

Abstract
A resurgent interest in the role of B cells following solid organ transplantation is being driven by clinical data suggesting that antibody mediated rejection (AMR) is a major cause of dysfunction and organ transplant failure. These observations suggest that, in a subset of patients, current immunotherapies are failing to control the development of alloantibody responses, or failing to reverse the production or the effects of alloantibodies. Quantification of donor-specific antibodies (DSA) has proven to be an imperfect predictor of AMR, and efforts to improve DSA quantification anticipate that this will result in improved predictive power. At the same time, attempts to control of ABMR have focused on the non-specific elimination of B cells, plasma cells (PCs) or circulating antibodies. In the past decade, there has been an improvement in our understanding of the processes that drive B cell differentiation into germinal center (GC)-dependent or GC-independent memory B cells and antibody-secreting PC. These insights are suggesting new ways to more specifically target the DSA response, which may lead to better long-term allograft survival outcomes while preserving protective immunity. In this review, new insights into processes that lead to antibody production upon primary and secondary antigen encounter are discussed, and the potential implications to DSA production and future areas of investigation to control AMR are discussed.
Introduction
Experimental data stemming from the early studies of skin graft rejection by Billingham and Medawar [1] set the stage for a paradigm underscoring a critical role of T cells and an unnecessary role for B cells and antibodies in allograft rejection [2]. In the past decade, clinical studies have challenged this T cell-centric paradigm, driven by seminal observations that the presence of preformed circulating donor-specific antibodies (DSA) is associated with high risk for acute rejection, and that de novo DSA generated after transplantation is associated with poor outcomes and vascular obliterative lesions [3]. Indeed, antibody mediated rejection (AMR), is now recognized as a significant, and possibly the major cause of chronic kidney transplant dysfunction and failure [4].
There has been an increase in the understanding of the mechanisms resulting in rapid antibody production following immunization of naïve and sensitized hosts. However, less is understood of the B cell responses that result in chronically sustained antibody production mediating chronic AMR and transplant failure. This review will summarize the processes that underlie the primary and recall phases of B cell activation and antibody production, and discuss how these insights made with model antigens or infections, may be applied to understanding the generation of DSA following solid organ transplantation.
Routes of antigen presentation to B cells
B cells have to encounter cognate antigen in order to start the process of differentiating into PCs producing high affinity antibody and memory B cells. While it has long been established that B cells can bind intact soluble antigen, there is increasing evidence suggest that optimal B cell activation occurs when the B cell receptor (BCR) engages intact antigen displayed on FDCs, B cells or macrophages (reviewed in [5]). Several strategies exist to increase the opportunities for B cells to encounter soluble and membrane-bound antigen in the draining lymph nodes (reviewed in [5; 6]; Figure 1). Mature B cells circulate though the lymph nodes approximately every 24 hours, by leaving the vascular system and entering the lymph nodes through specialized high endothelial venules (HEV), migrating along processes extending from follicular dendritic cell (FDCs) and following the chemokine CXCL13 gradient established by FDCs and fibroblastic reticular cells (FRCs). Eventually these B cells congregate within the cortical region near the subcapsular sinus where they may encounter soluble or particulate antigens that enter the draining lymph node via multiple routes depending on antigen size, the presence of circulating antigen-specific antibodies, and the deposition of complement on the antigen by the classical or alternative pathways. In addition, there may be additional contribution by migratory DCs that acquire antigen at the tissue site and transport them into the lymph node.
Figure 1:
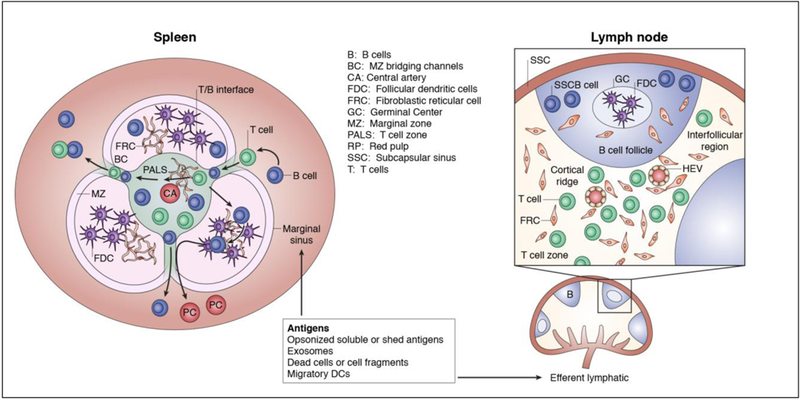
Potential routes of antigen entry into the B and T cell zones in the lymph node and spleen. The FRC network guides soluble antigens, dendritic cells and macrophages bearing antigen, as well and T and B cells, into the correct anatomical location to optimize the likelihood of cognate antigen encounter.
The humoral response can also arise from the spleen, which is divided into two distinct compartments: the red pulp comprising a network of venous sinuses that traps old or damaged erythrocytes by red pulp macrophages, and the white pulp involved in the initiation of immune responses (reviewed in [7]; Figure 1). The white pulp is comprised of the T cell zone (also referred to as the periarteriolar lymphoid sheath; PALS), B cell follicles and marginal zone (MZ), which is strategically situated at the interface of the red and white pulp. The MZ contains a large reservoir of resident cells that participate in capturing and shuttling the antigen to FDCs within the B cell follicles and cells that are capable of processing and presenting the antigen to T cells. The routes for antigen delivery to B cells in the spleen and lymph nodes, identified through the study of model antigens or pathogens, are applicable to antigens derived from solid organ transplantation.
Small soluble proteins (≤14 kD), such as those secreted by the transplanted organs, may gain direct access to the B cell via passive flow through FRC conduits or gaps in the sinus floor of the lymph node or in the marginal sinus in the spleen (reviewed in [5; 6], [8] ). These FRC conduits intersect with FDCs and thus provide direct connection for soluble antigen to be captured and presented on the FDC surface. In addition, natural polyreactive antibodies and DSA in naïve and sensitized recipients, respectively, can bind to soluble antigens and activate complement. These opsonized antigens are captured by complement receptor 3 (CR3; CD11b/CD18) expressed by subcapsular sinus macrophages (SSM) in draining lymph nodes. The localization of these CD169+CD11cloCD11b+MOMA-1+ SSMs, lining the sinus region of afferent lymphatic vessels, is dependent on lymphotoxin. Opsonized antigens captured and displayed by the SSM are relayed to non-antigen-specific naïve B cells in the underlying follicles via the complement receptors, CD21 (CR2) and CD35 (CR1), or FcRγIIB receptors. These B cells then transport opsonized antigens to FDCs that capture the immune complexes by CR1 binding C3b or C4d [9]. Likewise, opsonins draining into the spleen are captured in a C3d/CR2-dependent manner by MZ macrophages or B cells located just underneath the marginal sinus, which are then delivered to FDCs [5; 10].
