

Abstract
Myoblast fusion is critical for muscle growth, regeneration, and repair. We previously reported that the enzyme peptidyl‐prolyl cis–trans isomerase NIMA interacting 1 (Pin1) is involved in osteoclast fusion. The objective of this study was to investigate the possibility that Pin1 also inhibits myoblast fusion. Here, we show the increased number of nuclei in the Pin1+/− mice muscle fiber compared to that in wild‐type mice. Moreover, we show that low dose of the Pin1 inhibitor dipentamethylene thiuram monosulfide treatment caused enhanced fusion in C2C12 cells. The R‐Smads are well‐known mediators of muscle hypertrophy and hyperplasia as well as being substrates of Pin1. We found that Pin1 is crucial for maintaining the stability of Smad3 (homologues of the Drosophila protein, mothers against decapentaplegic (Mad) and the Caenorhabditis elegans protein Sma). Our results show that serine 204 within Smad3 is the key Pin1‐binding site during inhibition of myoblast fusion and that both the transforming growth factor‐β receptor and extracellular signal‐regulated kinase (ERK)‐mediated phosphorylation are required for the interaction of Pin1 with Smad3. These findings suggest that a precise level of Pin1 activity is essential for regulating myoblast fusion during myogenesis and muscle regeneration.
Keywords: cell fusion, ERK, myoblast, peptidyl‐prolyl cis–trans isomerase NIMA interacting 1, Smad3, TGF superfamily
1. INTRODUCTION
The main cellular components of skeletal muscle are multinucleated muscle cells, which are formed by the fusion of mononucleated myoblasts (Abmayr & Pavlath, 2012). A variety of extracellular, cell‐surface, and intracellular molecules finely coordinate the cellular and molecular events of mammalian myoblast fusion (Jansen & Pavlath, 2008). Myoblast fusion is a prerequisite for normal development and repair of damaged muscle fibers in adult life (Charge & Rudnicki, 2004). Therefore, myoblast fusion is very important for muscle mass maintenance and treatment of skeletal muscle atrophies during normal aging and under pathological conditions such as cancer, disuse after fracture, diabetes, and AIDS (Lecker, Goldberg, & Mitch, 2006). Either defects in the fusion of nascent satellite cells or lack of balance in muscle protein levels can lead to muscle atrophy. Thus, the treatment of muscle atrophy might require reversal of these processes (Hochreiter‐Hufford et al., 2013; Millay et al., 2013).
Myostatin, a member of the transforming growth factor‐β (TGF‐β) superfamily, has garnered a lot of attention as a negative regulator of skeletal muscle mass and a potential target of drugs to enhance muscle growth and function in patients with muscle loss (Clop et al., 2006; McPherron & Lee, 1997; McPherron, Lawler, & Lee, 1997; Schuelke et al., 2004). The TGF‐β superfamily directly activates the Smad signaling pathway, in addition to other Smad‐independent pathways, thus potently, yet reversibly, suppressing myoblast differentiation and fusion (Liu, Black, & Derynck, 2001). TGF‐β and myostatin induce the phosphorylation of receptor‐activated Smads (R‐Smads), Smad2 and Smad3. As adults advance in age, their muscles begin to produce excessive TGF‐β (but not myostatin), which upregulates pSmad3 and reduces the regenerative capacity of the muscle that is mainly mediated by fusion of satellite cells (Carlson, Hsu, & Conboy, 2008).
Peptidyl‐prolyl cis–trans isomerase NIMA interacting 1 (Pin1) is a highly conserved peptidyl‐prolyl isomerase (PPI) with high binding specificity. Pin1 catalyzes the isomerization of the peptide bond between a phosphorylated serine or threonine residue and a following proline residue (pSer–Thr‐Pro motif) in substrate proteins (Kamato et al., 2013). Proline residues in molecules assist in the adoption of the cis or trans conformation, often with a wide array of biological consequences, depending on configuration. Pin1 has been implicated in several disease states (Islam, Yoon, & Ryoo, 2016). The linker region of Smad3 was found to have multiple binding sites for Pin1, with different consequences depending on cell type (Matsuura et al., 2010; Nakano et al., 2009; Shen et al., 2012).
In cardiac muscle, the Pin1 protein seems to operate within a limited range, as both upregulation and downregulation of Pin1 attenuate cardiac hypertrophy (Toko et al., 2013). Apart from a few molecular experiments on osteoclasts (Islam et al., 2014), no studies have examined the role of this critical regulatory molecule in postmitotic cells in a physiological context. Pin1 was found to be involved in osteoclast fusion (Islam et al., 2014), a physiological process of syncytia formation similar to myoblast fusion. Considering the central role of proline‐directed Ser–Thr kinases in the control of skeletal muscle differentiation, coupled with emerging evidence that the enzymatic activity of Pin1 promotes the regulatory postphosphorylation of proteins at sites of proline‐dependent phosphorylation, we evaluated whether Pin1 also modulates myoblast fusion and influences the pathways regulating muscle mass.
2. MATERIALS AND METHODS
2.1. Animals and diets
Pin1+/− mice on a mixed genetic background (Fujimori et al., 1999) were bred to yield Pin1+/+, Pin1+/−, and Pin1−/− on a C57Bl6 background. Male Pin1+/− and littermate control Pin1+/+ mice on a C57Bl6 genetic background were analyzed at 10 weeks of age. All mice were housed in a mouse facility with a 12 hr light–dark cycle in a temperature‐controlled room. The animal care and study protocols were approved by the Animal Care Committee of Seoul National University (protocol SNU‐120327‐6‐3).
2.2. Cell culture
C2C12 (acquired from ATCC) myoblasts were grown and differentiated for 2–5 days in a differentiation medium consisting of Dulbecco’s modified eagle medium (DMEM) supplemented with 2% fetal bovine serum (FBS) and 1% antibiotics. Cells within passage eight were used for all experimentation.
2.3. Plasmids
Flag‐tagged CS2 Smad3‐WT, CS2 Smad3 T179V, CS2 Smad3 S204A, CS2 Smad3 S208A, and CS2 Smad3 S213A (Kretzschmar, Doody, Timokhina, & Massague, 1999) were gifts from Joan Massague (Addgene plasmid).
2.4. Cytotoxicity assay
The cell cytotoxicity was measured using the EZ‐Cytox cell viability assay kit (Daeillab Service, Seoul, Korea). Briefly, 100 μl of C2C12 cell suspension (3,000 cells per well) was added to each well of a 96‐well plate. After the required incubation with the Pin1 inhibitor in fusogenic media (DMEM with 2% FBS) for 48 hr, 10 μl of the EZ‐Cytox solution was added to each well of the plate and incubated at 37°C for 2–4 hr. The absorbance was measured by spectrophotometry at 450 nm.
2.5. Fluorescence microscopy and quantitative analysis of fusion
For the measurement of fusion, myoblasts were plated in growth medium on cover slips inside six‐well plates at 1.5 × 105 cells per well (C2C12). Myoblasts were induced to undergo myogenic differentiation as described above. Myoblasts or myotubes were washed and fixed with 4% PFA in phosphate‐buffered saline (PBS) for 10 min at room temperature and permeabilized in 0.1% Triton X‐100 in PBS for 3 min at room temperature. After being blocked with 2% bovine serum albumin (BSA)–PBS, the cells were stained with the antimyosin antibody (Ab) (Clone MF20, Cat #14‐6503‐82; Affymetrix Ebioscience, Vienna, Austria) at a 1:100 dilution overnight at 4 °C. After being washed with PBS, the cells were incubated with the Cy3‐labeled secondary Ab (1:400) for 1 hr at room temperature, stained with 4′,6‐diamidino‐2‐phenylindole dihydrochloride (DAPI, 1 µg/ml; Invitrogen, Carlsbad, CA) for 2 min at room temperature, washed, and mounted with the Vectashield mounting solution. Microscopy was performed using LSM700 and LSM800 (Carl Zeiss, Oberkochen, Germany) confocal laser scanning microscopes and ZEN Blue software for analysis.
For the quantitation of in vitro myoblast fusion, the fusion index was determined as described previously (Islam et al., 2014; Pajcini, Pomerantz, Alkan, Doyonnas, & Blau, 2008; Park et al., 2016; Sun et al., 2010). Briefly, fluorescent images of six random fields were captured per sample for each experiment, yielding a total of 18 images. ImageJ (National Institutes of Health, Bethesda, Maryland) cell counter plugin software was used to determine the number of nuclei. The fusion index (the percentage of nuclei within myosin‐positive cells versus the total number of nuclei) was calculated for each field. Fusion indices from each field for each sample were compiled and are depicted as the mean ± SD. In addition, the number of myosin‐positive myotubes per field with 2–4 nuclei (small myotubes) or 5 or more nuclei (large myotubes) was recorded as another means to quantify myoblast fusion.
