M (CD40 agonist) + Gemcitabine and Nab-Paclitaxel | NCT03214250 | |
| Durvalumab (anti-PD-L1) + Stereoactive Ablative Body Radiotherapy | NCT03245541 | |
| Nivolumab (anti-PD-1) + Entinostat (HDAC inhibitor) | NCT03250273 | |
| Durvalumab (anti-PD-L1) + Guadecitabine (DNA methyltransferase inhibitor) | NCT03257761 | |
| Pembrolizumab (anti-PD-1) + Paricalcitol (Vitamin D analog) | NCT03331562 | |
| Durvalumab (anti-PD-L1) + Radiation Therapy | NCT03490760 | |
| Nivolumab (anti-PD-1) + BMS-813160 (CCR2/CCR5 antagonist) + chemotherapies | NCT03496662 | |
| Oleclumab (anti-CD73) + Durvalumab (anti-PD-L1) + chemotherapies | NCT03611556 | |
| Pembrolizumab (anti-PD-1) + PEGPH20 (pegylated hyaluronidase) | NCT03634332 | |
| Pembrolizumab (anti-PD-1) + Pelareorep (Oncolytic Reovirus) | NCT03723915 | |
| Pembrolizumab (anti-PD-1) + Defactinib (FAK inhibitor) | NCT03727880 | |
| anti-PD-1/ vaccine | GVAX Vaccine (With CY) and CRS-207 Vaccine + Nivolumab (anti-PD-1) | NCT02243371 |
| Nivolumab (anti-PD-1) + GVAX Vaccine + Urelumab (anti-CD137) + Cyclophosphamide | NCT02451982 | |
| Peptide Vaccine + Pembrolizumab (anti-PD-1) | NCT02600949 | |
| GVAX Vaccine + Pembrolizumab (anti-PD-1) + Cyclophosphamide + SBRT | NCT02648282 | |
| Epacadostat (anti-IDO)+ Pembrolizmuab (anti-PD-1) + GVAX vaccine + CRS-207 vaccine | NCT03006302 | |
| Pembrolizumab (anti-PD-1), GVAX vaccine, IMC-CS4 (anti-CSF-1R) | NCT03153410 | |
| Nivolumab (anti-PD-1) + GVAX vaccine + SBRT + Cyclophosphamide | NCT03161379 | |
| GVAX Vaccine (With CY) and CRS-207 Vaccine + Nivolumab (anti-PD-1) + Ipilimumab (anti-CTLA4) | NCT03190265 | |
| Durvalumab (anti-PD-L1) + anti-CEA- & MUC1-vaccine | NCT03376659 | |
| GVAX Vaccine + Nivolumab (anti-PD-1) and BMS-813160 (CCR2/CCR5 antagonist) | NCT03767582 | |
| Vaccine | Allogenic GM-CSF plasmid- transfected tumor cell vaccine | NCT00389610 |
| Falimarev (anti-CEA vaccine) + Inalimarev (anti-CEA and MUC1 vaccine) + Sargramostim (recombinant GM-CSF) | NCT00669734 | |
| GVAX vaccine + Cyclophosphamide | NCT00727441 | |
| GVAX vaccine + Cyclophosphamide + SBRT + FOLFIRINOX | NCT01595321 | |
| NPC-1C vaccine + Gemcitabine + Nab-Paclitaxel | NCT01834235 | |
| Tumor derived gp96 vaccine | NCT02133079 | |
| anti-DC1 vaccine + Interferon alpha-2b + Rintatolimod (immunomodulatory RNA drug) | NCT02151448 | |
| ETBX-011 Vaccine + ALT-803 (IL-15 complex) | NCT03127098 | |
| ETBX-011 Vaccine + ALT-803 (IL-15 complex) | NCT03329248 | |
| MDC3/8 (dendritic cell KRAS vaccine) | NCT03592888 | |
| CAR-T cell therapy | Meso CAR T Cells | NCT01897415 |
| anti-CEA CAR-T cells + Sir-Spheres | NCT02416466 | |
| BPX-601 (anti PSCA CAR-T cells) + Rimiducid | NCT02744287 | |
| anti-CEA CAR-T cells | NCT02850536 | |
| antiCLD18 CAR-T cell | NCT03159819 | |
| anti-Meso CAR-T cells | NCT03323944 | |
