

Abstract
Background
Helicobacter pylori recurrence after successful eradication is an important problem. Children are particularly vulnerable to reinfection, by intrafamilial transmission which facilitates the acquisition or recombination of new genetic information by this bacterium. We investigated the evolutionary dynamics of 80 H. pylori strains isolated from two paediatric patients with recurrent infection (recrudescence and reinfection).
Results
We characterized the virulence genes vacA (s1, m1, s2, and m2), cagA, cagE, and babA2 and performed multilocus sequence typing (MLST) on 7 housekeeping genes (atpA, efp, ureI, ppa, mutY, trpC, and yphC) to infer the evolutionary dynamics of the H. pylori strains through phylogenetic and genealogic inference analyses, genetic diversity analysis and the exploration of recombination events during recurrent infections. The virulence genotype vacAs1m1/cagA+/cagE+/babA2 was present at a high frequency, as were the EPIYA motifs EPIYA-A, −B and -C. Furthermore, the housekeeping genes of the H. pylori strains exhibited high genetic variation, comprising 26 new alleles and 17 new Sequence Type (ST). In addition, the hpEurope (76.5%) and hspWAfrica (23.5%) populations predominated among the paediatric strains. All strains, regardless of their ancestral affiliation, harboured western EPIYA motifs.
Conclusions
This study provides evidence of the evolutionary dynamics of the H. pylori strains in two paediatric patients during recrudescence and reinfection events. In particular, our study shows that the strains changed during these events, as evidenced by the presence of different STs that emerged before and after treatment; these changes may be due to the accumulation of mutations and recombination events during the diversification process and recolonization of the patients by different genotypes.
Electronic supplementary material
The online version of this article (10.1186/s12866-019-1554-z) contains supplementary material, which is available to authorized users.
Keywords: Helicobacter pylori, Recurrent infection, Reinfection, Paedriatric patients, Genetic variability, Evolutionary relationship
Background
Helicobacter pylori is a gram-negative, pleomorphic, microaerophilic bacterium that has adapted to survive the extreme conditions of the human stomach [1]. Infection by this bacterium generally occurs during childhood and persists throughout the life of the host if it is not eradicated [2]. The infection is mostly asymptomatic and, to a lesser extent, is associated with chronic gastritis, gastroduodenal ulcers, mucosa-associated lymphoid tissue lymphoma and gastric cancer [3, 4].
Generally, bacteria exhibit clonal propagation during infection; however, H. pylori increases its adaptation potential by being highly recombinant [5]. The transmission from parents to offspring [6–8], horizontal gene transfer occurs through contaminated food, water, and non-parental caregivers [9], facilitates the acquisition or recombination of new genetic information by this specie.
H. pylori is an example of successful bacterial adaptation because it has evolved with its host [10, 11]. Phylogeographic studies performed with multilocus sequence typing (MLST) have shown that the dispersal of H. pylori throughout the world is associated with human evolution because H. pylori has infected humans since before the migration from Africa occurred [12]. This evolution of H. pylori is observed in the different recorded molecular variants associated with different geographical areas [11, 13–16].
One of the evolutionary features of H. pylori is its great capacity for recombination during infection in humans. A study performed to determine the sequence type (ST) diversification of H. pylori strains in an animal model showed that in only three months, the original strain ST181 accumulated both synonymous and non-synonymous mutations in different housekeeping genes, giving rise to new STs (ST2902 and ST2903) [17]. In addition, several changes occurred in the strains through allelic recombination in the cagA and babA2 genes, and nucleotide substitutions in the vacA gene, as well as through the development of chimeric vacAs2m1 strains, thus confirming that genetic variation, which is critical for adaptation to specific conditions or environments within the host [17].
Studies in humans have revealed the presence of dominant strains; however, most studies use only one isolate per patient, resulting in very few reports of mixed infections. In a previous study, we characterized the virulence genes cagA, cagE, vacA and babA2 in five isolates from each patient analyzed, and our results showed that 75% of patients had mixed infections, some with up to 5 different genotypes [18]. Other studies that have sequenced the complete genome have also found different genotypes apparently produced by recombination [19]. In contrast, studies employing MLST have detected patients infected with strains containing the same STs or with closely related strains [9, 20].