2.6. Quantitative real‐time PCR (qPCR)
Total RNA was extracted from either mouse tissue or cultured cells with TRIZOL (Invitrogen), and cDNA was synthesized with Superscript III reverse transcriptase and random hexamer primers (Invitrogen). Gene expression was assessed by standard qPCR approaches with Power SYBR Green (Takara). The primers used are listed in Supporting Information Table 1. We used a 7,500 Fast Real‐Time PCR machine (Applied Biosystems). Expression levels were normalized to the glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) level and represented as fold‐change.
2.7. Histology and immunohistochemistry
Muscles were mounted at a resting length, covered in OCT compound, frozen in liquid nitrogen‐cooled isopentane, and stored at −80°C until cryosectioning at 7 μm. For immunostaining, muscle sections were fixed in 4% paraformaldehyde (PFA), and epitope retrieval was performed with sodium citrate (10 mM, pH 6.5) at 60°C for 20 min. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide in PBS, followed by an additional blocking step with the Mouse‐on‐Mouse Blocking Reagent (Vector Laboratories, Burlingame, CA). Sections were costained with DAPI (10 nM; Invitrogen) and mounted with Vectashield fluorescent mounting media. For myonuclear counting, fresh‐frozen muscle cross‐sections were immunoreacted with dystrophin Ab, followed by Cy3‐conjugated secondary Ab or Alexa 488 to delineate the muscle fibers. Sections were postfixed in 4% paraformaldehyde and then stained with DAPI for visualization of nuclei.
Skeletal muscles or limbs were dissected, and the tissues were fixed in 10% neutral buffered formalin and processed for routine paraffin histology. Frozen and paraffin‐embedded sections were cut and stained with haemotoxylin and eosin using routine procedures. Immunohistochemistry was performed by fixation with 1% PFA–PBS; permeabilization with 0.2% Triton X‐100 in PBS; blocking with PBS/1% BSA, 1% heat‐inactivated rabbit serum, and 0.025% Tween 20; incubation with the primary Ab for at least 2 hr; incubation with secondary Alexa‐Fluor antibodies (Invitrogen) for 1 hr; and mounting with Vectashield‐containing DAPI (°C).
2.8. Subcellular fractionation and western blot analysis
To fractionate C2C12 cells into cytosolic and membrane fractions, we first washed a 10‐cm dish with cold PBS and lysed the cells by homogenization in hypotonic buffer (10 mM Tris pH 8.0, 1 mM ethylenediaminetetraacetic acid [EDTA]) supplemented with 1 mM phenylmethane sulfonyl fluoride (PMSF) and a protease inhibitor mix. The homogenate was centrifuged at 15,000 revolutions per minute (RPM) for 5 min to pellet nuclei and cell debris. The supernatant was centrifuged at 12,000 RPM for 20 min to pellet membrane structures. The supernatant from this step was the cytosolic fraction, and the membrane fraction was solubilized in an equal volume of hypotonic buffer + 1% n‐dodecyl β‐d‐maltoside (DDM; Sigma‐Aldrich, St. Louis, United States) for further analyses by immunoblotting.
For analysis of whole‐cell extracts, Radioimmunoprecipitation assay buffer (RIPA buffer) (10 mM Tris–Cl [pH 8.0], 1 mM EDTA, 0.5 mM egtazic acid (EGTA), 1% Triton X‐100, 0.1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 140 mM NaCl) supplemented with 1 mM PMSF and a protease inhibitor mix was used for cell lysate preparation.
For immunoblotting, equal protein amounts were separated by 12% SDS–polyacrylamide gel electrophoresis, transferred to a PVDF membrane (Millipore, MA), blocked in 5% milk in TBS‐tween, and incubated with primary antibodies.
2.9. Glutathione S‐transferase (GST) pull‐down assay and coimmunoprecipitation
For the GST pull‐down assay, cells were lysed with a single‐detergent lysis buffer (50 mm Tris–HCl, pH 8; 150 mm NaCl; 1% Triton X‐100) supplemented with a complete protease inhibitor mixture (Roche Applied Science, Basel, Switzerland) and phosphatase inhibitors (sodium orthovanadate and sodium fluoride). Equal amounts of each protein extract were incubated for 2 hr at 4°C with bacterially purified GST fusion proteins bound to agarose‐glutathione beads (Sigma‐Aldrich). After three washings, the 2 × Laemmli loading buffer was added to the beads and proteins were analyzed by Western blot analysis.
2.10. Study design and statistics
Studies were conducted in both blinded and nonblinded fashions. For all studies, the sample sizes are explicitly stated in the figure legends. Normal distribution was assessed first, and if the data were normally distributed, most differences were analyzed by Student’s t test or analysis of variance (using excel or Graphpad Prism software, CA). The results were considered significant at p < 0.05.
3. RESULTS
3.1. Pin1 regulates skeletal muscle fusion
According to previous reports, Pin1−/− mice exhibit neurodegenerative features in some brain regions in an age‐dependent manner but appear relatively normal until they are 3 months old (Fujimori, Takahashi, Uchida, & Uchida, 1999; Liou et al., 2002, 2003). We have not found visible differences in muscle bulk among Pin1+/+, Pin1+/−, and Pin1−/− mice before they are 1 month old. After 3 months, Pin1−/− mice gradually become smaller than Pin1+/+ and Pin1+/− mice (Liou et al., 2002). To the naked eye, Pin1+/− mice appear more muscular than their littermates from 8 weeks onward. Muscle bulk is at its optimum level at 10 weeks age. At this age, the onset of neurodegeneration might have already happened in the Pin1−/− mice. Pin1+/− mice have not been reported to have any neuropathology. Mouse embryonic fibroblasts (MEFs) from Pin1+/+ and Pin1+/− mice at passage 3 grow at similar rates, and Pin1+/− mice have been reported to be similar to normal mice (Fujimori et al., 1999).
Muscle biology is intimately related to neurobiology; to avoid confusion, we selected the Pin1+/− mice as the genetic model with which to analyze the effects of Pin1 on muscle. To identify myonuclei, we used dystrophin staining to label the fiber margins and determined whether each nucleus had its geometric center inside the dystrophin‐stained “ring” (Bruusgaard et al., 2012; McCarthy et al., 2011). In cross‐sections of muscle from 10‐week‐old Pin1+/− mice, there were more nuclei inside the myofibers when the cell surface area was defined with dystrophin immunostaining (Figure 1a). The ratio of myonuclei to fiber cross‐sectional area (CSA) was 1.067 ± 0.00693 myonuclei–fiber CSA in Pin1+/+ cross‐sections and 2.144 ± 0.0848 myonuclei–fiber CSA in Pin1+/− (Figure 1b). As the nuclei in each fiber represent the number of cells that fused to form that muscle fiber, we inferred that loss of Pin1 enhanced the fusion of myoblasts in vivo.
Figure 1.
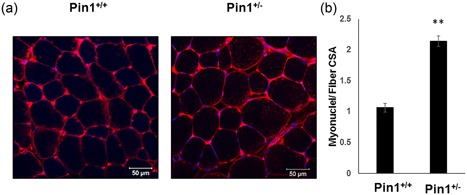
Effect of peptidyl‐prolyl cis–trans isomerase NIMA interacting 1 (Pin1) expression level on the number of myonuclei determined from cross‐sections and single fibers. (a) More myoneuclei observed inside each muscle fiber cross‐section demarcated by dystrophin staining from Pin1+/− mice compared to those in muscle sections from Pin1+/+. The representative muscle section from the tibialis anterior (TA) muscle is shown stained with dystrophin (red) and counterstained with 4′,6‐diamidino‐2‐phenylindole dihydrochloride (DAPI; blue). Scale bar= 50 μm. (b) Graph showing an increase in myoneuclei number inside each fiber. Number of myonuclei in each cross‐sectional area (CSA) from the TA muscle cross sections are shown in the graph, where the y‐axis shows the myonuclei number–fiber CSA and the x‐axis shows the mice genotype. Data are means ± SD. The data was calculated from four fields each from three mice of each genotype. p < 0.05 compared with control. Scale bar= 50 μm each [Color figure can be viewed at wileyonlinelibrary.com]
3.2. Loss of Pin1 enhances muscle fusion in vitro
As the identification of myonuclei on cross‐sections can sometimes be unreliable, we analyzed muscle fusion in vitro with a more controlled experimental design. The C2C12 embryonic skeletal muscle cell line is a well‐defined model for in vitro skeletal muscle differentiation. These cells proliferate as myoblasts at high calf‐serum concentrations (10–15%). When the cells reach confluence, they can be induced to differentiate if the calf‐serum concentration is reduced from 10% to 2% or if the medium is switched to contain 5% horse serum (Yaffe & Saxel, 1977).