| anti-Meso CAR-T cell vs anti-CD19 CAR-T cells + Cyclophosphamide | NCT03497819 | |
| Other T cell therapy | FOLFOX6 + Anti-Erbitux Activated T cells | NCT01420874 |
| Bispecific Antibody T cells + Aldesleukin (recombinant IL-2) + Sargramostim (recombinant GM-CSF) + chemotherapies | NCT02620865 | |
| Gemcitabine with CD8+NKG2D+ AKT cells | NCT02929797 | |
| Oncolytic virus | VCN-01 (Oncolytic Adenovirus) | NCT02045602 |
| LOAd703 (Oncolytic Adenovirus) | NCT03225989 | |
| CAdVEC Oncolytic Adenovirus | NCT03740256 | |
| Other | CD40 agonist monocolonal antibody + Gemcitabine | NCT00711191 |
| Indoximod (IDO inhibitor) + Nab-Paclitaxel + Gemcitabine | NCT02077881 | |
| TG01 injection + Gemcitabine | NCT02261714 | |
| CCX872-B (CCR2 antagonist) | NCT02345408 | |
| ALT-803 (IL-15 complex) + Gemcitabine + Nab-paclitaxel | NCT02559674 | |
| Ibrutinib (BTK inhibitor) + paclitaxel and gemcitiabine | NCT02562898 | |
| Ibrutinib (BTK inhibitor) | NCT02575300 | |
| Plerixafor (CXCR4 antagonist) | NCT03277209 | |
| CDX-1140 (anti-CD40) + CDX-301 (anti-flt3L) | NCT03329950 |
SBRT, stereotactic body radiation therapy; IRE, irreversible electroporesis; CSF-1R, colony-stimulating factor-1 receptor; GM-CSF, granulocyte-macrophage colony-stimulating factor.
Conclusions
Early genomic studies were used in PDAC to define mechanisms of tumorigenesis and chemoresistance. The application of genome-wide analysis to PDAC immunology has revealed that PDAC patients either possess effective tumor-antigen specific T cells for the cancer or have the capacity to generate an anti-tumor T cell response. However, we have also learned that PDAC can mount a robust immunosuppressive response at both the cellular and molecular level. Thus, to enhance the success of PDAC immunotherapy, treatment strategies will need to not only elicit immune responses, but also overwhelm the tumor’s defenses which counter them. In addition, the diversity of potential tumor antigens and defense mechanisms discovered in PDAC tumors yields a vast array of possible tumor phenotypes, each one with its own sensitivities and resistance to treatment. Genomic analysis can determine these key factors in an individual’s cancer and assist with guiding future therapy strategies.123 Pre-treatment genomic assessment of PDAC has already proven feasible35 and has the power to inform personalized immunotherapies strategies (Figure 3).
Figure 3.
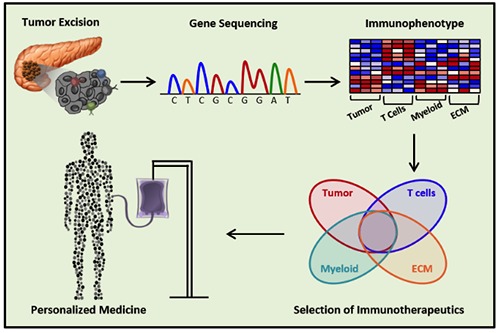
Developing personalized therapies for patients with pancreatic cancer. Sequencing of excised tumor or biopsies may one day allow physicians to categorize a patient’s cancer into therapeutic groups based on expression of tumor and immunologic markers. This will allow for the intelligent selection and design of combination immunotherapies for more successful treatment.