During recurrent H. pylori infection, even when patients are treated, signs and symptoms of disease reappear, resulting in two clinically important mechanisms: recrudescence and reinfection. Recrudescence is defined as the apparent elimination of infection due to bacterial suppression but not eradication [21, 22], while reinfection occurs when a patient is reinfected after successful eradication with a new strain or with the same strain at least one year after eradication [23, 24]. Both mechanisms of infection by H. pylori assume that the bacterial population is suppressed whether or not the treatment used is effective, thus affecting the clinical diagnosis.
The mechanisms of recurrence have effects on the H. pylori population before and after clinical treatment because the population can experience contraction and expansion events of genetic variation due to the reduction and increase in the population size. Therefore, to infer the evolutionary dynamics of H. pylori, we performed phylogenetic and genealogy inference analyses, genetic diversity analysis, and the exploration of possible recombination events in 80 H. pylori strains isolated from two paediatric patients with recurrent infection. The H. pylori strains were characterized before and after treatment in these patients, who were diagnosed by their symptoms as experiencing recrudescence or reinfection. In particular, we were interested in 1) determining whether the suppressed strains in the host are the same over time (recrudescence) and 2) determining whether the reinfection occurred with different strains.
Results
The analysis of 80 strains isolated from two paediatric patients with recurrent H. pylori infection (40 strains per patient), showed the antimicrobial susceptibility profile. In patient one, 100% of the strains involved in the first event were sensitive to CLA (0.015 μg/mL) and AMX (0.25 μg/mL) but resistant to MTZ (16 μg/mL); during the second event, the MICs of the strains were different but they did not show changes in the sensitivity, remaining sensitive to CLA (0.015–0.031 μg/mL) and AMX (0.015 μg/mL) but resistant to MTZ (32–128 μg/mL). Similarly, in patient two, 100% of the strains involved in the first event were sensitive to CLA (< 0.0078–0.031 μg/mL) and AMX (0.015 μg/mL) but resistant to MTZ (16–32 μg/mL); during the second event, the MICs of the strains were different, without changes in the sensitivity 100% of these strains were sensitive to CLA (0.015–0.031 μg/mL) and AMX (0.0078–0.015 μg/mL) but resistant to MTZ (8–32 μg/mL) (Table 1).
Table 1.
Susceptibility profile of the Helicobacter pylori strains identified from paedriatic patients during different events
| Strain | CLA Event |
AMX Event |
MTZ Event |
TC Event |
||||
|---|---|---|---|---|---|---|---|---|
| First | Second | First | Second | First | Second | First | Second | |
| [μg/mL] | [μg/mL] | [μg/mL] | [μg/mL] | |||||
| Patient One | ||||||||
| 1 | 0.015 | 0.031 | 0.25 | 0.015 | 16 | 64 | 0.125 | 0.062 |
| 2 | 0.015 | 0.015 | 0.25 | 0.015 | 16 | 64 | 0.5 | 0.062 |
| 3 | 0.015 | 0.031 | 0.25 | 0.015 | 16 | 32 | 0.5 | 0.062 |
| 4 | 0.015 | 0.015 | 0.25 | 0.015 | 16 | 64 | 0.5 | 0.062 |
| 5 | 0.015 | 0.015 | 0.25 | 0.015 | 16 | 128 | 0.25 | 0.062 |
| 6 | 0.015 | 0.031 | 0.25 | 0.015 | 16 | 64 | 0.25 | 0.062 |
| 7 | 0.015 | 0.015 | 0.25 | 0.015 | 16 | 64 | 0.5 | 0.062 |