Previously, it was observed that Pin1 undergoes differential subcellular relocalization from the nucleus to the cytoplasm during the early phases of myogenic differentiation (Magli et al., 2010b). We differentiated C2C12 cells in the presence of different doses (0.001–10 µm) of the Pin1 inhibitor dipentamethylene thiuram monosulfide (DTM; Tatara, Lin, Bamba, Mori, & Uchida, 2009; Figure 2a). To quantitate in vitro myoblast fusion, we determined the fusion index as described previously. Minimal inhibition of Pin1 with 0.01 μM DTM most significantly enhanced fusion, whereas excessive inhibition (with 10 μM DTM) did not enhance fusion (Figure 2b,c), although it did very slightly increase cytotoxicity (Supporting Information Figure S1a). Inhibition of Pin1 with the most effective dose of DTM (0.01 μM) also increased the myofiber width (Figure 2d) and length (Figure 2e), indicating that Pin1 has a role in hyperplasia and fusion.
Figure 2.
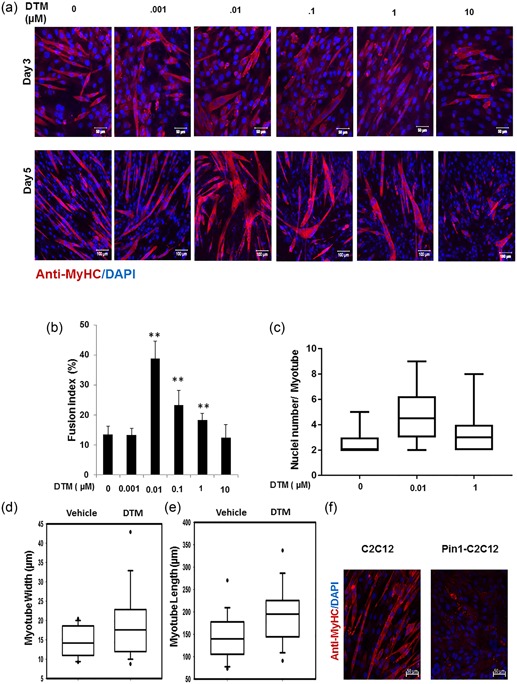
Loss of peptidyl‐prolyl cis–trans isomerase NIMA interacting 1 (Pin1) enhances muscle fusion in vitro. (a) Low dose of the Pin1 inhibitor dipentamethylene thiuram monosulfide (DTM; 0.01 μM) treatment caused more enhanced fusion than higher dosage (1 μM) in C2C12 cells. C2C12 cells were differentiated for 5 days with different dosage of Pin1 inhibitor DTM ranging from 0.001 to 10 μM. Cells were then immunostained for myosin heavy chain (MyHC) with anti‐MyHC antibody and Cy3‐conjugated secondary antimouse antibody. Scale bar= 50 μM in Day 3 images and scale bar= 100 μM in Day 5 images. (b) DTM treatment at 0.01 μM concentration shows highest fusion index. Graph showing the fusion index calculated by dividing the total number of nuclei in multinucleated cells (which have more than two nuclei) in each field by the total number of nuclei and making percentage from cells treated with DTM or without DTM (**p < 0.001). DTM treatment at 0.01 μM concentration shows the highest fusion index. (c) Nuclei number in each myotube (x‐axis in the graph) of cells treated with different dosages of DTM. Statistics was calculated with GraphPad Prism software, and p < 0.01. (d) Myotube width and (e) length shown graphically in a box plot made with data derived from DTM (0.01 μM)‐treated C2C12 cells after 3 days of differentiation. Pin1 inhibition increases both the width and length of myotubes. (f) Overexpression of Pin1 inhibiting myotube formation. Pin1‐overexpressing stable C2C12 cells were allowed to differentiate for 3 days in DMEM containing 2% FBS and were immunostained with antibody against MyHC and secondary anti‐mouse antibody that was conjugated with Cy3 [Color figure can be viewed at wileyonlinelibrary.com]
C2C12 cells stably overexpressing Pin1 displayed no signs of fusion until the third day of differentiation (Figure 2f) and very little afterward.
We also examined the expression of differentiation markers. Myogenin (MyoG) is an essential basic‐helix–loop‐helix myogenic transcription factor involved in skeletal muscle development in vivo. Pin1 inhibition did not induce the expression of Myog transcripts in the initial stages of differentiation and only slightly increased their expression by Day 3 (Supporting Information Figure S1b). These data indicate that the muscle phenotype might be regulated by Pin1 with a certain threshold limit and that Pin1 limits excessive fusion.
3.3. Smad3 level in cells of muscle origin regulated by Pin1
The R‐Smad proteins are thought to compose one of the main groups of transcription factors during myoblast differentiation and fusion. The balance between muscle atrophy and hypertrophy also depends on the preferential recruitment of Smad4 to the myostatin pathway versus the bone morphogenetic protein (BMP) pathway (Sartori et al., 2013). Previously, we identified Pin1 as a critical molecular switch in the determination of Smad1 fate (Yoon et al., 2015). Because R‐Smads 1, 2, and 3 are well‐known targets of Pin1 (Yang et al., 2014; Yoon et al., 2015), we opted to evaluate their expression and phosphorylation status in C2C12 cells. With minimal Pin1 inhibition, only p‐Smad3 expression was reduced (Figure 3a and Supporting Information Figure S2), whereas phosphorylated Smad2 and Smad1 levels did not change significantly. Despite their extensive sequence similarity and functional overlaps, Smad2 and Smad3 play distinct roles in the regulation of certain genes (Petersen et al., 2010) and might be differentially regulated (Brown, Pietenpol, & Moses, 2007), perhaps even in muscle. Thus, we focused exclusively on Smad3 from this point onward. In cells overexpressing Pin1, the transcriptionally active tail‐phosphorylated Smad3 was more highly detected inside nuclei than the cytoplasm (Figure 3b).
Figure 3.
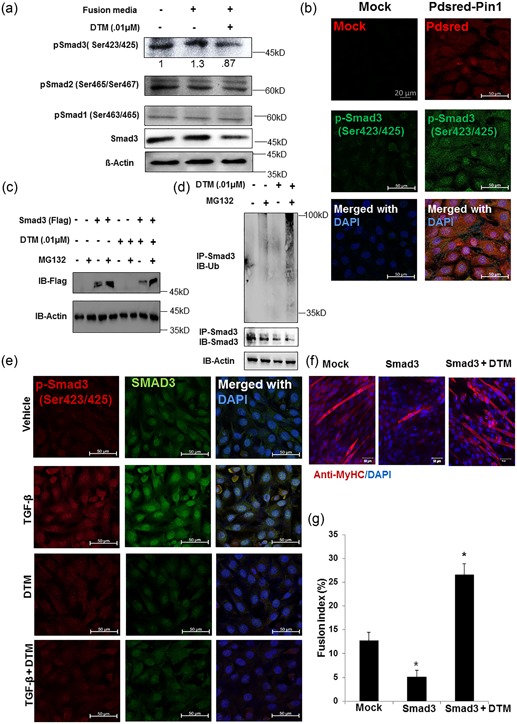
Smad3 level in muscle is regulated by peptidyl‐prolyl cis–trans isomerase NIMA interacting 1 (Pin1). (a) Phosphorylated Smad 3 was reduced with low‐dose dipentamethylene thiuram monosulfide (DTM) (0.01 μM) treatment in the fusion medium. Western blot analysis was done to observe C‐terminal tail‐phosphorylated R‐Smad protein (pSmad3, pSmad2, and pSmad1) levels in the C2C12 cell treated with DTM (0.01 μM) for 24 hr in fusogenic media. β‐Actin was used as loading control. The average pSmad3 band intensity after normalization is shown below the band inside the figure. Statistics of the densitometry data was calculated using GraphPad Prism software. p< .05 as determined by One way analysis of variance. (b) Pin1 overexpression increases the amount of transcriptionally active phosphorylated Smad3. Expression levels of tail‐phosphorylated Smad3 (active) in C2C12 cells transfected with PDsRed‐tagged Pin1 were observed. Cells were harvested 24 hr posttransfection. The expression of pSmad 3 was detected with the Phospho‐Smad3 (Ser423/425; C25A9) Rabbit monoclonal antibody and Alexa 488‐conjugated (green) secondary antibody (Scale bar= 20 μM). (c) Inhibition of Pin1 increased proteasomal degradation of Smad3. The flag‐tagged Smad3 protein level was observed with western blot analysis after 4 hr of 50 μM MG132 (proteasome inhibitor) treatment with and without DTM. Cells were transfected transiently with CS2 Flag‐Smad3 and treated with or without DTM for 24 hr post transfection. (d) DTM treatment increased the ubiquitination of Smad3 protein in C2C12 cells. Ubiquitination assay to observe ubiquitin binding to the endogenous Smad3 protein with DTM treatment. C2C12 cells were treated with 50 μM MG132 for 4 hr before harvest. (e) Total pSmad3 and tail‐phosphorylated Smad3 levels after 1 hr of induction of TGF‐β treatment with and without DTM treatment. Nuclear pSmad3 and total Smad3 levels are reduced with the DTM treatment even after induction of TGF‐β signaling. DTM was treated for 2 hr considering the half‐life of this small‐molecule inhibitor and TGF‐β for 1 hr in DMEM with 5% FBS. Scale bar = 20 μM. (f) CS2 Flag‐Smad3 was overexpressed in C2C12 cells and allowed to differentiate for 5 days with one group being treated with DTM 48 hr before harvest. Scale bar= 50 μM. (g) Fusion index calculated as the percentage of the number of nuclei in multinucleated cells among total nuclei. *p < 0.05 compared with Control [Color figure can be viewed at wileyonlinelibrary.com]
To determine whether the degradation of Smad3 in cells treated with the Pin1 inhibitor was mediated by the proteasome, we cultured C2C12 cells in the presence of proteasome inhibitor Mg132 and Pin1 inhibitor DTM. Upon proteasomal inhibition, Smad3 was not degraded, even in the presence of DTM (Figure 3c). Inhibition of Pin1 also increased the polyubiquitination of Smad3 (Figure 3d). The total Smad3 level and tail‐phosphorylated Smad3 level were reduced with the DTM treatment, even in the presence of TGF‐β (Figure 3e and Supporting Information Figure S2). The immunostaining experiment indicates that the interaction between Pin1 and Smad3 occurs more in the cytosol (Figure S3).