Antigens not bound by antibodies can bind to macrophages and DCs located in the lymph nodes, via innate recognition proteins such as natural IgM; C-type lectins such as MBL (mannose binding lectin) and ficolins; and pentraxins, including C-reactive protein. These DCs and macrophages in the lymph node, together with CD8- DCs, MZ metallophilic macrophages and MZ macrophages in the spleen capture antigens via SIGLEC1, SIGNR1, TLR, type 1 scavenger receptor MARCO and C-type lectin, SIGNR1 (reviewed [7; 11]). Antigens displayed on the surface of these cells activate complement, and then follow the route of opsonized antigen delivery to CR1 on FDCs.
In addition to antigens secreted and shed from the allograft, a new type of alloantigen delivery has become increasingly appreciated ([12], reviewed [13; 14]). Donor-derived intact MHC complexes carried by extracellular vesicles or exosomes released by the allograft can accumulate in the draining lymph node and spleen. These exosomes are then captured by SIGN-R1+ MZ macrophages, F4/80+ macrophages in the red pulp of the spleen, SCS macrophages and DCs. Alternatively, a relatively limited number of donor DCs migrate from the graft into draining lymph nodes or spleen, where they deliver exosome to resident DCs. Donor MHC delivered by exosomes to recipient DCs, results in cross-dressed DCs that have the ability to stimulate recipient CD8+ T cells via the direct pathway [15; 16]. Curiously, recipient APCs cross-dressed with donor MHC II cannot prime efficiently CD4+ T cells [17]. The exosome surface also bears numerous glycoproteins decorated with sialic acid that allow their capture by sialoadhesin (CD169) expressed on SSM and MZ macrophages [18]. These macrophages then deliver exosomes bearing intact donor MHC to FDCs within the B cell zone [19].
Early B cell activation and the extrafollicular T/B cell interaction
When the BCR binds to antigen displayed on FDCs, SSMs or non-antigen-specific B cells, it triggers B cell activation, the upregulation of CCR7 and migration to the T/B border (reviewed in [5; 6]). Concurrently, B cells internalize the BCR-bound antigen, process and present the peptides derived from the antigen on MHC Class II molecules, in preparation for interaction with antigen-specific T cell receptors on CD4+ T cells. It is notable that while alloreactive T cells can directly recognize intact donor MHC or indirectly recognize processed donor MHC presented on recipient MHC, only CD4+ T cells that have indirect specificity for donor-derived antigens presented by the MHC molecules expressed by recipient B cells are capable of driving the differentiation of recipient B cells into PCs producing DSA [20]. This cognate interaction between T and B cells initiates the T-dependent DSA response.
T cells in the lymph node exit via HEVs located in the cortical ridge, and migrate along a network of reticular fibers to a gradient of CCL19 and CCL2 to localize in the T cell zone. Likewise, a FRC network exist within the PALS and connects the PALS to specific regions (bridging channels) of the MZ; T cells exiting the blood into the spleen use these FRC networks to enter the PALS [21; 22]. The activation of T cells requires an initial encounter with migratory DCs that had acquired antigen from the allograft and migrated via afferent lymph and along the same FRC network in the lymph node or spleen to reach the T cell zone. These DCs arrive between 12–18 hours to 5 days after antigen encounter in the tissues, and it has been argued that such delivery would be asynchronous to the more rapid delivery of antigen to FDCs and B cell activation, and that there has to be another more rapid route of antigen delivery to DCs in the T cell zone. Indeed, small antigens can gain rapid access to lymph node and spleen resident DCs within minutes of subcutaneous injection [8], via lymph and through FRC conduits that provide an efficient network for delivering small antigens, cytokine and chemokines to resident DCs that are localized along the FRC conduits in the T cell area. The FRC conduits have an outer diameter of 1–2 μm, are composed of tightly packed type I collagen fibers with spacing of 5–8 nm, resulting in a size exclusion of proteins over 60–70 kDa. For larger antigens, Gerner et al. [23] reported that strategically localized CD11b+DCs within the lymphatic sinus endothelium scan the lymph with motile dendrites, and capture particulate vaccine antigens, pathogens and potentially, allograft-derived exosomes and membrane fragments. These DCs process and present captured antigens to T cells, thereby inducing T cell responses more rapidly than, and independently of, migratory DCs.
The initial encounter of T cells with cognate antigen presented on DCs result in a subset acquiring T follicular helper (Tfh) cell properties [24; 25; 26] (Figure 2). How CD4+ T cell fates are determined has been the subject of a number of investigations and remains incompletely resolved. Fazilleau et al. [27] and Tubo et al. [28] independently concluded that increasing T cell receptor (TCR) strength results in more Tfh at the expense of Th1 cells, consistent with our findings that optimum Tfh responses were elicited by higher doses of T cell epitopes compared to Th1/Th2 responses [29]. In contrast, Keck et al. [30] reported that antigen affinity significantly controlled Th1 accumulation whereas Tfh cells were less affected by affinity. Krishnamoorty et al. [31] reported that low-affinity TCR signaling, resulting in lower levels of IRF4 were linked preferentially to a Tfh over Th1 gene program; findings recently corroborated by Snook et al. [32]. The reasons for the conflicting impact of TCR signaling strength are unclear; nevertheless, it is clear that the differentiation into Tfh cells require specific signals derived from the TCR, co-stimulation and inflammatory milieu that are distinct from those facilitating differentiation into the other CD4+ T effector cell lineages. Understanding these differences may lead to the identification ways to more precisely prevent and treat AMR versus T cell-mediated rejection.
Figure 2.

B cell responses in naïve and sensitized recipients, and their control by CTLA-4Ig. Allografts transplanted into naïve recipients elicit a T cell-dependent B cell response that results in a germinal center (GC) reaction, whereas allografts in sensitized recipients elicit a T cell-dependent but GC-independent donor-specific antibody (DSA) response. CTLA-4Ig is inhibitory at multiple stages during primary and recall antibody responses.
Tfh cells can be divided into two subsets: early extrafollicular mantel Tfh and GC Tfh cells [33]. Both Tfh subsets are characterized by the upregulated expression of CXCR5 and the transcription factor, Bcl-6. CXCR5 together with the G-protein-coupled receptor EBI2 promotes the initial localization of mantel Tfh cells towards the B cell follicle and T zone (T/B) interface [34] [35], where they interact with specialized activated DCs expressing inducible co-stimulator (ICOS) ligand and CD25. Membrane and soluble CD25 quench T cell-derived IL-2, together with ICOS engagement and IL-21 produced by Tfh cells, promote Bcl-6-dependent Tfh cell differentiation [36] [37].