| 8 | 0.015 | 0.015 | 0.25 | 0.015 | 16 | 32 | 0.25 | 0.062 |
| 9 | 0.015 | 0.015 | 0.25 | 0.015 | 16 | 64 | 0.25 | 0.062 |
| 10 | 0.015 | 0.015 | 0.25 | 0.015 | 16 | 128 | 0.125 | 0.062 |
| 11 | 0.015 | 0.031 | 0.25 | 0.015 | 16 | 64 | 0.5 | 0.062 |
| 12 | 0.015 | 0.015 | 0.25 | 0.015 | 16 | 64 | 0.5 | 0.062 |
| 13 | 0.015 | 0.031 | 0.25 | 0.015 | 16 | 32 | 0.5 | 0.062 |
| 14 | 0.015 | 0.015 | 0.25 | 0.015 | 16 | 64 | 0.25 | 0.062 |
| 15 | 0.015 | 0.015 | 0.25 | 0.015 | 16 | 128 | 0.25 | 0.062 |
| 16 | 0.015 | 0.015 | 0.25 | 0.015 | 16 | 64 | 0.5 | 0.062 |
| 17 | 0.015 | 0.015 | 0.25 | 0.015 | 16 | 64 | 0.25 | 0.062 |
| 18 | 0.015 | 0.031 | 0.25 | 0.015 | 16 | 64 | 0.25 | 0.062 |
| 19 | 0.015 | 0.015 | 0.25 | 0.015 | 16 | 128 | 0.5 | 0.062 |
| 20 | 0.015 | 0.015 | 0.25 | 0.015 | 16 | 64 | 0.25 | 0.062 |
| Patient Two | ||||||||
| 1 | < 0.078 | 0.031 | 0.015 | 0.0078 | 32 | 8 | 0.062 | 0.062 |
| 2 | < 0.078 | 0.015 | 0.015 | 0.062 | 32 | 8 | 0.062 | 0.25 |
| 3 | < 0.078 | 0.031 | 0.015 | 0.015 | 32 | 16 | 0.031 | 0.062 |
| 4 | 0.031 | 0.031 | 0.015 | 0.0078 | 16 | 32 | 0.062 | 0.062 |
| 5 | < 0.078 | 0.031 | 0.015 | 0.031 | 64 | 8 | 0.062 | 0.062 |
| 6 | < 0.078 | 0.015 | 0.015 | 0.062 | 32 | 8 | 0.062 | 0.25 |
| 7 | < 0.078 | 0.015 | 0.015 | 0.062 | 32 | 8 | 0.031 | 0.062 |
| 8 | < 0.078 | 0.031 | 0.015 | 0.015 | 32 | 16 | 0.062 | 0.062 |
| 9 | < 0.078 | 0.015 | 0.015 | 0.062 | 32 | 8 | 0.062 | 0.25 |
| 10 | < 0.078 | 0.031 | 0.015 | 0.015 | 32 | 16 | 0.031 | 0.062 |
| 11 | 0.031 | 0.031 | 0.015 | 0.0078 | 16 | 32 | 0.062 | 0.062 |
| 12 | < 0.078 | 0.031 | 0.015 | 0.031 | 64 | 8 | 0.062 | 0.062 |
| 13 | < 0.078 | 0.015 | 0.015 | 0.062 | 32 | 8 | 0.062 | 0.25 |
| 14 | < 0.078 | 0.031 | 0.015 | 0.015 | 32 | 16 | 0.031 | 0.062 |
| 15 | 0.031 | 0.031 | 0.015 | 0.0078 | 16 | 32 | 0.062 | 0.062 |
| 16 | < 0.078 | 0.031 | 0.015 | 0.031 | 64 | 8 | 0.125 | 0.031 |
| 17 | < 0.078 | 0.015 | 0.015 | 0.062 | 32 | 8 | 0.062 | 0.25 |
| 18 | < 0.078 | 0.031 | 0.015 | 0.015 | 32 | 16 | 0.031 | 0.062 |
| 19 | 0.031 | 0.031 | 0.015 | 0.0078 | 16 | 32 | 0.062 | 0.062 |
| 20 | < 0.078 | 0.031 | 0.015 | 0.031 | 64 | 8 | 0.062 | 0.25 |
CLA: Clarithromycin (Resistant ≥1 μg/mL CLSI 2015) AMX: Amoxicillin (Resistant ≥4 g/mL Torres et al., 2001) MTZ: Metronidazole (Sensitive < 2 μg/mL; Intermediate 2 to 4 μg/mL; Low Resistant 8 to 16 μg/mL, Moderate Resistant 32 to 64 μg/mL; High Resistant ≥128 μg/mL Poon et al., 2009; Maggi et al., 2000 TC: Tetracycline ≥4 μg/mL Gerrits et al., 2003)
Likewise, the 80 strains exhibited variable frequencies of the virulence genes within the mosaic vacA (s1, s2, m1, and m2) structure (Tables 2 and 3). For vacA, allele s1 was the most frequent, at 77.5% (62/80), while alleles s2, at 20% (16/80), and m2, at 1% (1/80), were the least frequent. The frequencies of the cagA and cagE genes, which belong to the cag-PAI (Pathogenicity Island), were 97.5% (78/80) and 98.75% (79/80), respectively. The babA2 gene was present in 25% (20/80) of strains. The following EPIYA motifs were identified in the polymorphic region of the CagA protein: 41.25% (33/80) type ABC, 26.25% (21/80) type ABCC, 26.25% (21/80) type ABCCC, 1.25% (1/80) type AAB&C, 1.25% (1/80) type AB&C and 1.25% (1/80) type ABC& (Additional file 1: Table S1). Six different genotypes were identified among the paediatric strains.