Similar to previous studies, we also found a reduced fusion index in C2C12 cells overexpressing Smad3; however, 48 hr after differentiation, the DTM treatment for an additional 48 hr reversed this and caused myotube formation (Figure 3f), as indicated by the fusion index (Figure 3g). These data demonstrate that Smad3 expression is regulated by Pin1 in myoblasts in a posttranslational manner. Pin1 stabilizes Smad3 and protects it from proteasomal degradation, whereas inhibition of Pin1 rescues fusion in Smad3‐overexpressing cells.
3.4. Pin1 functions downstream of myostatin and TGF‐β‐mediated TGF signaling in muscle
Myostatin is the best‐known cytokine in the muscle hypertrophy pathway (Welle, 2009), and Smad3 is one of the main transcriptional regulators of this pathway. Because TGF‐β is released in aged myotubes (Carlson et al., 2008), we concentrated on the role of Pin1 in the TGF‐β pathway. We differentiated C2C12 cells in the presence of TGF‐β and the Pin1 inhibitor DTM. The inhibition of Pin1 induced multicellular myotube formation in TGF‐β‐treated cells (Figure 4a) and restored the fusion index to a nearly normal level (Figure 4b). The inhibition of Pin1 also restored myotube formation in C2C12 cells treated with myostatin (Figure 4c). It is evident that the inhibition of Pin1 can rescue fusion in TGF‐β‐treated and myostatin‐treated differentiating C2C12 cells.
Figure 4.
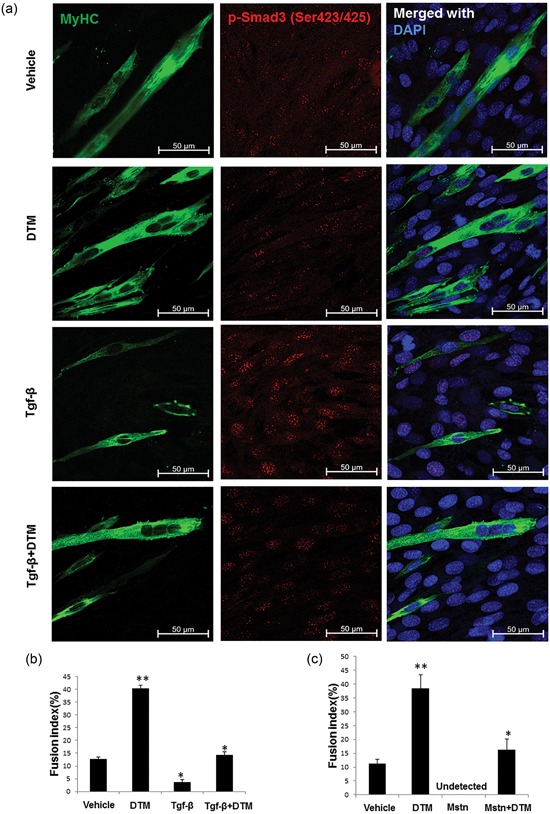
Peptidyl‐prolyl cis–trans isomerase NIMA interacting 1 (Pin1) function on muscle is downstream of myostatin and transforming growth factor‐β (TGF‐β)–mediated TGF signaling. (a) Pin1 inhibition rescuing TGF‐β‐induced suppression of fusion. C2C12 cells were differentiated for 3 days in the presence of TGF‐β with and without dipentamethylene thiuram monosulfide (DTM) in fusogenic media containing 2% DMEM. The cells were then immunostained with the anti–myosin heavy chain antibody and anti‐Smad3 antibody. Scale bar= 50 μM. (b) Fusion index calculation for Pin1‐overexpressing cells treated in media containing TGF‐β with or without Pin1 inhibition by DTM. (c) Graph showing fusion index calculations for images captured from cells treated with DTM and myostatin (10 ng/ml) for 3 days in a differentiating medium (*p < 0.05, **p < 0.001) [Color figure can be viewed at wileyonlinelibrary.com]
3.5. Expression of muscle atrophy–related genes downstream of Smad3 regulated by Pin1
We deduced from our data that Pin1 modulates Smad3 expression. Smad3 also regulates muscle bulk by altering the expression of atrogenes (muscle atrophy–related genes; Bollinger, Witczak, Houmard, & Brault, 2014; Goodman, McNally, Hoffmann, & Hornberger, 2013). For instance, Smad3 is needed to upregulate the E3‐ubiquitin ligase F‐box only protein 32 (also known as atrogin‐1, Fbxo32, and MAFbx), a well‐known skeletal muscle atrogene (Trendelenburg et al., 2009; Waning et al., 2015; Welle, 2009). Atrogin‐1 and Trim63 (MuRF1) are upregulated during muscle atrophy and tend to degrade myofibrillar elements, MyoD, and components of the muscle hypertrophy pathway and translational machinery (Lecker et al., 2004).
We found that both atrogin‐1 and Murf‐1 mRNA levels were reduced when Pin1 was inhibited in vitro, even in the presence of TGF‐β (Figure 5a). Murf‐1 (Figure 5b) and Maf (Figure S4) expressions were also reduced in the Pin1+/− tibialis anterior muscle. Atrogin‐1 was detected in Pin1+/+ samples but was undetectable in Pin1+/− samples (Figure 5b). Musa‐1, a newly identified denervation atrophy–related gene (Sartori et al., 2013), was upregulated in Pin1+/− samples (Figure S4), probably due to the downregulation of Smad1/5 resulting from the reduced Pin1 level (Yoon et al., 2015). These observations indicate that Pin1 regulates atrogenes that are downstream of Smad3 and suggest that the inhibition of Pin1 and subsequent degradation of Smad3 promote hypertrophy not only in terms of the number of nuclei but also in terms of the total muscle fiber content. Indeed, we confirmed that Pin1+/− muscle had more fibers with larger CSAs than Pin1+/+ muscle (Figure 1).
Figure 5.