B cell priming at the T/B interface results in the generation of early PCs, memory B cells as well as B cells that are destined to migrate back to the follicle to initiate GC responses (Reviewed in [38]). These pre-GC responses tend to generate PCs that are of lower affinity than post-GC PCs, but can be class-switched [39]. Ballesteros-Tato et al. [37] reported that bcl-6-expression in T cells is essential for the extrafollicular production of IgG1 in response to 2 different model antigens, and of IgG2 specific to Salmonella enterica. This early antibody production has been shown to be critical for constraining infection, while waiting for the higher affinity post-GC antibody response to develop. The relative importance of the pre-GC response to DSA production following solid organ transplantation, and the contribution of this response to AMR, has not been delineated.
Germinal center response
When extrafollicular B cells receive costimulation from extrafollicular Tfh cells, they down-regulate EBI2 EBI2, which releases T cells from the outer follicle, and increase expression of the chemorepulsive receptor S1PR2, which repels cells from the S1P-rich lymph in the subcapsular sinus toward the follicle center [40; 41]. In addition, the retention of Tfh cells in the GC is regulated by contact-dependent repulsion by Ephrin B1-expressing GC B cells, which may additionally prevent Tfh cell exhaustion by providing respite from interacting with GC B cells and preserving the ability of the GC-Tfh cells to secrete IL-12 [42]. The GC-Tfh cells express higher levels of Cxcr5, Pdcd1 and IL-21 compared to extrafollicular Tfh.
The GC is divided into the light zone comprising stroma-derived FDCs and Tfh cells and the dark zone, which is devoid of FDCs. Light zone B cells express the activation markers, CD86 and CD83 [43; 44] and gene signatures associated with signaling from the BCR, CD40 and Myc. These B cells engage immobilized antigen presented on FDCs, and receive survival signals from GC-Tfh cells (reviewed in [38; 45]). Survival signals include those provided by the interaction between costimulatory molecules, CD28-B7, CD40-CD154 and ICOS-ICOSL, by cytokines such as IL-21 and BAFF, and signals downstream of SAP-SLAM intracellular interactions. Positively selected B cells upregulate CXCR4 and migrate into the dark zone to undergo cell proliferation and BCR diversification mediated by activation-induced cytidine deaminase (AID) [44; 46], [33; 47]. In the absence of T cell survival signals, B cells undergo apoptosis in the light zone, while B cells with damaged BCR genes as a result of AID activity also undergo apoptosis in the dark zone. Dark zone B cells with intact BCR downregulate CXCR4 and reenter the light zone where their newly generated BCR are tested for binding to antigen and access to T cell help [47]. Within each GC, and following the selection for B cells with high affinity BCR for antigens presented by the FDCs, post-GC B cells emerge as PCs (PC) that are ultimately responsible for persistent circulating antibodies, or as quiescent memory B cells that are responsible for the recall antibody response upon antigen reencounter (reviewed in [38]).
We, and others, have used MHC Class I and Class II tetramers to track the fate of alloreactive B cells in mice immunized with donor spleen cells or following heart allograft transplantation [48; 49; 50; 51; 52] (Figure 2). Within 7 days, donor-specific B cells acquire an activated phenotype (downregulated IgD and upregulated Class II and CD86), and differentiate into GC B cells or PC. The importance of CD40:CD154 interactions between B cells and T cells in the initial activation of T cells and in the maintenance of the GC response is well documented [53; 54]. In contrast, the necessity of CD28:B7 interactions in sustaining the GC response was less clear. Because there is currently no FDA approved drug that targets the CD40:CD154 interaction, while CTLA-4Ig is an FDA approved immunosuppressive agent for preventing kidney rejection, it was important to test the effect of CTLA-4Ig on established B cell responses. Additionally, observations that CTLA-4 on Tfr limited the expansion of antigen-specific Tfh cells and reduce antigen-specific antibody responses raised the possibility that CTLA-4Ig may in fact, promote humoral responses [55; 56]. Chen et al. [49] compared the ability of anti-CD154 and of CTLA-4Ig to reverse established GC response in mice. Both anti-CD154 and CTLA-4Ig administered at day 7 post-immunization comparably collapsed established GC B cell responses and prevented further DSA increase. Furthermore, delayed treatment with CTLA-4Ig significantly diminished the frequency of memory alloreactive B cells generated, and the recall DSA response upon re-immunization in the absence CTLA-4Ig [50]. The efficacy of CTLA-4Ig in controlling ongoing GC B cell responses and memory B cells in experimental models are congruent with reports of belatacept, a high affinity mutant of CTLA-4Ig, preventing DSA development in kidney transplant recipients despite high rates of acute rejection [57] [58]. Indeed, Leibler et al. [59] reported that belatacept was able to control humoral responses in humans by inhibiting B cell-Tfh interactions, thereby preventing B cell differentiation into PC. Whether this superiority of belatacept in controlling humoral responses compared to cyclosporine, will be maintained when compared to tacrolimus, which is currently more extensively used for kidney transplant recipients, requires further investigation.
Germinal Center responses to multiple complex antigens
For most part, the events shaping affinity maturation within the GC is based on the analysis following immunization with single haptens or small proteins, however most organ transplantation involves multiple MHC and non-MHC antigen incompatibilities. Therefore, it is critical that we understand how the B cell and antibody repertoire is shaped following the simultaneous introduction of multiple complex antigens. For select pathogens such as malaria, most naïve individuals generate antibodies directed to a variety of epitopes, whereas for other pathogens such as dengue, the majority of antibodies target an immunodominant epitope [60; 61]. How the immune response responds to multiple antigens to culminate in these diametrically opposite outcomes remains unresolved, and could be due to accessibility of binding sites on the antigen, stochastic founding events, genetic predisposition affecting the helper T cell response, and chance mutations during affinity maturation. Childs et al. [62] used computational modeling approaches to explain how different patterns of antibody responses arise. Their study suggested that upon initial antigen encounter, B cell clones with different antigenic specificity and affinity compete for stimulation during rounds of somatic hypermutation within GCs. As a result, the presence of many antigenic epitopes tends to reduce the relative breadth of the antibody repertoire. Their model also predicted that the initial affinity of the generated antibodies to multiple antigens would be reduced, due to a reduction in the number of GCs and B cells available for each epitope, which compromises affinity selection. When the initial repertoire of circulating antibodies is generated, these antibodies will bind to the same antigenic epitopes driving their production, resulting either in the selection of B cells with higher affinity than circulating antibodies, or in the selection of B cells with specificity for new epitopes. If there are no alternative immunogenic epitopes, the B cell response will terminate with a relatively restricted repertoire. Thus circulating antibodies may increase the avidity, but either increase or decrease the breadth of the antibody repertoire.
In the majority of kidney transplant recipients, recipient B cells will most likely encounter multiple complex antigens. Under current immunosuppression, approximately 15% of renal transplant recipients develop de novo DSA within 4.6 years of transplantation, with some patients responding with a limited DSA repertoire while others more broadly [63]). Using a computation method to evaluate HLA epitope mismatch, Wiebe et al. [64] reported that the development of antibodies directed at donor Class II was most frequent in renal transplantation recipients with a higher degree of HLA epitope mismatch. Understanding how the alloantibody repertoire is selected in response to multiple antigen-mismatched allografts, how this repertoire evolves over time, and the impact of immunosuppression on these process, will result in a better understanding of when pathogenic versus non-pathogenic DSA develops.