References
- 1.Simard EP, Ward EM, Siegel R, Jemal A. Cancers with increasing incidence trends in the United States: 1999 through 2008. CA Cancer J Clin 2012;62:118-28. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74:2913-21. [DOI] [PubMed] [Google Scholar]
- 3.Worni M, Guller U, White RR, et al. Modest improvement in overall survival for patients with metastatic pancreatic cancer: a trend analysis using the surveillance, epidemiology, and end results registry from 1988 to 2008. Pancreas 2013;42:1157-63. [DOI] [PubMed] [Google Scholar]
- 4.Vincent A, Herman J, Schulick R, et al. Pancreatic cancer. Lancet 2011;378:607-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Griffin JF, Poruk KE, Wolfgang CL. Pancreatic cancer surgery: past, present, and future. Chinese J Cancer Res = Chung-kuo yen cheng yen chiu 2015;27:332-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McDowell BD, Chapman CG, Smith BJ, et al. Pancreatectomy predicts improved survival for pancreatic adenocarcinoma: results of an instrumental variable analysis. Ann of Surg 2015;261:740-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817-25. [DOI] [PubMed] [Google Scholar]
- 9.Neoptolemos JP, Palmer DH, Ghaneh P, et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet 2017;389:1011-24. [DOI] [PubMed] [Google Scholar]
- 10.Van Cutsem E, van de Velde H, Karasek P, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol 2004;22:1430-8. [DOI] [PubMed] [Google Scholar]
- 11.Middleton G, Palmer DH, Greenhalf W, et al. Vandetanib plus gemcitabine versus placebo plus gemcitabine in locally advanced or metastatic pancreatic carcinoma (ViP): a prospective, randomised, double-blind, multicentre phase 2 trial. Lancet Oncol 2017;18:486-99. [DOI] [PubMed] [Google Scholar]
- 12.Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single- agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 2010;28:3167-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 2015;373:23-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fyfe G, Fisher RI, Rosenberg SA, et al. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J Clin Oncol 1995;13:688-96. [DOI] [PubMed] [Google Scholar]
- 15.Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J Sci Am 2000;6:S11-4. [PubMed] [Google Scholar]
- 16.Royal RE, Levy C, Turner K, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother (Hagerstown, Md: 1997). 2010;33:828-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aglietta M, Barone C, Sawyer MB, et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann Oncol 2014;25:1750-5. [DOI] [PubMed] [Google Scholar]
- 19.Feng M, Xiong G, Cao Z, et al. PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett 2017;407:57-65. [DOI] [PubMed] [Google Scholar]
- 20.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7-30. [DOI] [PubMed] [Google Scholar]
- 21.Almoguera C, Shibata D, Forrester K, et al. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988;53:549-54. [DOI] [PubMed] [Google Scholar]
- 22.Smit VT, Boot AJ, Smits AM, et al. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucl Acids Res 1988;16:7773-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The Cancer Genome Atlas Research Network. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017;32:185-203.e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bournet B, Buscail C, Muscari F, et al. Targeting KRAS for diagnosis, prognosis, and treatment of pancreatic cancer: hopes and realities. Eur J Cancer (Oxford, England: 1990) 2016;54:75-83. [DOI] [PubMed] [Google Scholar]
- 25.Delpu Y, Hanoun N, Lulka H, et al. Genetic and epigenetic alterations in pancreatic carcinogenesis. Curr Genom 2011;12:15-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang H, Daniluk J, Liu Y, et al. Oncogenic K-Ras requires activation for enhanced activity. Oncogene 2014;33:532-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mueller S, Engleitner T, Maresch R, et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 2018;554:62-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dal Molin M, Zhang M, de Wilde RF, et al. Very long-term survival following resection for pancreatic cancer is not explained by commonly mutated genes: results of wholeexome sequencing analysis. Clin Cancer Res 2015;21:1944-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science (New York, NY). 2008;321:1801-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012;491:399-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collisson EA, Sadanandam A, Olson P, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nature Med 2011;17:500-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moffitt RA, Marayati R, Flate EL, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nature Genet 2015;47:1168-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531:47-52. [DOI] [PubMed] [Google Scholar]
- 34.Connor AA, Denroche RE, Jang GH, et al. Association of distinct mutational signatures with correlates of increased immune activity in pancreatic ductal adenocarcinoma. JAMA Oncol 2017;3:774-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aung KL, Fischer SE, Denroche RE, et al. Genomics-driven precision medicine for advanced pancreatic cancer: early results from the COMPASS trial. Clin Cancer Res 2017. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nones K, Waddell N, Song S, et al. Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2 and MET signaling. Int J Cancer 2014;135:1110-8. [DOI] [PubMed] [Google Scholar]