Expression of muscle atrophy–related genes downstream of Smad3 is regulated by peptidyl‐prolyl cis–trans isomerase NIMA interacting 1 (Pin1). (a) Dipentamethylene thiuram monosulfide (DTM) treatment suppressing the mRNA expression of atrogin‐1 and MuRF‐1 even in the presence of transforming growth factor‐β (TGF‐β) signaling. C2C12 cells treated with DTM and TGF‐β in myogenic media for 12 hr were harvested, and RNA was extracted from them. Quantitative real‐time PCR (qPCR) was performed for Atrogin‐1 and MuRF‐1 (*p < .05, **p < .001). (b) Pin1+/− muscle also shows reduced expression of Atrogin‐1 and MuRF‐1 at 10 weeks of age. Pin1+/+ and Pin1+/− tibialis anterior muscles were homogenized, and mRNA was extracted from them. qPCR was performed for atrogenes, Atrogin‐1 and MuRF‐1 (*p < 0.05)
3.6. Binding of Pin1 to S204 might have a role in myoblast fusion
Phosphorylation of the Smad linker region is considered to be a centerpiece in the function, regulation, and connectivity of Smad transcription factors. The TGF‐β signal transduction machinery is regulated by a combination of Smad phosphorylation and degradation processes (Massague, 2012). What determines the use of one process over the other in a particular cell context and the specific roles of each phospho‐residue in each physiological setting remain to be fully elucidated. In each cell type, all steps in the Smad pathway are tightly controlled to ensure that Smad3 transmits signals from the plasma membrane to the nucleus in a manner that reflects the strength and duration of ligand stimulation in a cell‐context–dependent manner (Massague, 2012; Wrighton, Lin, & Feng, 2009). We focused on differentiating myoblasts undergoing fusion.
In addition to the C‐tail, three (S/T)‐P sites in the Smad3 linker region—Ser208, Ser204, and Thr179—are phosphorylated in response to TGF‐β (Kamato et al., 2013). Linker phosphorylation peaks 1 hr after TGF‐β treatment, following the peak of C‐tail phosphorylation (Wang, Matsuura, He, & Liu, 2009). Previously, ITC titrations revealed that the affinity of the Pin1 WW domain was twice as great for pT179 within a Smad3 176–193 peptide (Kilodalton; kDa = 12.6 ± 1 μM) than for pS204/pS208 within a Smad3 202–211 peptide (kDa= 23.4 ± 7 μM) and that Pin1 did not bind to S213 in the Smad3 linker region (Aragon et al., 2011). We found that Pin1 bound more strongly to S204 and S208 in the Smad linker region than to T179 after 1 hr of TGF‐β stimulation in C2C12 cells (Figure 6a).
Figure 6.

Peptidyl‐prolyl cis–trans isomerase NIMA interacting 1 (Pin1)–binding serine 204 residue in the smad3 linker region is crucial for fusion. (a) Pin1 binds to serine 204 and serine 208 of the Smad3 linker region in postmitotic C2C12 cells. Smad3 wild type and SP/TP site mutants in the linker region of Smad3 (CS2 Smad3‐WT, CS2 Smad3 T179V, CS2 Smad3 S204A, CS2 Smad3 S208A, and CS2 Smad3 S213A) were coexpressed with PCNDA3.1 HA‐Pin1. 24 hr posttransfection and postculture in a differentiation medium, cells were harvested and Immunoprecipitatioin (IP) was done with the anti‐HA antibody. Later, western blot analysis was done for IP samples and input samples followed by immunoblotting for the Flag tag and actin. (b) Cells overexpressing CS2 Smad3 S204A only showed rescue of myotube fusion, and cells overexpressing CS2 Smad3 T179V showed many cells with two nuclei. C2C12 cells were transfected with constructs for Smad3 wild type and SP–TP site mutants in the linker region of Smad3 (CS2 Smad3‐WT, CS2 Smad3 S204A, CS2 Smad3 S208A, and CS2 Smad3 T179V). The cells were allowed to differentiate for 4 days before fixation and immunostained with the Anti‐MyHC antibody and Anti‐Flag antibody. Scale bar= 50 μM [Color figure can be viewed at wileyonlinelibrary.com]
To determine whether these residues were involved in myoblast fusion and differentiation, we transiently transfected C2C12 cells with wild‐type and mutant constructs of Smad3 and allowed them to differentiate. Different expression patterns were apparent after 4 days of differentiation: wild‐type Smad3 was expressed strongly in nuclear regions, whereas Smad3 S204A was mostly expressed in the cytosol. Cells overexpressing wild‐type Smad3 and the Smad3 S208A mutant were found to be mononuclear with MyHC staining, whereas cells expressing the Smad3 S204A mutant displayed multinucleated myotubes. Overexpression of Smad3 T179V did not generate multinucleated cells containing more than two nuclei but resulted in the formation of many elongated cells containing two nuclei (Figure 6b). Thus, S204 in the Smad3 linker region might have a specific role in myoblast fusion and is a Pin1‐binding site after TGF‐β signal activation in myoblasts. We also found that cells overexpressing Smad3 with all of its linker sites mutated (with intact C‐terminal phosphorylation) also showed formation of multinucleated myotubes (Supporting Information Figure S5).
3.7. ERK‐mediated phosphorylation is required for the binding of Pin1 to Smad3 after TGF‐β stimulation in myoblasts
During or directly prior to the assembly of Smad proteins into transcriptional complexes, linker phosphorylation is mediated by cytoplasmic MAPK or by CDK8 and CDK9 (Alarcon et al., 2009). However, in muscle cells that are old or have been treated with TGF‐β, CDK inhibitors are expressed (Carlson et al., 2008). We measured the expression of the CDK inhibitor CDKN1A in postmitotic C2C12 cells and found that TGF‐β increased its transcript level (Supporting Information Figure S7).
We found that with Pin1 overexpression the interaction between total Smad3 and GSK3α/β is reduced (Supporting Information Figure S2). A candidate for S204 phosphorylation is GSK3β (Aragon et al., 2011), but it is known that only nonactive Smad3 physically interacts with GSK3β (Guo et al., 2008). The most likely candidate for S204 phosphorylation in myoblasts in the presence of TGF‐β and CDK inhibitors is ERK. The effect of ERK inhibition on Smad signaling is cell type specific (Hayashida, de Caestecker, & Schnaper, 2003), so we inhibited ERK in C2C12 cells after TGF‐β signal activation. Inhibition of ERK with U0126 reduced the phosphorylation of Smad3 at S204 after TGF‐β stimulation (Figure 7a) and increased the ubiquitination of Smad3 (Figure 7b). U0126 treatment also inhibited the binding of Pin1 to Smad3 (Figure 7c). Upon expression of constitutively active MEK (CA‐MEK), the binding between Smad3 S204A and Pin1 did not increase to the same level seen for wild‐type Smad3 upon CA‐MEK overexpression (Figure 7d). From our data, it is evident that, in muscle, ERK‐mediated phosphorylation of the Smad3 linker region has a specific regulatory role in Pin1 binding and stabilization of Smad3 (Figure 7e).
Figure 7.

Smad3 requires ERK‐mediated phosphorylation at S204 to bind peptidyl‐prolyl cis–trans isomerase NIMA interacting 1 (Pin1). (a) Serine 204 phosphorylation within the Smad3 linker region is reduced with ERK inhibition posttransforming growth factor‐β (TGF‐β) signaling. Levels of phosphorylated S204, S208, S213, and T179 were observed in C2C12 cells 1 hr after treatment with U0126 following TGF‐β exposure. (b) Smad3 ubiquitination is increased with ERK inhibition. Flag‐tagged Smad3 and HA‐tagged ubiquitin (Ub) were overexpressed, and then the cells were treated with U0126 and MG132 for 4 hr. IP was done with the Flag antibody followed by immunoblotting with the HA antibody. (c) Binding of Smad3 with GST–Pin1 isreduced with ERK inhibition after TGF‐β treatment. C2C12 cells were treated for 1 hr with TGF‐β in the presence or absence of kinase inhibitors U0126. The GST pulldown experiment was performed with GST and Pin1–GST proteins to observe the binding affinity (G and P depict GST and GST–Pin1, respectively). (d) Pin1 binding is not increased as highly as in CS2 Smad3 overexpressing cells with over expression ofconstitutively active MEK, in CS2 Smad3 S204A overexpressing cells. C2C12 cells were transfected with CS2 Smad3 WT and CS2 Smad3 S204A with or without CA‐MEK. All cells were treated with TGF‐β 1 hr before harvest. IP was done with the anti‐Pin1 antibody followed by immunoblotting with the Flag antibody for both IP and input. (e) Schematic figure showing Pin1 binding to Smad3 that has been phosphorylated at the tail by TGF‐β and in the linker region at S204 by ERK. This Pin1‐bound stabilized Smad3 inhibits myoblast fusion and myotube formation [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
Although associated with viral infections and neoplasia, spontaneous cell–cell fusion, or syncytialization, is a key part of normal development in tissues such as muscle (Abmayr & Pavlath, 2012) and bone (Soe et al., 2011) and is crucial for their proper functioning. Myoblast fusion is a critical aspect of muscle differentiation, and the control of fusion might occur through different pathways.