Regulating the germinal center response
A subset of CD4+ T cells that express the regulatory T (Treg) cell master regulator Foxp3, and sharing many phenotypic characteristics of Tfh cells, has recently been described [65; 66; 67]. These T follicular regulatory (TFr) cells express CXCR5, PD-1, Bcl-6. Blimp-1, FoxP3, GITR, ICOS, CTLA-4 and IL-10, but not CD25, and function by controlling GC responses. Early studies utilizing adoptive transfer or mixed bone marrow chimeras showed that Tfr are located in the GC, and that they suppress self-reactive GC B cells that may have inadvertently arose during the GC response [66]. Using Bcl6flox/floxFoxp3cre mice (where bcl6 is deleted in FoxP3+ Tregs) that lack Tfr, Botta et al. [68] recently reported that Tfr cells expanded during the contraction phase of an anti-influenza response, when IL-2 levels were reduced. These Trf cells migrated to the B cell follicles, and suppressed self-reactive B inadvertently generated in GCs during infection [69]. In the absence of Tfr, these self-reactive B cells differentiated into PC that produced anti-histone and anti-nuclear antibodies. Interestingly, Tfr cells did not inhibit the expansion of influenza-specific B cells or their differentiation into PC, and had no significant effect on influenza-specific antibody responses. In contrast, other studies have reported that Tfr cells inhibit antigen-specific GC B cells and antibody production [65; 66; 67].
Using multiplexed quantitative imaging of human mesenteric lymph node, and functional assays, Sayin et al. [70] recently reported that the majority of CD3+FOXP3+ Tfr cells expressing CD25 but low levels of CD5, resided at the T/B or GC-mantle borders, with very few located in the GC. The median Tfh/Tfr ratio was 1.3:1; 2:1 and 24:1 at the T-B border, B cell follicle and GC, respectively, leading the authors to suggest an alternative model wherein Tfr cells regulate antibody responses by preventing long-lived interactions at the T/B border. Inhibition at this early stage of B cell response, would prevent further activation of Tfh cells and entry into GCs, as well as B cell differentiation into GC B cells, and post-GC memory B cells or PCs. Suppression of B cells by Tfr was independent of PD-1 expression, and correlated with elevated expression of CD69, IL-10, CTLA-4, and GARP, a protein critical for the surface expression of latent TGF-ß [71]. These observations led the authors to speculate that TGF-ß production may be the mechanism by which Tfr inhibit B cell responses.
Generation of memory B cells and PCs
The appropriate and rapid generation of PCs is essential for the successful control of infection, and of memory cells and long-lived PCs for the protection against reinfection. Factors that determine the PC fate during primary antigen encounter have been extensively investigated, with critical roles for BCR affinity and Tfh (reviewed by [72]). Differentiation into PCs at the T:B border and within GCs is facilitated by high BCR affinity [73; 74; 75]. Sciammas and colleagues [76; 77; 78] reported that graded expression of interferon regulatory factor-4 (IRF4) is an early measure of BCR signaling intensity: high levels of IRF4 induced Blimp-1 expression, promoted the PC program and shut down the expression of Bach2 controlling GC B cell fate [78; 79], while modest levels of IRF4 promote differentiation into GC B cells [76; 78]. Within GCs, PC differentiation was induced in a discrete subset of high-affinity B cells residing within the light zone upon BCR engagement with antigen presented on FDC, while Tfh cells provided the subsequent signals essential for completing the PC differentiation and driving their migration out of the GC [80].
Memory B cells are also generated in two distinct phases: early pre-GC memory B cells with lower affinity and enriched for IgM, and later post-GC memory B cells with higher affinity and expressing IgM or IgG [54; 81]. Relative to PCs, memory B cells are derived from cells receiving lower-affinity BCR signaling and reduced T cell help, which in turn, induces reduced IRF-4 and higher Bach2, respectively [82]. Gitlin et al. [83] reported that class-switching to membrane IgG1, which signals more efficiently compared to IgM [84], biased the fate choice to PC over memory fate. Furthermore, GC B cells that lack CXCR4 and therefore failed to enter the DZ were more likely to enter the memory compartment, confirming that BCR-signaling and T cell help is sufficient to drive memory differentiation, without the need for further proliferation or somatic hypermutation [85]. These features of memory B cell versus plasma cell differentiation are consistent with memory B cells being preferentially generated in the pre-GC and early GC period, and long-lived PC emerging significantly later [86].
The differences in affinity and kinetics of memory B cell versus PC generation suggest that successful treatment of acute AMR and reduction of circulating DSA may not have prevented memory donor-specific B cell generation. Indeed, in a recent meeting report by the Sensitization in Transplantation: Assessment of Risk 2017 working group [87], a major finding was that the absence of donor-specific antibody does not equate to an absence of sensitization or of memory B cells. There may be other explanations for humoral sensitization without detectable DSA in transplant recipients, including the absorption of DSA by the allograft, the loss of shorter-lived PC the generation of a repertoire of memory B cells that is not identical to the long-lived PC [88]. In the absence of a clinical test of donor-specific memory B cells, the STAR recommendation is that an accurate patient history be obtained, and only patients without DSA and also without HLA sensitizing events such as pregnancies, transfusions, previous transplantation and implants should be considered to be immunologically low risk for alloimmune memory.
Recall B cell responses
Memory B cells upon antigen reencounter will either differentiate directly into PCs, generating a faster, high-titer and class-switched recall antibody response compared to a primary response, or they will differentiate into GC B cells that generate new, higher‐affinity and class‐switched PCs. However, the rules that predict memory B cell fate in the recall response remain to be fully clarified. Dogan et al. [89] and Pape et al. [81] reported that IgM+ memory B cells preferentially differentiated into GC cells, whereas the IgG1+ memory B cells gave rise primarily to PCs. Furthermore, Pape et al. [81] reported that the IgG+ memory B cells predominated over IgM+ memory B cells in the presence of circulating antibodies, because these antibodies outcompeted the low-affinity IgM BCR for access to limiting amounts of antigens. Shlomchik and colleagues [90] reported that memory B cells that express CD80, PD‐L2, and CD73 were mostly IgM+ and were mostly likely to produce PC. Most recently, Krishnamurthy et al. [91] investigated the memory B cell responses in mice infected with the malaria parasite, Plasmodium chabaudi. Upon challenge with P. Chabaudi-infected red blood cells, high‐affinity IgM+ memory B cells rapidly differentiated (≤3 days) into IgM+ and IgG+ PC. Collectively these data suggest that the fate of memory B cells upon reencounter with antigens is dependent, in part, on how and type of memory B cell was generated and the conditions of the antigen-reencounter.