Table 2.
Characteristics of the Helicobacter pylori strains identified from patient one during different events
| Patient one | ||||
|---|---|---|---|---|
| Date of biopsy | First event | |||
| Strain | ST | Virulence genes | EPIYA motif | |
| August 2006 | 1 | 2888 | vacAs1+ / cagA+ / cagE+ / babA2+ | ABC |
| 2 | 2889 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCC | |
| 3 | 2890 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCC | |
| 4 | 313 | vacAs2m1 / cagA+ / cagE+ / babA2- | ABCC | |
| 5 | 313 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCC | |
| 6 | 2891 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCCC | |
| 7 | 288 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCC | |
| 8 | 313 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCC | |
| 9 | 288 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCC | |
| 10 | 313 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCC | |
| 11 | 313 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCC | |
| 12 | 313 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCC | |
| 13 | 313 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCC | |
| 14 | 313 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCC | |
| 15 | 313 | vacAm1+ / cagA+ / cagE+ / babA2- | ABCC | |
| 16 | 2892 | vacAs2m1 / cagA+ / cagE+ / babA2- | ABCC | |
| 17 | 313 | vacAs2m1 / cagA+ / cagE+ / babA2- | ABCC | |
| 18 | 313 | vacAs2m1 / cagA+ / cagE+ / babA2- | ABCC | |
| 19 | 2893 | vacAs2m1 / cagA+ / cagE+ / babA2- | ABCC | |
| 20 | 313 | vacAs2m1 / cagA+ / cagE+ / babA2- | ABCC | |
| October 2007 | Second event | |||
| Strain | ST | Virulence genes | EPIYA motif | |
| 1 | 288 | vacAs2m1 / cagA+ / cagE+ / babA2- | ABCCC | |
| 2 | 288 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCCC | |
| 3 | 288 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCCC | |
| 4 | 288 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCCC | |
| 5 | 313 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCCC | |
| 6 | 288 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCCC | |
| 7 | 288 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCCC | |
| 8 | 288 | vacA- / cagA+ / cagE+ / babA2- | ABCCC | |
| 9 | 288 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCC | |
| 10 | 288 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCCC | |
| 11 | 288 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCCC | |
| 12 | 288 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCCC | |
| 13 | 813 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCCC | |
| 14 | 313 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCCC | |
| 15 | 288 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCCC | |
| 16 | 288 | vacAs2m1 / cagA+ / cagE+ / babA2- | ABCCC | |
| 17 | 288 | vacAs2m1 / cagA+ / cagE+ / babA2- | ABCCC | |
| 18 | 2887 | vacAs2m1 / cagA+ / cagE+ / babA2- | ABCCC | |
| 19 | 313 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCCC | |
| 20 | 288 | vacAs2m1 / cagA+ / cagE+ / babA2- | ABCCC | |
ST: Sequence Type
Table 3.