Pin1 is an important and ubiquitous protein that maintains the balance of many biological processes (Liou et al., 2002). The elevation of Pin1 in cancer cells suggests that this protein regulates cellular growth and proliferation (Bao et al., 2004). Unlike transformed proliferative cells, mature myotubes are postmitotic, highlighting the context‐dependent nature of Pin1 function. Our findings support a role for Pin1 as a regulator of skeletal muscle fusion at both the cellular and organ levels (Figures 1, 2). The small increase in the myogenin transcript level only on the third day after treatment with the Pin1 inhibitor DTM during differentiation (Supporting Information Figure 2) suggests that Pin1 inhibition does not stimulate all steps of differentiation and that, during myoblast fusion, Pin1 might also have a role independent of the differentiation process. Although Smad3 was identified as a target of the Pin1‐mediated inhibition of muscle fusion, it might not be the only one, as the Pin1 inhibitor treatment after Smad3 overexpression further enhanced the fusion index even after restoring it to a normal level (Figure 3). Thus, other substrates might be regulated by Pin1 during muscle fusion. From our observation, we believe that Smad3 is one of the most important candidates in the regulation of myoblast fusion by Pin1.
The conditions that induce muscle atrophy also activate the myostatin–TGF‐β pathway. Myostatin and other TGF‐β family members bind activin type II receptors, resulting in Smad2/3 phosphorylation and a reduction in muscle mass (Egerman & Glass, 2014; McFarlane et al., 2006; Ruegg & Glass, 2011). Thus, inhibition of TGF‐β improves muscle strength (Waning et al., 2015). With age, there is a dramatic and constant upregulation of TGF‐β (inactive precursor plus bioactive protein) and pSmad3 (Carlson et al., 2008). Smad3 is a critical regulator of overall muscle bulk. Smad3 knockout mice exhibit atrophy (Ge et al., 2011), whereas Smad3 inhibition increases muscle bulk (Burks & Cohn, 2011; Sartori et al., 2009). The Smad3 knockout phenotype is similar to the phenotypes seen in Pin1−/− mice. The Pin1+/− mice in which Smad3 is functionally inhibited may mimic models of Smad3 inhibition. In the case of Smad3, this paradox is sometimes attributed to an increase in availability of Smad4 to the BMP pathway when Smad3 is inhibited (Sartori et al., 2013). As Pin1 targets Smad3, it might explain why inhibition of Pin1 at low doses shows more fusion. Analysis of the distribution of muscle fiber CSA demonstrated that Pin1+/− muscle had not only a higher nuclear content but also larger myofibrils (Figure 1). Although Pin1+/− muscle cross‐sections exhibited rounded hypertrophic fibers, they did not display signs of myopathy such as angulated atrophic myofibers, fiber‐type grouping, group atrophy, target fibers, nuclear clumps, myofiber necrosis, myophagocytosis, regenerating fibers, myofiber splitting, increases in centrally positioned or internal nuclei, or nuclear inclusions in histology.
Binding of Pin1 to Smad3 has different consequences in different contexts and cell types, such as promoting TGF‐β signaling without changing the protein level of Smad3 (Matsuura et al., 2010) or inducing the degradation of Smad3 (Nakano et al., 2009). It was suggested in an earlier report that the conformational status and final consequences of R‐Smad can vary according to the physiological or pathological setting (Wrighton et al., 2009). Pin1 is also known to be required for the TGF‐β‐induced phosphorylation, nuclear translocation, and transcriptional activity of Smad3 (Yang et al., 2014). Externalization of Pin1 into the cytosol (Magli et al., 2010a) might be an important part of the myogenic program in which Pin1 binds to the master regulator Smad3, mediates its conformational change in the cytosol, and governs the consequences of BMP–TGF signaling. Pin1 interacts with Smad2/3 in a TGF‐β1‐dependent manner, and Pin1 is actively involved in the TGF‐β1‐stimulated migration and invasion of PC3 prostate cancer cells (Matsuura et al., 2010). Both Smad‐dependent gene transcription and Smad2/3 phosphorylation were found to be lower in Pin1−/− MEFs than in Pin1+/+ MEFs (Yang et al., 2014). According to previous reports, CDK8/9‐mediated phosphorylation of pSmad3 creates binding sites for Pin1, resulting in transcriptional activation of pSmad3 (Aragon et al., 2011; Gao et al., 2009). From our experimental data, it can be inferred that Pin1 is biologically important for maintaining the balance in Smad signaling during fusion (Figures 5, 6) to regulate muscle bulk and that ERK‐mediated phosphorylation is required for the interaction between Pin1 and Smad3 after TGF‐β activation (Figure 7).
We demonstrated that Pin1 modulates the muscle fiber size by stabilizing Smad3 while simultaneously upregulating the expression of growth‐limiting genes such as Atrogin‐1 and Trim63 (MuRF1). The increase in Musa‐1 might have been due to the downregulation of Smad1/5 resulting from the decrease in the Pin1 protein (Islam et al., 2016; Yoon et al., 2015). A major limitation in our understanding of TGF‐β biology is the lack of knowledge as for a direct link between TGF‐β “Smad signaling” and “non‐Smad” pathways in different cells, such as the one mediated by ERK. Considering the functional diversity of TGF‐βs, these pathways might play a major role in crosstalk with Smads, thus contributing to the multitude of observed TGF‐β effects. In this study on myoblasts, we demonstrated that the ERK and TGF pathways interact and are not independent (Smad vs. non‐Smad), as they modulate myogenic cellular responses together, downstream of TGF‐β. According to our data, downstream of TGF‐β signaling, the Smad and ERK pathways converge in the Pin1‐mediated isomerization of Smad3 (Figure 7), and this isomerization induces further downstream consequences. The downstream effectors of this unique interaction are yet to be fully understood.
Serine 204 phosphorylation in the Smad3 linker region is an important ERK‐mediated step in TGF‐β‐stimulated collagen expression (Browne, Liu, Schnaper, & Hayashida, 2013). Our data indicate that ERK‐mediated phosphorylation of Smad3 is required for Pin1 binding (Figure 7). Thus, serine 204 in the Smad3 linker region and its isomerization by Pin1 might be promising drug targets in relevant pathological settings.
Cancer‐related muscle atrophy (cachexia), which is characterized by the progressive loss of skeletal muscle mass, affects up to 80% of patients with advanced cancer (Tisdale, 2002) and is estimated to be responsible for ~25% of the deaths in these patients (Tse, 2010). Recently, Smad3 was also implicated as a major player in cancer cachexia (Waning et al., 2015). Therapeutic strategies to prevent muscle wastage are limited, and the underlying mechanisms are unclear. Discovering the mechanism of Pin1 inhibition–based muscle fusion could facilitate the discovery of drug targets for the same purpose.
The excessive inhibition or total absence of Pin1 might have adverse effects, such as neurodegeneration or osteoporosis (Islam et al., 2014; Shin et al., 2016). However, in our study, only a low dose of the Pin1 inhibitor enhanced muscle fusion. Tight control of the fusion process through Pin1 might be a natural strategy to prevent the fusion of inappropriate cell types and the formation of abnormal, nonfunctional syncytia. As only a small modification of the Pin1 level generously enhanced muscle fusion (Figure 2), Pin1 could be a very useful target in the development of muscle bulk‐enhancing drugs with reduced side effects.
Our results identify Pin1 as an important determinant of muscle mass through modulation of Smad3 and as a potential regulator of the transcriptional link between cellular metabolism and muscle growth.
CONFLICTS OF INTERESTS
The authors declare no conflict of interests.
SUMMARY STATEMENT
Myoblast fusion is pivotal for muscle development. We found that postphosphorylation isomerization of Smad3 by Pin1 is a crucial mechanism that governs myoblast fusion and maintains muscle mass.
AUTHOR CONTRIBUTIONS
R.I. conducted and designed the experiments, analyzed the data, and wrote the manuscript. H.Y. assisted with the experiments and analyzed the data. H.R.S, H.S.B, and B.S.K helped with the animal experiments and in vitro experiment set up. Y.S.L., W.J.Y., K.M.W., and J.H.B edited the manuscript and gave valuable input for modification of the experimental design. W.J.Y., H.S.B., and H.M.R. acquired funding for the project. H.M.R. supervised the project, designed the experiments, and edited the manuscript.