When memory alloreactive B cells in sensitized murine recipients reencounter alloantigen following heart transplantation, they generate a recall DSA response that is largely dependent on the rapid differentiation into PC, with minimal GC responses [50] [48] (Figure 2). Nevertheless, the recall response remains dependent on T cell help, and co-stimulation blockade with CTLA-4Ig completely prevented memory B cell differentiation into PC and the recall DSA response. In addition, long-lived PC express CD28 that provides survival signals by engaging CD80/CD86 expressed by bone marrow stromal cells [92; 93; 94]. As a result, inhibition of the CD28-CD80/CD86 interaction with CTLA-4Ig may result in long-lived PC depletion and explain the recently described ability of belatacept to control and reverse DSA responses in sensitized recipients [95].
Conclusion
Despite an emerging appreciation of the limits of DSA in predicting ABMR and sensitization, the appearance de novo DSA remains a strong predictor of allograft loss [96; 97; 98]. In this review, we have provided a summary of the cellular processes that generate an antibody response, but for brevity, have avoided discussion of non-antibody producing roles of B cells, including their roles as antigen-presenting cells and as immune modulators to T cell responses. Insights into the processes that lead to antibody production and humoral memory have arisen largely from the study of model antigens in reductionist mouse models, and how these processes are altered by the unique features of organ transplantation require further investigation. Following solid organ transplantation, the recipient is exposed to an enormous diversity of antigenic epitopes, large amounts of antigens that are persistent, as well as pharmacological immunosuppression. These factors will impact on the cellular responses that generate PCs, and the quality of the DSA they produce, as well as quality of memory donor-specific B cells, all of which remain formidable barriers to successful transplantation. Finally, defining the mechanistic differences between the immune responses to model antigens, pathogens and transplantation antigens may lead to new ways to control DSA production while preserving protective immunity.
Acknowledegements
This was supported in part by a grant (1R01AI110513) from the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health.
References
- [1].Billingham RE, Krohn PL, and Medawar PB, Effect of cortisone on survival of skin homografts in rabbits. Br Med J 1 (1951) 1157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Benichou G, Yamada Y, Yun SH, Lin C, Fray M, and Tocco G, Immune recognition and rejection of allogeneic skin grafts. Immunotherapy 3 (2011) 757–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Pouliquen E, Koenig A, Chen CC, Sicard A, Rabeyrin M, Morelon E, Dubois V, and Thaunat O, Recent advances in renal transplantation: antibody-mediated rejection takes center stage. F1000Prime Rep 7 (2015) 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sellares J, de Freitas DG, Mengel M, Reeve J, Einecke G, Sis B, Hidalgo LG, Famulski K, Matas A, and Halloran PF, Understanding the causes of kidney transplant failure: the dominant role of antibody-mediated rejection and nonadherence. Am J Transplant 12 (2012) 388–99. [DOI] [PubMed] [Google Scholar]
- [5].Heesters BA, van der Poel CE, Das A, and Carroll MC, Antigen Presentation to B Cells. Trends Immunol 37 (2016) 844–854. [DOI] [PubMed] [Google Scholar]
- [6].Gonzalez SF, Degn SE, Pitcher LA, Woodruff M, Heesters BA, and Carroll MC, Trafficking of B cell antigen in lymph nodes. Annu Rev Immunol 29 (2011) 215–33. [DOI] [PubMed] [Google Scholar]
- [7].Hey YY, and O’Neill HC, Murine spleen contains a diversity of myeloid and dendritic cells distinct in antigen presenting function. J Cell Mol Med 16 (2012) 2611–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pape KA, Catron DM, Itano AA, and Jenkins MK, The humoral immune response is initiated in lymph nodes by B cells that acquire soluble antigen directly in the follicles. Immunity 26 (2007) 491–502. [DOI] [PubMed] [Google Scholar]
- [9].Heesters BA, Chatterjee P, Kim YA, Gonzalez SF, Kuligowski MP, Kirchhausen T, and Carroll MC, Endocytosis and recycling of immune complexes by follicular dendritic cells enhances B cell antigen binding and activation. Immunity 38 (2013) 1164–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kranich J, and Krautler NJ, How Follicular Dendritic Cells Shape the B-Cell Antigenome. Frontiers in immunology 7 (2016) 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Land WG, Agostinis P, Gasser S, Garg AD, and Linkermann A, Transplantation and Damage-Associated Molecular Patterns (DAMPs). Am J Transplant 16 (2016) 3338–3361. [DOI] [PubMed] [Google Scholar]
- [12].Wakim LM, and Bevan MJ, Cross-dressed dendritic cells drive memory CD8+ T-cell activation after viral infection. Nature 471 (2011) 629–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zeng F, and Morelli AE, Extracellular vesicle-mediated MHC cross-dressing in immune homeostasis, transplantation, infectious diseases, and cancer. Semin Immunopathol (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nakayama M, Antigen Presentation by MHC-Dressed Cells. Frontiers in immunology 5 (2014) 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Harper IG, Ali JM, Harper SJ, Wlodek E, Alsughayyir J, Negus MC, Qureshi MS, Motalleb-Zadeh R, Saeb-Parsy K, Bolton EM, Bradley JA, Clatworthy MR, Conlon TM, and Pettigrew GJ, Augmentation of Recipient Adaptive Alloimmunity by Donor Passenger Lymphocytes within the Transplant. Cell Rep 15 (2016) 1214–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sivaganesh S, Harper SJ, Conlon TM, Callaghan CJ, Saeb-Parsy K, Negus MC, Motallebzadeh R, Bolton EM, Bradley JA, and Pettigrew GJ, Copresentation of intact and processed MHC alloantigen by recipient dendritic cells enables delivery of linked help to alloreactive CD8 T cells by indirect-pathway CD4 T cells. Journal of immunology 190 (2013) 5829–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Markey KA, Koyama M, Gartlan KH, Leveque L, Kuns RD, Lineburg KE, Teal BE, MacDonald KP, and Hill GR, Cross-dressing by donor dendritic cells after allogeneic bone marrow transplantation contributes to formation of the immunological synapse and maximizes responses to indirectly presented antigen. Journal of immunology 192 (2014) 5426–33. [DOI] [PubMed] [Google Scholar]
- [18].Saunderson SC, Dunn AC, Crocker PR, and McLellan AD, CD169 mediates the capture of exosomes in spleen and lymph node. Blood 123 (2014) 208–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Naslund TI, Gehrmann U, Qazi KR, Karlsson MC, and Gabrielsson S, Dendritic cell-derived exosomes need to activate both T and B cells to induce antitumor immunity. Journal of immunology 190 (2013) 2712–9. [DOI] [PubMed] [Google Scholar]