Characteristics of the Helicobacter pylori strains identified from patient two during different events
| Patient two | ||||
|---|---|---|---|---|
| Date of biopsy | First event | |||
| Strain | ST | Virulence genes | EPIYA motif | |
| October 2007 | 1 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABC& |
| 2 | 2895 | vacAs1m1 / cagA+ / cagE+ / babA2+ | ABC | |
| 3 | 2896 | vacAs1m1 / cagA+ / cagE+ / babA2+ | AAB&C | |
| 4 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABC | |
| 5 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCC | |
| 6 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2+ | ABCC | |
| 7 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABC | |
| 8 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABC | |
| 9 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABC | |
| 10 | 2894 | vacAs1m1 / cagA- / cagE+ / babA2+ | NEGATIVE | |
| 11 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2+ | ABC | |
| 12 | 2897 | vacAs1+ / cagA+ / cagE- / babA2- | ABC | |
| 13 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABC | |
| 14 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2+ | ABC | |
| 15 | 2898 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABC | |
| 16 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABC | |
| 17 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABC | |
| 18 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABC | |
| 19 | 2898 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABC | |
| 20 | 2898 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABC | |
| June 2008 | Second event | |||
| Strain | ST | Virulence genes | EPIYA motif | |
| 1 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABC | |
| 2 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2+ | AB&C | |
| 3 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2+ | ABC | |
| 4 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2+ | ABC | |
| 5 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABC | |
| 6 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2+ | ABC | |
| 7 | 2894 | vacAs2m1 / cagA+ / cagE+ / babA2+ | ABC | |
| 8 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2+ | ABC | |
| 9 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2+ | ABC | |
| 10 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2+ | ABC | |
| 11 | 2894 | vacAs1m1 / cagA- / cagE+ / babA2- | NEGATIVE | |
| 12 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2+ | ABC | |
| 13 | 2899 | vacAs2m1 / cagA+ / cagE+ / babA2+ | ABC | |
| 14 | 2894 | vacAs2m1 / cagA+ / cagE+ / babA2+ | ABC | |
| 15 | 2894 | vacAs2m1 / cagA+ / cagE+ / babA2+ | ABC | |
| 16 | 2900 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABC | |
| 17 | 2894 | vacAs2m1 / cagA+ / cagE+ / babA2+ | ABC | |
| 18 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABC | |
| 19 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABC | |
| 20 | 2894 | vacAs1m1 / cagA+ / cagE+ / babA2- | ABCCC | |
ST: Sequence Type; &: Amino acid change in EPIYA motif
During the first event, patient one harboured the genotypes vacAs1m1/cagA+/cagE+/babA2- and vacAs2m1/cagA+/cagE+/babA2- in 12/20 and 6/20 strains, respectively. In addition, this patient had a greater number of strains containing the EPIYA-ABCC motif (18/20) than the other motifs. The same genotypes were present during the second event (14/20 and 5/20 strains, respectively), although a higher number of strains containing the EPIYA-ABCCC motif (19/20) was observed (Table 2). Patient two harboured the genotypes vacAs1m1/cagA+/cagE+/babA2-, vacAs1m1/cagA+/cagE+/babA2+ and vacAs1m1/cagA−/cagE+/babA2+ in 13/20, 5/20 and 1/20 strains, respectively, during the first event. In addition, patient two harboured greater numbers of strains containing the EPIYA-ABC motif (15/20) and the EPIYA-ABC& and EPIYA-AAB&C motifs. The genotypes vacAs1m1/cagA+/cagE+/babA2-, vacAs1m1/cagA+/cagE+/babA2+, vacAs2m1/cagA+/cagE+/babA2+ and vacAs1m1/cagA−/cagE+/babA2- (6/20, 8/20, 5/20 and 1/20 strains, respectively) were present during the second event. In addition, there were more EPIYA-ABC (17/20) and EPIYA-AB&C motifs during the second event (Table 3).
The analysis of seven housekeeping genes in the H. pylori strains revealed 26 new alleles (atpA, 4 alleles; efp, 3 alleles; mutY, 5 alleles; ppa, 2 alleles; trpC, 4 alleles; ureI, 4 alleles; and yphC, 4 alleles) (Additional file 2: Table S2). The genetic diversity, as measured by the π and θ indices, was high in all genes, with trpC in the first event in both patients presenting the greatest diversity. In most of the analysed genes, it was found that the θ values were higher than the π values, which indicates that there are some haplotypes that are very divergent. The number of haplotypes at each locus ranged from one to four (Table 4).
Table 4.