Supporting information
Supporting information
ACKNOWLEDGEMENTS
CS2 Flag‐Smad3 and mutant plasmids were a gift from Dr. Joan Massague and acquired through Addgene (Cambridge, MA). This study was supported by the Bio & Medical Technology Development Program (20100030015), the General Researcher Program (20100010590), the Korean–Japanese International Research Program (K20802001314‐09B1200‐11110), and the MEST (NRF‐2014R1A2A2A01004865) of the National Research Foundation (NRF) of Korea.
Islam R, Yoon H, Shin H-R et al. Peptidyl‐prolyl cis–trans isomerase NIMA interacting 1 regulates skeletal muscle fusion through structural modification of Smad3 in the linker region. J Cell Physiol. 2018;233:9390–9403. 10.1002/jcp.26774
References
REFERENCES
- Abmayr, S. M. , & Pavlath, G. K. (2012). Myoblast fusion: Lessons from flies and mice. Development, 139, 641–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcon, C. , Zaromytidou, A. I. , Xi, Q. , Gao, S. , Yu, J. , Fujisawa, S. , … Macias, M. J. (2009). Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF‐beta pathways. Cell, 139, 757–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragon, E. , Goerner, N. , Zaromytidou, A. I. , Xi, Q. , Escobedo, A. , Massague, J. , & Macias, M. J. (2011). A smad action turnover switch operated by WW domain readers of a phosphoserine code. Genes and Development, 25, 1275–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, L. , Kimzey, A. , Sauter, G. , Sowadski, J. M. , Lu, K. P. , & Wang, D. G. (2004). Prevalent overexpression of prolyl isomerase Pin1 in human cancers. American Journal of Pathology, 164, 1727–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollinger, L. M. , Witczak, C. A. , Houmard, J. A. , & Brault, J. J. (2014). SMAD3 augments FoxO3‐induced MuRF‐1 promoter activity in a DNA‐binding‐dependent manner. American Journal of Physiology‐Cell Physiology, 307, C278–C287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, K. A. , Pietenpol, J. A. , & Moses, H. L. (2007). A tale of two proteins: Differential roles and regulation of Smad2 and Smad3 in TGF‐beta signaling. Journal of Cellular Biochemistry, 101, 9–33. [DOI] [PubMed] [Google Scholar]
- Browne, J. A. , Liu, X. , Schnaper, H. W. , & Hayashida, T. (2013). Serine‐204 in the linker region of Smad3 mediates the collagen‐I response to TGF‐beta in a cell phenotype‐specific manner. Experimental Cell Research, 319, 2928–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruusgaard, J. C. , Egner, I. M. , Larsen, T. K. , Dupre‐Aucouturier, S. , Desplanches, D. , & Gundersen, K. (2012). No change in myonuclear number during muscle unloading and reloading. Journal of Applied Physiology, 113, 290–296. [DOI] [PubMed] [Google Scholar]
- Burks, T. N. , & Cohn, R. D. (2011). Role of TGF‐beta signaling in inherited and acquired myopathies. Skeletal Muscle, 1, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson, M. E. , Hsu, M. , & Conboy, I. M. (2008). Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature, 454, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charge, S. B. , & Rudnicki, M. A. (2004). Cellular and molecular regulation of muscle regeneration. Physiological Reviews, 84, 209–238. [DOI] [PubMed] [Google Scholar]
- Clop, A. , Marcq, F. , Takeda, H. , Pirottin, D. , Tordoir, X. , Bibe, B. , … Eychenne, F. (2006). A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nature Genetics, 38, 813–818. [DOI] [PubMed] [Google Scholar]
- Egerman, M. A. , & Glass, D. J. (2014). Signaling pathways controlling skeletal muscle mass. Critical Reviews in Biochemistry and Molecular Biology, 49, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimori, F. , Takahashi, K. , Uchida, C. , & Uchida, T. (1999). Mice lacking Pin1 develop normally, but are defective in entering cell cycle from G(0) arrest. Biochemical and Biophysical Research Communications, 265, 658–663. [DOI] [PubMed] [Google Scholar]
- Gao, S. , Alarcon, C. , Sapkota, G. , Rahman, S. , Chen, P. Y. , Goerner, N. , … Massague, J. (2009). Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF‐beta signaling. Molecular Cell, 36, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge, X. J. , McFarlane, C. , Vajjala, A. , Lokireddy, S. , Ng, Z. H. , Tan, C. K. , … Kambadur, R. (2011). Smad3 signaling is required for satellite cell function and myogenic differentiation of myoblasts. Cell Research, 21, 1591–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman, C. A. , McNally, R. M. , Hoffmann, F. M. , & Hornberger, T. A. (2013). Smad3 induces atrogin‐1, inhibits mTOR and protein synthesis, and promotes muscle atrophy in vivo. Molecular Endocrinology, 27, 1946–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, X. , Ramirez, A. , Waddell, D. S. , Li, Z. , Liu, X. , & Wang, X. F. (2008). Axin and GSK3‐ control Smad3 protein stability and modulate TGF‐signaling. Genes and Development, 22, 106–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashida, T. , de Caestecker, M. , & Schnaper, H. W. (2003). Cross‐talk between ERK MAP kinase and Smad‐signaling pathways enhances TGF‐beta dependent responses in human mesangial cells. FASEB Journal, 17, 1576–8. [DOI] [PubMed] [Google Scholar]
- Hochreiter‐Hufford, A. E. , Lee, C. S. , Kinchen, J. M. , Sokolowski, J. D. , Arandjelovic, S. , Call, J. A. , … Ravichandran, K. S. (2013). Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature, 497, 263–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam, R. , Bae, H. S. , Yoon, W. J. , Woo, K. M. , Baek, J. H. , Kim, H. H. , … Ryoo, H. M. (2014). Pin1 regulates osteoclast fusion through suppression of the master regulator of cell fusion DC‐STAMP. Journal of Cellular Physiology, 229, 2166–2174. [DOI] [PubMed] [Google Scholar]
- Islam, R. , Yoon, W. J. , & Ryoo, H. M. (2016). Pin1, the master orchestrator of bone cell differentiation. Journal of Cellular Physiology, 232, 2339–2347. [DOI] [PubMed] [Google Scholar]
- Jansen, K. M. , & Pavlath, G. K. (2008). Molecular control of mammalian myoblast fusion. Methods in Molecular Biology, 475, 115–133. [DOI] [PubMed] [Google Scholar]
- Kamato, D. , Burch, M. L. , Piva, T. J. , Rezaei, H. B. , Rostam, M. A. , Xu, S. , … Osman, N. (2013). Transforming growth factor‐beta signalling: Role and consequences of Smad linker region phosphorylation. Cellular Signalling, 25, 2017–2024. [DOI] [PubMed] [Google Scholar]
- Kretzschmar, M. , Doody, J. , Timokhina, I. , & Massague, J. (1999). A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes and Development, 13, 804–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecker, S. H. , Goldberg, A. L. , & Mitch, W. E. (2006). Protein degradation by the ubiquitin‐proteasome pathway in normal and disease states. Journal of the American Society of Nephrology, 17, 1807–1819. [DOI] [PubMed] [Google Scholar]
- Lecker, S. H. , Jagoe, R. T. , Gilbert, A. , Gomes, M. , Baracos, V. , Bailey, J. , … Goldberg, A. L. (2004). Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB Journal, 18, 39–51. [DOI] [PubMed] [Google Scholar]
- Liou, Y. C. , Ryo, A. , Huang, H. K. , Lu, P. J. , Bronson, R. , Fujimori, F. , … Lu, K. P. (2002). Loss of Pin1 function in the mouse causes phenotypes resembling cyclin D1‐null phenotypes. Proceedings of the National Academy of Sciences of the United States of America, 99, 1335–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou, Y. C. , Sun, A. , Ryo, A. , Zhou, X. Z. , Yu, Z. X. , Huang, H. K. , … Li, X. (2003). Role of the prolyl isomerase Pin1 in protecting against age‐dependent neurodegeneration. Nature, 424, 556–561. [DOI] [PubMed] [Google Scholar]