- [20].Conlon TM, Saeb-Parsy K, Cole JL, Motallebzadeh R, Qureshi MS, Rehakova S, Negus MC, Callaghan CJ, Bolton EM, Bradley JA, and Pettigrew GJ, Germinal center alloantibody responses are mediated exclusively by indirect-pathway CD4 T follicular helper cells. Journal of immunology 188 (2012) 2643–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bajenoff M, Egen JG, Koo LY, Laugier JP, Brau F, Glaichenhaus N, and Germain RN, Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity 25 (2006) 989–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bajenoff M, Glaichenhaus N, and Germain RN, Fibroblastic reticular cells guide T lymphocyte entry into and migration within the splenic T cell zone. Journal of immunology 181 (2008) 3947–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gerner MY, Torabi-Parizi P, and Germain RN, Strategically localized dendritic cells promote rapid T cell responses to lymph-borne particulate antigens. Immunity 42 (2015) 172–85. [DOI] [PubMed] [Google Scholar]
- [24].Fazilleau N, Mark L, McHeyzer-Williams LJ, and McHeyzer-Williams MG, Follicular helper T cells: lineage and location. Immunity 30 (2009) 324–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, Wang YH, and Dong C, Bcl6 mediates the development of T follicular helper cells. Science 325 (2009) 1001–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Linterman MA, Rigby RJ, Wong RK, Yu D, Brink R, Cannons JL, Schwartzberg PL, Cook MC, Walters GD, and Vinuesa CG, Follicular helper T cells are required for systemic autoimmunity. The Journal of experimental medicine 206 (2009) 561–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Fazilleau N, McHeyzer-Williams LJ, Rosen H, and McHeyzer-Williams MG, The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nature immunology 10 (2009) 375–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tubo NJ, Pagan AJ, Taylor JJ, Nelson RW, Linehan JL, Ertelt JM, Huseby ES, Way SS, and Jenkins MK, Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell 153 (2013) 785–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pompano RR, Chen J, Verbus EA, Han H, Fridman A, McNeely T, Collier JH, and Chong AS, Titrating T-cell epitopes within self-assembled vaccines optimizes CD4+ helper T cell and antibody outputs. Adv Healthc Mater 3 (2014) 1898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Keck S, Schmaler M, Ganter S, Wyss L, Oberle S, Huseby ES, Zehn D, and King CG, Antigen affinity and antigen dose exert distinct influences on CD4 T-cell differentiation. Proc Natl Acad Sci U S A 111 (2014) 14852–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Krishnamoorthy V, Kannanganat S, Maienschein-Cline M, Cook SL, Chen J, Bahroos N, Sievert E, Corse E, Chong A, and Sciammas R, The IRF4 Gene Regulatory Module Functions as a Read-Write Integrator to Dynamically Coordinate T Helper Cell Fate. Immunity 47 (2017) 481–497 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Snook JP, Kim C, and Williams MA, TCR signal strength controls the differentiation of CD4(+) effector and memory T cells. Sci Immunol 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Suan D, Nguyen A, Moran I, Bourne K, Hermes JR, Arshi M, Hampton HR, Tomura M, Miwa Y, Kelleher AD, Kaplan W, Deenick EK, Tangye SG, Brink R, Chtanova T, and Phan TG, T follicular helper cells have distinct modes of migration and molecular signatures in naive and memory immune responses. Immunity 42 (2015) 704–18. [DOI] [PubMed] [Google Scholar]
- [34].Crotty S, T follicular helper cell differentiation, function, and roles in disease. Immunity 41 (2014) 529–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ramiscal RR, and Vinuesa CG, T-cell subsets in the germinal center. Immunological reviews 252 (2013) 146–55. [DOI] [PubMed] [Google Scholar]
- [36].Li J, Lu E, Yi T, and Cyster JG, EBI2 augments Tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nature 533 (2016) 110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ballesteros-Tato A, Randall TD, Lund FE, Spolski R, Leonard WJ, and Leon B, T Follicular Helper Cell Plasticity Shapes Pathogenic T Helper 2 Cell-Mediated Immunity to Inhaled House Dust Mite. Immunity 44 (2016) 259–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mesin L, Ersching J, and Victora GD, Germinal Center B Cell Dynamics. Immunity 45 (2016) 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pape KA, Kouskoff V, Nemazee D, Tang HL, Cyster JG, Tze LE, Hippen KL, Behrens TW, and Jenkins MK, Visualization of the genesis and fate of isotype-switched B cells during a primary immune response. The Journal of experimental medicine 197 (2003) 1677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Moriyama S, Takahashi N, Green JA, Hori S, Kubo M, Cyster JG, and Okada T, Sphingosine-1-phosphate receptor 2 is critical for follicular helper T cell retention in germinal centers. The Journal of experimental medicine 211 (2014) 1297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yi T, Wang X, Kelly LM, An J, Xu Y, Sailer AW, Gustafsson JA, Russell DW, and Cyster JG, Oxysterol gradient generation by lymphoid stromal cells guides activated B cell movement during humoral responses. Immunity 37 (2012) 535–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ma CS, and Phan TG, Here, there and everywhere: T follicular helper cells on the move. Immunology 152 (2017) 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Victora GD, Dominguez-Sola D, Holmes AB, Deroubaix S, Dalla-Favera R, and Nussenzweig MC, Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood 120 (2012) 2240–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, and Nussenzweig MC, Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 143 (2010) 592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tarlinton D, and Victora G, Editorial overview: Germinal centers and memory B-cells: from here to eternity. Current opinion in immunology 45 (2017) v–viii. [DOI] [PubMed] [Google Scholar]