Polymorphisms and features of housekeeping genes of Helicobacter pylori strains obtained from paediatric patients
| Gene | First event | Second event | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Polymorphic Sites | Haplotype | Hd | Π | Θ | Polymorphic Sites | Haplotype | Hd | Π | Θ | ||
| Patient one | atpA | 23 | 2 | 0.100 | 0.0036 | 0.0104 | 0 | 1 | 0 | 0 | 0 |
| efp | 17 | 3 | 0.279 | 0.0087 | 0.0116 | 0 | 1 | 0 | 0 | 0 | |
| mutY | 32 | 3 | 0.195 | 0.0084 | 0.0214 | 0 | 1 | 0 | 0 | 0 | |
| ppa | 10 | 2 | 0.189 | 0.0047 | 0.0070 | 20 | 3 | 0.353 | 0.00960 | 0.01416 | |
| trpC | 45 | 4 | 0.284 | 0.0124 | 0.0290 | 0 | 1 | 0 | 0 | 0 | |
| ureI | 31 | 4 | 0.284 | 0.0081 | 0.0159 | 14 | 2 | 0.100 | 0.00239 | 0.00675 | |
| yphC | 0 | 1 | 0.000 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | |
| Concatenated | 158 | 8 | 0.647 | 0.00640 | 0.0134 | 34 | 4 | 0.432 | 0.00153 | 0.00281 | |
| Patient two | atpA | 17 | 3 | 0.195 | 0.00357 | 0.0076 | 23 | 2 | 0.100 | 0.00367 | 0.1034 |
| efp | 9 | 2 | 0.100 | 0.00220 | 0.0061 | 9 | 2 | 0.100 | 0.00220 | 0.00619 | |
| mutY | 37 | 3 | 0.195 | 0.01051 | 0.0248 | 0 | 1 | 0 | 0 | 0 | |
| ppa | 18 | 2 | 0.100 | 0.00452 | 0.0127 | 0 | 1 | 0 | 0 | 0 | |
| trpC | 50 | 3 | 0.195 | 0.01412 | 0.0315 | 0 | 1 | 0 | 0 | 0 | |
| ureI | 18 | 2 | 0.100 | 0.00308 | 0.0086 | 18 | 2 | 0.100 | 0.00308 | 0.00867 | |
| yphC | 19 | 3 | 0.353 | 0.00406 | 0.1050 | 21 | 2 | 0.100 | 0.00412 | 0.01161 | |
| Concatenated | 168 | 5 | 0.505 | 0.00577 | 0.0139 | 71 | 3 | 0.195 | 0.00208 | 0.00588 | |
Π: Nucleotide diversity per site; Θ: Average number of nucleotide differences per site
Both paediatric patients demonstrated infection recurrence with 17 new ST sequences. In patient one, ST313 (12/20) predominated in the first infection event, followed by ST288, ST2888, ST2889, ST2890, ST2891, ST2892 and ST2893. ST288 (15/20) predominated during the second infection event, and two new STs, ST813 and ST2887, were present (Table 2). In patient two, we identified ST2894, ST2895, ST2896, ST2897 and ST2898 during the first infection event, while ST2894 prevailed in 18 strains; two new STs, ST2899 and ST2900, emerged during the second infection event (Table 3).
The different STs identified during each event in the two paediatric patients were used as genotyping data in the PHYLOViZ platform. The goeBURST algorithm was used at the TLV level for the dataset analysis. The most frequent alleles among the 80 paediatric strains were atpA 2336 (49.38%), efp 901 (48.15%), mutY 2341 (46.91%) ppa 2199 (74.07%), trpC 2371 and 2413 (46.91% each), ureI 2386 (50%), and yphC 2590 (50.62%). Patient one demonstrated one genotypic signature including five (ST288, ST2890, ST2891, ST2892, ST2893) and three (ST313, ST813, ST2887) linked STs derived from the first and second infection events, respectively (Fig. 1a, b). Patient two demonstrated one genotypic signature comprising two (ST2897, ST2898) and one (ST2900) linked STs derived from the first and second event, respectively, as well as individual unlinked STs (Fig. 2a, b). The genealogy of the H. pylori strains in each paediatric patient was determined using the neighbour-net algorithm and showed recombinant ST networks in both patient one (Fig. 1c) and patient two (Fig. 2c) during each infection event, with bootstrap values of > 84 and 85% for patient one and patient two, respectively. In addition, the STs identified during the second event in each patient were related to those identified in the first event, because they occurred on the same side of the network. In patient one, ST288 (17/40 strains) was the most frequent, followed by ST313 (15/40 strains); ST813, ST2887, ST2888, ST2889, ST2890, ST2891, ST2892, and ST2893 were each present in only a single strain. In patient two, ST2894 (32/40 strains) was the most frequent, followed by ST2898 (3/40 strains); ST2895, ST2896, ST2897, ST2899, and ST2900 were each present in only a single strain.