- 37.Lu C, Yang D, Sabbatini ME, et al. Contrasting roles of H3K4me3 and H3K9me3 in regulation of apoptosis and gemcitabine resistance in human pancreatic cancer cells. BMC Cancer 2018;18:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hao S, Yao L, Huang J, et al. Genome-wide analysis identified a number of dysregulated long noncoding RNA (lncRNA) in human pancreatic ductal adenocarcinoma. Technol Cancer Res Treat 2018;17:1533034617748429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, Li M, Wang H, et al. Profiling of 95 microRNAs in pancreatic cancer cell lines and surgical specimens by realtime PCR analysis. World J Surg 2009;33:698-709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rachagani S, Kumar S, Batra SK. MicroRNA in pancreatic cancer: pathological, diagnostic and therapeutic implications. Cancer Lett 2010;292:8-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feigin ME, Garvin T, Bailey P, et al. Recurrent noncoding regulatory mutations in pancreatic ductal adenocarcinoma. Nature Genet 2017;49:825-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woerner SM, Tosti E, Yuan YP, et al. Detection of coding microsatellite frameshift mutations in DNA mismatch repairdeficient mouse intestinal tumors. Mol Carcinogen 2015;54:1376-86. [DOI] [PubMed] [Google Scholar]
- 43.Germano G, Lamba S, Rospo G, et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017;552:116-20. [DOI] [PubMed] [Google Scholar]
- 44.Wang L, Tsutsumi S, Kawaguchi T, et al. Whole-exome sequencing of human pancreatic cancers and characterization of genomic instability caused by MLH1 haploinsufficiency and complete deficiency. Genome Res 2012;22:208-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nature Rev Clin Oncol 2010;7:153-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (New York, NY) 2015;348:124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (New York, NY) 2017;357:409-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu ZI, Shia J, Stadler ZK, et al. Evaluating mismatch repair deficiency in pancreatic adenocarcinoma: challenges and recommendations. Clin Cancer Res 2018. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu C, Hart SN, Polley EC, et al. Association between inherited germline mutations in cancer predisposition genes and risk of pancreatic cancer. JAMA 2018;319:2401-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang J, Wolfgang CL, Zheng L. Precision immuno-oncology: prospects of individualized immunotherapy for pancreatic cancer. Cancers 2018;10:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kieler M, Unseld M, Bianconi D, Prager G. Challenges and perspectives for immunotherapy in adenocarcinoma of the pancreas: the cancer immunity cycle. Pancreas 2018;47:142-57. [DOI] [PubMed] [Google Scholar]
- 53.Samstein RM, Lee CH, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nature Genet 2019;51:202-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Balachandran VP, Luksza M, Zhao JN, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017;551:512-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bailey P, Chang DK, Forget MA, et al. Exploiting the neoantigen landscape for immunotherapy of pancreatic ductal adenocarcinoma. Sci Rep 2016;6:35848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pauken KE, Sammons MA, Odorizzi PM, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science (New York, NY). 2016;354:1160-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Poschke I, Faryna M, Bergmann F, et al. Identification of a tumor-reactive T-cell repertoire in the immune infiltrate of patients with resectable pancreatic ductal adenocarcinoma. Oncoimmunology 2016;5:e1240859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ikeda Y, Kiyotani K, Yew PY, et al. Clinical significance of T cell clonality and expression levels of immune-related genes in endometrial cancer. Oncol Rep 2017;37:2603-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McGranahan N, Furness AJ, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science (New York, NY). 2016;351:1463-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seliger B, Harders C, Wollscheid U, et al. Suppression of MHC class I antigens in oncogenic transformants: association with decreased recognition by cytotoxic T lymphocytes. Exper Hematol 1996;24:1275-9. [PubMed] [Google Scholar]
- 62.Atkins D, Breuckmann A, Schmahl GE, et al. MHC class I antigen processing pathway defects, ras mutations and disease stage in colorectal carcinoma. Int J Cancer 2004;109:265-73. [DOI] [PubMed] [Google Scholar]
- 63.Ding G, Zhou L, Qian Y, et al. Pancreatic cancer-derived exosomes transfer miRNAs to dendritic cells and inhibit RFXAP expression via miR-212-3p. Oncotarget 2015;6:29877-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cioffi M, Trabulo S, Hidalgo M, et al. Inhibition of CD47 effectively targets pancreatic cancer stem cells via dual mechanisms. Clin Cancer Res 2015;21:2325-37. [DOI] [PubMed] [Google Scholar]