- [46].Victora GD, and Mesin L, Clonal and cellular dynamics in germinal centers. Current opinion in immunology 28 (2014) 90–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mayer CT, Gazumyan A, Kara EE, Gitlin AD, Golijanin J, Viant C, Pai J, Oliveira TY, Wang Q, Escolano A, Medina-Ramirez M, Sanders RW, and Nussenzweig MC, The microanatomic segregation of selection by apoptosis in the germinal center. Science 358 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chen J, Wang Q, Yin D, Vu V, Sciammas R, and Chong AS, Cutting Edge: CTLA-4Ig Inhibits Memory B Cell Responses and Promotes Allograft Survival in Sensitized Recipients. Journal of immunology 195 (2015) 4069–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chen J, Yin H, Xu J, Wang Q, Edelblum KL, Sciammas R, and Chong AS, Reversing endogenous alloreactive B cell GC responses with anti-CD154 or CTLA-4Ig. Am J Transplant 13 (2013) 2280–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yang J, Chen J, Young JS, Wang Q, Yin D, Sciammas R, and Chong AS, Tracing Donor-MHC Class II Reactive B cells in Mouse Cardiac Transplantation: Delayed CTLA4-Ig Treatment Prevents Memory Alloreactive B-Cell Generation. Transplantation 100 (2016) 1683–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Young JS, Chen J, Miller ML, Vu V, Tian C, Moon JJ, Alegre ML, Sciammas R, and Chong AS, Delayed Cytotoxic T Lymphocyte-Associated Protein 4-Immunoglobulin Treatment Reverses Ongoing Alloantibody Responses and Rescues Allografts From Acute Rejection. Am J Transplant 16 (2016) 2312–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kwun J, Oh BC, Gibby AC, Ruhil R, Lu VT, Kim DW, Page EK, Bulut OP, Song MQ, Farris AB, Kirk AD, Knechtle SJ, and Iwakoshi NN, Patterns of de novo allo B cells and antibody formation in chronic cardiac allograft rejection after alemtuzumab treatment. Am J Transplant 12 (2012) 2641–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kaji T, Ishige A, Hikida M, Taka J, Hijikata A, Kubo M, Nagashima T, Takahashi Y, Kurosaki T, Okada M, Ohara O, Rajewsky K, and Takemori T, Distinct cellular pathways select germline-encoded and somatically mutated antibodies into immunological memory. The Journal of experimental medicine 209 (2012) 2079–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Taylor JJ, Pape KA, and Jenkins MK, A germinal center-independent pathway generates unswitched memory B cells early in the primary response. The Journal of experimental medicine 209 (2012) 597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wing JB, Ise W, Kurosaki T, and Sakaguchi S, Regulatory T Cells Control Antigen-Specific Expansion of Tfh Cell Number and Humoral Immune Responses via the Coreceptor CTLA-4. Immunity 41 (2014) 1013–25. [DOI] [PubMed] [Google Scholar]
- [56].Sage PT, Paterson AM, Lovitch SB, and Sharpe AH, The coinhibitory receptor ctla-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity 41 (2014) 1026–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Vincenti F, Belatacept and Long-Term Outcomes in Kidney Transplantation. N Engl J Med 374 (2016) 2600–1. [DOI] [PubMed] [Google Scholar]
- [58].Bray RA, Gebel HM, Townsend R, Roberts ME, Polinsky M, Yang L, Meier-Kriesche HU, and Larsen CP, De novo donor-specific antibodies in belatacept-treated vs cyclosporine-treated kidney-transplant recipients: Post hoc analyses of the randomized phase III BENEFIT and BENEFIT-EXT studies. Am J Transplant (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Leibler C, Thiolat A, Henique C, Samson C, Pilon C, Tamagne M, Pirenne F, Vingert B, Cohen JL, and Grimbert P, Control of Humoral Response in Renal Transplantation by Belatacept Depends on a Direct Effect on B Cells and Impaired T Follicular Helper-B Cell Crosstalk. J Am Soc Nephrol 29 (2018) 1049–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Crompton PD, Kayala MA, Traore B, Kayentao K, Ongoiba A, Weiss GE, Molina DM, Burk CR, Waisberg M, Jasinskas A, Tan X, Doumbo S, Doumtabe D, Kone Y, Narum DL, Liang X, Doumbo OK, Miller LH, Doolan DL, Baldi P, Felgner PL, and Pierce SK, A prospective analysis of the Ab response to Plasmodium falciparum before and after a malaria season by protein microarray. Proc Natl Acad Sci U S A 107 (2010) 6958–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Parameswaran P, Liu Y, Roskin KM, Jackson KK, Dixit VP, Lee JY, Artiles KL, Zompi S, Vargas MJ, Simen BB, Hanczaruk B, McGowan KR, Tariq MA, Pourmand N, Koller D, Balmaseda A, Boyd SD, Harris E, and Fire AZ, Convergent antibody signatures in human dengue. Cell Host Microbe 13 (2013) 691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Childs LM, Baskerville EB, and Cobey S, Trade-offs in antibody repertoires to complex antigens. Philos Trans R Soc Lond B Biol Sci 370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wiebe C, Gibson IW, Blydt-Hansen TD, Karpinski M, Ho J, Storsley LJ, Goldberg A, Birk PE, Rush DN, and Nickerson PW, Evolution and clinical pathologic correlations of de novo donor-specific HLA antibody post kidney transplant. Am J Transplant 12 (2012) 1157–67. [DOI] [PubMed] [Google Scholar]
- [64].Wiebe C, Rush DN, Nevins TE, Birk PE, Blydt-Hansen T, Gibson IW, Goldberg A, Ho J, Karpinski M, Pochinco D, Sharma A, Storsley L, Matas AJ, and Nickerson PW, Class II Eplet Mismatch Modulates Tacrolimus Trough Levels Required to Prevent Donor-Specific Antibody Development. J Am Soc Nephrol 28 (2017) 3353–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, Wang YH, Lim H, Reynolds JM, Zhou XH, Fan HM, Liu ZM, Neelapu SS, and Dong C, Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med 17 (2011) 983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, Srivastava M, Divekar DP, Beaton L, Hogan JJ, Fagarasan S, Liston A, Smith KG, and Vinuesa CG, Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med 17 (2011) 975–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wollenberg I, Agua-Doce A, Hernandez A, Almeida C, Oliveira VG, Faro J, and Graca L, Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. Journal of immunology 187 (2011) 4553–60. [DOI] [PubMed] [Google Scholar]
- [68].Botta D, Fuller MJ, Marquez-Lago TT, Bachus H, Bradley JE, Weinmann AS, Zajac AJ, Randall TD, Lund FE, Leon B, and Ballesteros-Tato A, Dynamic regulation of T follicular regulatory cell responses by interleukin 2 during influenza infection. Nature immunology 18 (2017) 1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Victora GD, and Nussenzweig MC, Germinal centers. Annu Rev Immunol 30 (2012) 429–57. [DOI] [PubMed] [Google Scholar]
- [70].Sayin I, Radtke AJ, Vella LA, Jin W, Wherry EJ, Buggert M, Betts MR, Herati RS, Germain RN, and Canaday DH, Spatial distribution and function of T follicular regulatory cells in human lymph nodes. The Journal of experimental medicine 215 (2018) 1531–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, and Shevach EM, GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A 106 (2009) 13445–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Shinnakasu R, and Kurosaki T, Regulation of memory B and plasma cell differentiation. Current opinion in immunology 45 (2017) 126–131. [DOI] [PubMed] [Google Scholar]
- [73].Phan TG, and Tangye SG, Memory B cells: total recall. Current opinion in immunology 45 (2017) 132–140. [DOI] [PubMed] [Google Scholar]