Fig. 1.

Evolutionary history among the STs of Helicobacter pylori identified in patient one with recurrent infection. a and b show the clonal relationships among the STs of H. pylori during the first and second infection events, respectively. Each line represents a different allele with mutational changes. PHYLOViZ (goeBURST algorithm) was used to define the clonal relationships [25]. a The main clonal complex in the first event was ST313 (12 strains), with five linked STs and two unlinked STs. b The main clonal complex in the second event was ST288 (15 strains), with three STs. c) Evolutionary relationships among the STs of H. pylori during both events. The neighbour-net graph defines the evolutionary relationships [26]; the black circles indicate the STs identified during the first infection event, and the red circles indicate the STs identified during the second infection event. Bootstrap values > 84% are indicated on the paths in the network. The highly branched network structure is indicative of possible recombination events among the STs
Fig. 2.

Evolutionary history among the STs of Helicobacter pylori identified in patient two with recurrent infection. a and b show the clonal relationships among the STs of H. pylori during the first and second infection events, respectively. Each line represents a different allele with mutational changes. PHYLOViZ (goeBURST algorithm) was used to define the clonal relationships [25]. a The main clonal complex in the first event was ST2894 (14 strains), with two linked STs and two unlinked STs. b The main clonal complexes in the second event were again ST2894 (18 strains), with only one ST, as well as ST2899, which was unlinked. c) Evolutionary relationships among the STs of H. pylori during both events. The neighbour-net graph defines the evolutionary relationships [26]; the black circles indicate the STs identified during the first infection event, and the blue circles indicate the STs identified during the second infection event. Bootstrap values > 85% are indicated on the paths in the network. The highly branched network structure is indicative of possible recombination events among the STs
Our eBURST findings provided an overview of the different clonal complexes. Clusters of related isolates and individual unlinked STs are shown as a tree, defining category zero for the seven shared alleles. The central part contains the major clonal complexes, the linked triples and doubles, and the following individual unlinked STs: ST2888, ST2889, ST2895, ST2896 and ST2899 (individually isolated) (Fig. 3). The ST313 and ST2894 clonal complexes (15/80 and 32/80 isolates, respectively) (Fig. 3) were the main founders (blue) (bootstraps: 1000). These complexes (ST313 and ST2894) were present in at least six alleles that also contained individual STs. The ST313 clonal complex contained six SLVs (yellow): ST288 (17/80 isolates) and ST2887, ST2890, ST2891, ST2892 and ST2893 (1/80 isolates each). In addition, in patient one, the SLV288 clonal complex contained TLV813 (1/80 isolates), along with the unlinked STs ST2888 and ST2889. The ST2894 clonal complex was found in patient two, and it contained only three SLVs: ST2898 (3/80 isolates), ST2897 and ST2900 (1/80 isolates each), as well as the unlinked STs ST2895, ST2896 and ST2899. Importantly, the spacing between the unlinked STs and the clonal complexes provides no information about genetic distances.
Fig. 3.