- 65.Michaels AD, Newhook TE, Adair SJ, et al. CD47 blockade as an adjuvant immunotherapy for resectable pancreatic cancer. Clin Cancer Res 2018;24:1415-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Murakami S, Shahbazian D, Surana R, et al. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene 2017;36:1232-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stromnes IM, Brockenbrough JS, Izeradjene K, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014;63:1769-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu J, Du W, Yan F, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol (Baltimore, Md: 1950) 2013;190:3783-97. [DOI] [PubMed] [Google Scholar]
- 69.Litzenburger UM, Opitz CA, Sahm F, et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget 2014;5:1038-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang Y, Wang Y, Wu G, et al. Prolonged skin grafts survival time by IFN-gamma in allogeneic skin transplantation model during acute rejection through IFN-gamma/STAT3/IDO pathway in epidermal layer. Biochem Biophys Res Commun 2018;496:436-42. [DOI] [PubMed] [Google Scholar]
- 71.Bi WW, Zhang WH, Yin GH, et al. Analysis of indoleamine 2-3 dioxygenase (IDO) and EGFR co-expression in breast cancer tissue by immunohistochemistry. Asian Pacific APJCP 2014;15:5535-8. [DOI] [PubMed] [Google Scholar]
- 72.Liu H, Shen Z, Wang Z, et al. Increased expression of IDO associates with poor postoperative clinical outcome of patients with gastric adenocarcinoma. Sci Rep 2016;6:21319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pan K, Wang H, Chen MS, et al. Expression and prognosis role of indoleamine 2,3-dioxygenase in hepatocellular carcinoma. J Cancer Res Clin Oncol 2008;134:1247-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang T, Tan XL, Xu Y, et al. Expression and prognostic value of indoleamine 2,3-dioxygenase in pancreatic cancer. Chinese Med J 2017;130:710-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lu J, Liu X, Liao YP, et al. Nano-enabled pancreas cancer immunotherapy using immunogenic cell death and reversing immunosuppression. Nature Commun 2017;8:1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lu C, Paschall AV, Shi H, et al. The MLL1-H3K4me3 Axis- Mediated PD-L1 Expression and pancreatic cancer immune evasion. J Natl Cancer Inst 2017;109:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Herter-Sprie GS, Koyama S, Korideck H, et al. Synergy of radiotherapy and PD-1 blockade in Kras-mutant lung cancer. JCI Insight 2016;1:e87415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen N, Fang W, Lin Z, et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol Immun 2017;66:1175-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lutz ER, Wu AA, Bigelow E, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res 2014;2:616-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Soares KC, Rucki AA, Wu AA, et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother (Hagerstown, Md: 1997) 2015;38:1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kadaba R, Birke H, Wang J, et al. Imbalance of desmoplastic stromal cell numbers drives aggressive cancer processes. J Pathol 2013;230:107-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.von Bernstorff W, Voss M, Freichel S, et al. Systemic and local immunosuppression in pancreatic cancer patients. Clinical Cancer Res 2001;7:925s-32s. [PubMed] [Google Scholar]
- 83.Hwang RF, Moore T, Arumugam T, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res 2008;68:918-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Erkan M, Adler G, Apte MV, et al. StellaTUM: current consensus and discussion on pancreatic stellate cell research. Gut 2012;61:172-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zeitouni D, Pylayeva-Gupta Y, Der CJ, Bryant KL. KRAS mutant pancreatic cancer: no lone path to an effective treatment. Cancers 2016;8:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Muerkoster S, Wegehenkel K, Arlt A, et al. Tumor stroma interactions induce chemoresistance in pancreatic ductal carcinoma cells involving increased secretion and paracrine effects of nitric oxide and interleukin-1beta. Cancer Res 2004;64:1331-7. [DOI] [PubMed] [Google Scholar]
- 87.Zhang D, Li L, Jiang H, et al. Tumor-stroma IL1beta-IRAK4 feedforward circuitry drives tumor fibrosis, chemoresistance, and poor prognosis in pancreatic cancer. Cancer Res 2018;78:1700-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jaffee EM, Hruban RH, Biedrzycki B, et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J Clin Oncol 2001;19:145-56. [DOI] [PubMed] [Google Scholar]
- 89.Thomas AM, Santarsiero LM, Lutz ER, et al. Mesothelin-specific CD8(+) T cell responses provide evidence of in vivo cross-priming by antigen-presenting cells in vaccinated pancreatic cancer patients. J Exper Med 2004;200:297-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Le DT, Lutz E, Uram JN, et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother (Hagerstown, Md: 1997) 2013;36:382-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mehrotra S, Britten CD, Chin S, et al. Vaccination with poly(IC:LC) and peptide-pulsed autologous dendritic cells in patients with pancreatic cancer. J Hematol Oncol 2017;10:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sorensen AL, Reis CA, Tarp MA, et al. Chemoenzymatically synthesized multimeric Tn/STn MUC1 glycopeptides elicit cancer-specific anti-MUC1 antibody responses and override tolerance. Glycobiology 2006;16:96-107. [DOI] [PubMed] [Google Scholar]
- 93.Rong Y, Qin X, Jin D, et al. A phase I pilot trial of MUC1-peptide- pulsed dendritic cells in the treatment of advanced pancreatic cancer. Clin Exper Med 2012;12:173-80. [DOI] [PubMed] [Google Scholar]