- [74].Paus D, Phan TG, Chan TD, Gardam S, Basten A, and Brink R, Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. The Journal of experimental medicine 203 (2006) 1081–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Basten A, and Brink R, High affinity germinal center B cells are actively selected into the plasma cell compartment. The Journal of experimental medicine 203 (2006) 2419–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ochiai K, Maienschein-Cline M, Mandal M, Triggs JR, Bertolino E, Sciammas R, Dinner AR, Clark MR, and Singh H, A self-reinforcing regulatory network triggered by limiting IL-7 activates pre-BCR signaling and differentiation. Nature immunology 13 (2012) 300–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, and Singh H, Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity 25 (2006) 225–36. [DOI] [PubMed] [Google Scholar]
- [78].Ochiai K, Maienschein-Cline M, Simonetti G, Chen J, Rosenthal R, Brink R, Chong AS, Klein U, Dinner AR, Singh H, and Sciammas R, Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity 38 (2013) 918–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Muto A, Ochiai K, Kimura Y, Itoh-Nakadai A, Calame KL, Ikebe D, Tashiro S, and Igarashi K, Bach2 represses plasma cell gene regulatory network in B cells to promote antibody class switch. EMBO J 29 (2010) 4048–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Krautler NJ, Suan D, Butt D, Bourne K, Hermes JR, Chan TD, Sundling C, Kaplan W, Schofield P, Jackson J, Basten A, Christ D, and Brink R, Differentiation of germinal center B cells into plasma cells is initiated by high-affinity antigen and completed by Tfh cells. The Journal of experimental medicine 214 (2017) 1259–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Pape KA, Taylor JJ, Maul RW, Gearhart PJ, and Jenkins MK, Different B cell populations mediate early and late memory during an endogenous immune response. Science 331 (2011) 1203–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, Takahashi Y, Fukuyama H, Okada T, and Kurosaki T, Regulated selection of germinal-center cells into the memory B cell compartment. Nature immunology 17 (2016) 861–9. [DOI] [PubMed] [Google Scholar]
- [83].Gitlin AD, von Boehmer L, Gazumyan A, Shulman Z, Oliveira TY, and Nussenzweig MC, Independent Roles of Switching and Hypermutation in the Development and Persistence of B Lymphocyte Memory. Immunity 44 (2016) 769–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Martin SW, and Goodnow CC, Burst-enhancing role of the IgG membrane tail as a molecular determinant of memory. Nature immunology 3 (2002) 182–8. [DOI] [PubMed] [Google Scholar]
- [85].Bannard O, Horton RM, Allen CD, An J, Nagasawa T, and Cyster JG, Germinal center centroblasts transition to a centrocyte phenotype according to a timed program and depend on the dark zone for effective selection. Immunity 39 (2013) 912–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Weisel FJ, Zuccarino-Catania GV, Chikina M, and Shlomchik MJ, A Temporal Switch in the Germinal Center Determines Differential Output of Memory B and Plasma Cells. Immunity 44 (2016) 116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Tambur AR, Campbell P, Claas FH, Feng S, Gebel HM, Jackson AM, Mannon RB, Reed EF, Tinckam K, Askar M, Chandraker A, Chang PP, Colvin M, Demetris AJ, Diamond JM, Dipchand AI, Fairchild RL, Ford ML, Friedewald J, Gill RG, Glotz D, Goldberg H, Hachem R, Knechtle S, Kobashigawa J, Levine DJ, Levitsky J, Mengel M, Milford E, Newell KA, O’Leary JG, Palmer S, Randhawa P, Smith J, Snyder L, Starling RC, Sweet S, Taner T, Taylor CJ, Woodle S, Zeevi A, and Nickerson P, Sensitization in Transplantation: Assessment of Risk (STAR) 2017 Working Group Meeting Report. Am J Transplant (2018). [DOI] [PubMed] [Google Scholar]
- [88].Lavinder JJ, Horton AP, Georgiou G, and Ippolito GC, Next-generation sequencing and protein mass spectrometry for the comprehensive analysis of human cellular and serum antibody repertoires. Curr Opin Chem Biol 24 (2015) 112–20. [DOI] [PubMed] [Google Scholar]
- [89].Dogan I, Bertocci B, Vilmont V, Delbos F, Megret J, Storck S, Reynaud CA, and Weill JC, Multiple layers of B cell memory with different effector functions. Nature immunology 10 (2009) 1292–9. [DOI] [PubMed] [Google Scholar]
- [90].Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, Good-Jacobson KL, and Shlomchik MJ, CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nature immunology 15 (2014) 631–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Krishnamurty AT, Thouvenel CD, Portugal S, Keitany GJ, Kim KS, Holder A, Crompton PD, Rawlings DJ, and Pepper M, Somatically Hypermutated Plasmodium-Specific IgM(+) Memory B Cells Are Rapid, Plastic, Early Responders upon Malaria Rechallenge. Immunity 45 (2016) 402–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Rozanski CH, Arens R, Carlson LM, Nair J, Boise LH, Chanan-Khan AA, Schoenberger SP, and Lee KP, Sustained antibody responses depend on CD28 function in bone marrow-resident plasma cells. The Journal of experimental medicine 208 (2011) 1435–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Rozanski CH, Utley A, Carlson LM, Farren MR, Murray M, Russell LM, Nair JR, Yang Z, Brady W, Garrett-Sinha LA, Schoenberger SP, Green JM, Boise LH, and Lee KP, CD28 Promotes Plasma Cell Survival, Sustained Antibody Responses, and BLIMP-1 Upregulation through Its Distal PYAP Proline Motif. Journal of immunology 194 (2015) 4717–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Halliley JL, Tipton CM, Liesveld J, Rosenberg AF, Darce J, Gregoretti IV, Popova L, Kaminiski D, Fucile CF, Albizua I, Kyu S, Chiang KY, Bradley KT, Burack R, Slifka M, Hammarlund E, Wu H, Zhao L, Walsh EE, Falsey AR, Randall TD, Cheung WC, Sanz I, and Lee FE, Long-Lived Plasma Cells Are Contained within the CD19(−)CD38(hi)CD138(+) Subset in Human Bone Marrow. Immunity 43 (2015) 132–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Bray RA, Gebel HM, Townsend R, Roberts ME, Polinsky M, Yang L, Meier-Kriesche HU, and Larsen CP, Posttransplant reduction in preexisting donor-specific antibody levels after belatacept- versus cyclosporine-based immunosuppression: Post hoc analyses of BENEFIT and BENEFIT-EXT. Am J Transplant (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Wiebe C, Gibson IW, Blydt-Hansen TD, Pochinco D, Birk PE, Ho J, Karpinski M, Goldberg A, Storsley L, Rush DN, and Nickerson PW, Rates and Determinants of Progression to Graft Failure in Kidney Allograft Recipients With De Novo Donor-Specific Antibody. Am J Transplant 15 (2015) 2921–30. [DOI] [PubMed] [Google Scholar]
- [97].Cooper JE, Gralla J, Cagle L, Goldberg R, Chan L, and Wiseman AC, Inferior kidney allograft outcomes in patients with de novo donor-specific antibodies are due to acute rejection episodes. Transplantation 91 (2011) 1103–9. [DOI] [PubMed] [Google Scholar]
- [98].Ho EK, Vlad G, Vasilescu ER, de la Torre L, Colovai AI, Burke E, Deng M, Schwartz J, Marboe C, Mancini D, Opelz G, and Suciu-Foca N, Pre- and posttransplantation allosensitization in heart allograft recipients: major impact of de novo alloantibody production on allograft survival. Hum Immunol 72 (2011) 5–10. [DOI] [PubMed] [Google Scholar]