- Liu, D. , Black, B. L. , & Derynck, R. (2001). TGF‐beta inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes & Development, 15, 2950–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magli, A. , Angelelli, C. , Ganassi, M. , Baruffaldi, F. , Matafora, V. , Battini, R. , … Del Sal, G. (2010a). Proline isomerase Pin1 represses terminal differentiation and myocyte enhancer factor 2C function in skeletal muscle cells. Journal of Biological Chemistry, 285, 34518–34527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magli, A. , Angelelli, C. , Ganassi, M. , Baruffaldi, F. , Matafora, V. , Battini, R. , … Del Sal, G. (2010b). Proline isomerase Pin1 represses terminal differentiation and myocyte enhancer factor 2C function in skeletal muscle cells. Journal of Biological Chemistry, 285, 34518–34527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague, J. (2012). TGFbeta signalling in context. Nature Reviews Molecular Cell Biology, 13, 616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura, I. , Chiang, K. N. , Lai, C. Y. , He, D. M. , Wang, G. N. , Ramkumar, R. , … Liu, F. (2010). Pin1 Promotes Transforming Growth Factor‐beta‐induced Migration and Invasion. Journal of Biological Chemistry, 285, 1754–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy, J. J. , Mula, J. , Miyazaki, M. , Erfani, R. , Garrison, K. , Farooqui, A. B. , … Keller, C. (2011). Effective fiber hypertrophy in satellite cell‐depleted skeletal muscle. Development, 138, 3657–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane, C. , Plummer, E. , Thomas, M. , Hennebry, A. , Ashby, M. , Ling, N. , … Kambadur, R. (2006). Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF‐kappa B‐independent, FoxO1‐dependent mechanism. Journal of Cellular Physiology, 209, 501–514. [DOI] [PubMed] [Google Scholar]
- McPherron, A. C. , Lawler, A. M. , & Lee, S. J. (1997). Regulation of skeletal muscle mass in mice by a new TGF‐beta superfamily member. Nature, 387, 83–90. [DOI] [PubMed] [Google Scholar]
- McPherron, A. C. , & Lee, S. J. (1997). Double muscling in cattle due to mutations in the myostatin gene. Proceedings of the National Academy of Sciences of the United States of America, 94, 12457–12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millay, D. P. , O'Rourke, J. R. , Sutherland, L. B. , Bezprozvannaya, S. , Shelton, J. M. , Bassel‐Duby, R. , & Olson, E. N. (2013). Myomaker is a membrane activator of myoblast fusion and muscle formation. Nature, 499, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano, A. , Koinuma, D. , Miyazawa, K. , Uchida, T. , Saitoh, M. , Kawabata, M. , … Miyazono, K. (2009). Pin1 down‐regulates transforming growth factor‐beta (TGF‐beta) signaling by inducing degradation of Smad proteins. Journal of Biological Chemistry, 284, 6109–6115. [DOI] [PubMed] [Google Scholar]
- Pajcini, K. V. , Pomerantz, J. H. , Alkan, O. , Doyonnas, R. , & Blau, H. M. (2008). Myoblasts and macrophages share molecular components that contribute to cell‐cell fusion. Journal of Cell Biology, 180, 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S. Y. , Yun, Y. , Lim, J. S. , Kim, M. J. , Kim, S. Y. , Kim, J. E. , & Kim, I. S. (2016). Stabilin‐2 modulates the efficiency of myoblast fusion during myogenic differentiation and muscle regeneration. Nature Communications, 7, 10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, M. , Pardali, E. , van der Horst, G. , Cheung, H. , van den Hoogen, C. , van der Pluijm, G. , & ten Dijke, P. (2010). Smad2 and Smad3 have opposing roles in breast cancer bone metastasis by differentially affecting tumor angiogenesis. Oncogene, 29, 1351–1361. [DOI] [PubMed] [Google Scholar]
- Ruegg, M. A. , & Glass, D. J. (2011). Molecular mechanisms and treatment options for muscle wasting diseases. Annual Review of Pharmacology and Toxicology, 51, 373–395. [DOI] [PubMed] [Google Scholar]
- Sartori, R. , Milan, G. , Patron, M. , Mammucari, C. , Blaauw, B. , Abraham, R. , & Sandri, M. (2009). Smad2 and 3 transcription factors control muscle mass in adulthood. American Journal of Physiology: Cell Physiology, 296, C1248–C1257. [DOI] [PubMed] [Google Scholar]
- Sartori, R. , Schirwis, E. , Blaauw, B. , Bortolanza, S. , Zhao, J. , Enzo, E. , … Ferry, A. (2013). BMP signaling controls muscle mass. Nature Genetics, 45, 1309–1318. [DOI] [PubMed] [Google Scholar]
- Schuelke, M. , Wagner, K. R. , Stolz, L. E. , Hubner, C. , Riebel, T. , Komen, W. , … Lee, S. J. (2004). Myostatin mutation associated with gross muscle hypertrophy in a child. New England Journal of Medicine, 350, 2682–2688. [DOI] [PubMed] [Google Scholar]
- Shen, Z. J. , Braun, R. K. , Hu, J. , Xie, Q. , Chu, H. , Love, R. B. , … Sorkness, R. L. (2012). Pin1 protein regulates Smad protein signaling and pulmonary fibrosis. Journal of Biological Chemistry, 287, 23294–23305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, H. R. , Islam, R. , Yoon, W. J. , Lee, T. , Cho, Y. D. , Bae, H. S. , … Ryoo, H. M. (2016). Pin1‐mediated modification prolongs the nuclear retention of beta‐catenin in Wnt3a‐induced osteoblast differentiation. Journal of Biological Chemistry, 291, 5555–5565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soe, K. , Andersen, T. L. , Hobolt‐Pedersen, A. S. , Bjerregaard, B. , Larsson, L. I. , & Delaisse, J. M. (2011). Involvement of human endogenous retroviral syncytin‐1 in human osteoclast fusion. Bone, 48, 837–846. [DOI] [PubMed] [Google Scholar]
- Sun, Y. , Ge, Y. , Drnevich, J. , Zhao, Y. , Band, M. , & Chen, J. (2010). Mammalian target of rapamycin regulates miRNA‐1 and follistatin in skeletal myogenesis. Journal of Cell Biology, 189, 1157–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatara, Y. , Lin, Y. C. , Bamba, Y. , Mori, T. , & Uchida, T. (2009). Dipentamethylene thiuram monosulfide is a novel inhibitor of Pin1. Biochemical and Biophysical Research Communications, 384, 394–398. [DOI] [PubMed] [Google Scholar]
- Tisdale, M. J. (2002). Cachexia in cancer patients. Nature Reviews Cancer, 2, 862–871. [DOI] [PubMed] [Google Scholar]
- Toko, H. , Konstandin, M. H. , Doroudgar, S. , Ormachea, L. , Joyo, E. , Joyo, A. Y. , … Volkers, M. (2013). Regulation of cardiac hypertrophic signaling by prolyl isomerase Pin1. Circulation Research, 112, 1244–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trendelenburg, A. U. , Meyer, A. , Rohner, D. , Boyle, J. , Hatakeyama, S. , & Glass, D. J. (2009). Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. American Journal of Physiology: Cell Physiology, 296, C1258–C1270. [DOI] [PubMed] [Google Scholar]
- Tse, M. T. (2010). Muscle disorders: Preventing wastage. Nature Reviews. Drug Discovery, 9, 763–763. [DOI] [PubMed] [Google Scholar]
- Wang, G. N. , Matsuura, I. , He, D. M. , & Liu, F. (2009). Transforming Growth Factor‐beta‐inducible Phosphorylation of Smad3. Journal of Biological Chemistry, 284, 9663–9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waning, D. L. , Mohammad, K. S. , Reiken, S. , Xie, W. , Andersson, D. C. , John, S. , … Niewolna, M. (2015). Excess TGF‐beta mediates muscle weakness associated with bone metastases in mice. Nature Medicine, 21, 1262–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welle, S. L. (2009). Myostatin and muscle fiber size. Focus on “Smad2 and 3 transcription factors control muscle mass in adulthood” and “Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size”. American Journal of Physiology: Cell Physiology, 296, C1245–C1247. [DOI] [PubMed] [Google Scholar]
- Wrighton, K. H. , Lin, X. , & Feng, X. H. (2009). Phospho‐control of TGF‐beta superfamily signaling. Cell Research, 19, 8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe, D. , & Saxel, O. (1977). A myogenic cell line with altered serum requirements for differentiation. Differentiation, 7, 159–166. [DOI] [PubMed] [Google Scholar]
- Yang, J. W. , Hien, T. T. , Lim, S. C. , Jun, D. W. , Choi, H. S. , Yoon, J. H. , … Kang, K. W. (2014). Pin1 induction in the fibrotic liver and its roles in TGF‐beta1 expression and Smad2/3 phosphorylation. Journal of Hepatology, 60, 1235–1241. [DOI] [PubMed] [Google Scholar]
- Yoon, W. J. , Islam, R. , Cho, Y. D. , Ryu, K. M. , Shin, H. R. , Woo, K. M. , … Ryoo, H. M. (2015). Pin1 plays a critical role as a molecular switch in canonical BMP signaling. Journal of Cellular Physiology, 230, 640–647. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information