- 94.Cook K, Daniels I, Symonds P, et al. Citrullinated alpha-enolase is an effective target for anti-cancer immunity. Oncoimmunol 2018;7:e1390642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Furukawa K, Tanemura M, Miyoshi E, et al. A practical approach to pancreatic cancer immunotherapy using resected tumor lysate vaccines processed to express alpha-gal epitopes. PLoS One 2017;12:e0184901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 2011;17:4550-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 2011;3:95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dudley ME, Gross CA, Somerville RP, et al. Randomized selection design trial evaluating CD8+-enriched versus unselected tumor-infiltrating lymphocytes for adoptive cell therapy for patients with melanoma. J Clin Oncol 2013;31:2152-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371:1507-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Garfall AL, Maus MV, Hwang WT, et al. Chimeric antigen receptor T Cells against CD19 for multiple myeloma. N Engl J Med 2015;373:1040-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Goff SL, Dudley ME, Citrin DE, et al. Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J Clin Oncol 2016;34:2389-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.June CH. Adoptive T cell therapy for cancer in the clinic. J Clin Invest 2007;117:1466-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Knochelmann HM, Smith AS, Dwyer CJ, et al. CAR T cells in solid tumors: blueprints for building effective therapies. Front Immunol 2018;9:1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 2015;33:540-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gardner RA, Finney O, Annesley C, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017;129:3322-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang QJ, Yu Z, Griffith K, et al. Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol Res 2016;4:204-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stromnes IM, Schmitt TM, Hulbert A, et al. T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Cancer Cell 2015;28:638-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Posey AD, Schwab RD, Boesteanu AC, et al. Engineered CAR T cells targeting the cancer-associated Tn-Glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity 2016;44:1444-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Abate-Daga D, Lagisetty KH, Tran E, et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum Gene Ther 2014;25:1003-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Beatty GL, O’Hara MH, Lacey SF, et al. Activity of mesothelin- specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterol 2018;155:29-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Golubovskaya V, Berahovich R, Zhou H, et al. CD47-CAR-T cells effectively kill target cancer cells and block pancreatic tumor growth. Cancers 2017;9:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Blair AB, Kleponis J, Thomas Ii DL, et al. IDO1 inhibition potentiates vaccine-induced immunity against pancreatic adenocarcinoma. J Clin Investig 2019. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Beatty GL, Winograd R, Evans RA, et al. Exclusion of T cells from pancreatic carcinomas in mice is regulated by Ly6C(low) F4/80(+) extratumoral macrophages. Gastroenterol 2015;149:201-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science (New York, NY). 2011;331:1612-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Beatty GL, Torigian DA, Chiorean EG, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res 2013;19:6286-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Stokes JB, Adair SJ, Slack-Davis JK, et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther 2011;10:2135-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jiang H, Hegde S, Knolhoff BL, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nature Med 2016;22:851-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A 2013;110:20212-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lo A, Wang LC, Scholler J, et al. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res 2015;75:2800-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Smith JP, Wang S, Nadella S, et al. Cholecystokinin receptor antagonist alters pancreatic cancer microenvironment and increases efficacy of immune checkpoint antibody therapy in mice. Cancer Immunol Immunother 2018;67:195-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nishida S, Ishikawa T, Egawa S, et al. Combination gemcitabine and WT1 peptide vaccination improves progressionfree survival in advanced pancreatic ductal adenocarcinoma: a phase II randomized study. Cancer Immunol Res 2018. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 122.Hirooka Y, Kawashima H, Ohno E, et al. Comprehensive immunotherapy combined with intratumoral injection of zoledronate- pulsed dendritic cells, intravenous adoptive activated T lymphocyte and gemcitabine in unresectable locally advanced pancreatic carcinoma: a phase I/II trial. Oncotarget 2018;9:2838-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Knudsen ES, Vail P, Balaji U, et al. Stratification of pancreatic ductal adenocarcinoma: combinatorial genetic, stromal, and immunologic markers. Clin Cancer Res 2017;23:4429-40. [DOI] [PMC free article] [PubMed] [Google Scholar]