Population “snapshot” of Helicobacter pylori STs found in the paediatric strains and clonal complexes ST313 and ST2894. Clusters of related isolates and individual unlinked STs found in the MLST database for H. pylori are presented as a single eBURST tree [http://www.phyloviz.net/goeburst/] defining category zero and comprising seven shared alleles. Unions link isolates that correspond to clonal complexes. Primary founders (blue) are located in the centre of the group, and the founders of the subgroups are shown for ST2888, ST2889, ST2895, ST2896 and ST2899 (red circles); the labels for the other STs [http://pubmlst.org//helicobacter/] have been removed for clarity. The predicted primary founders are ST313 and ST2894 (bootstrap value: 1000). The primary founders (blue) are located in the centre of the group, and the founders of the subgroups are shown in yellow
Phylogenetic analyses of the concatenated housekeeping genes showed that the STs in this study clustered in the hpEurope population (76.5%), as did STs from other Latin American countries (Fig. 4). However, the STs also clustered within the hspWAfrica subpopulation (23.5%). In addition, all STs from patient one were grouped in the hpEurope population, while the STs from patient two were distributed between the hpEurope and hspWAfrica populations.
Fig. 4.
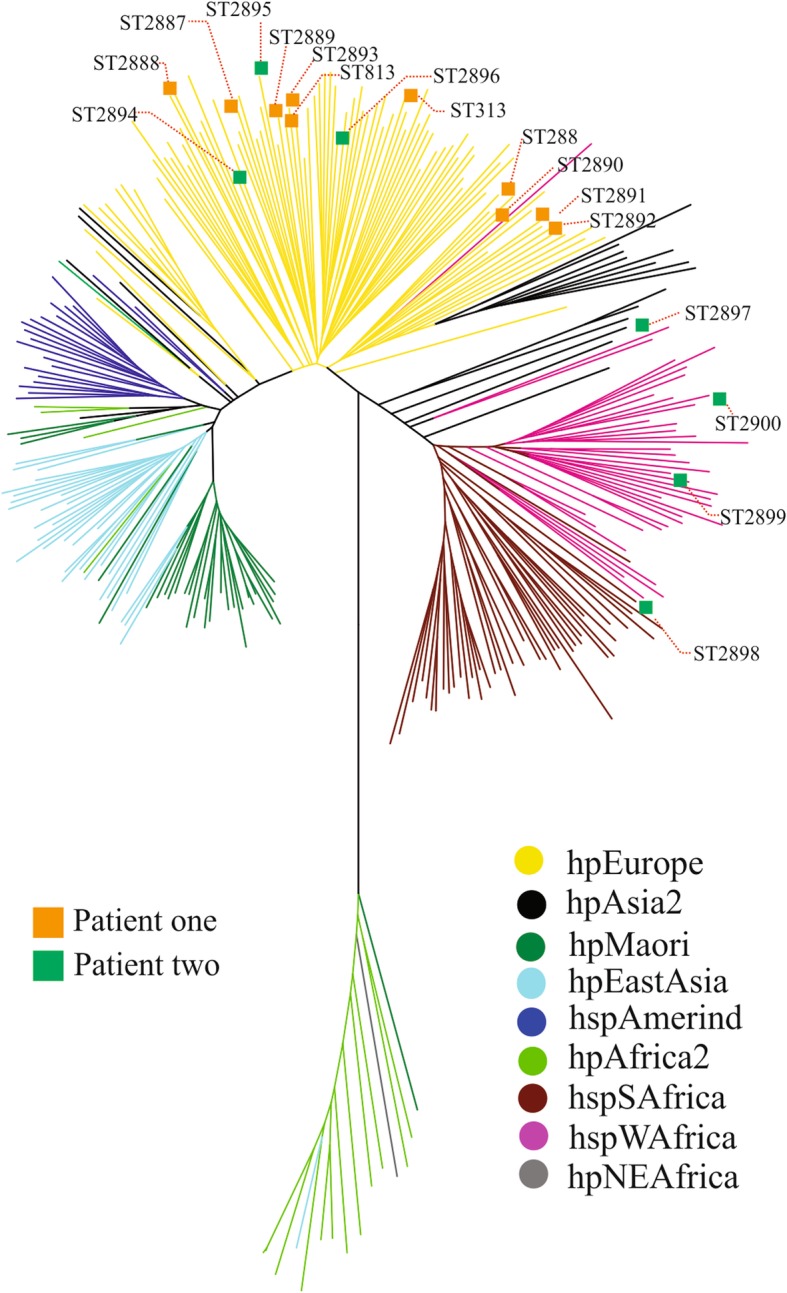
Phylogeography of the analysed STs. The phylogeography was inferred using the neighbour-joining method [27]. The optimal tree with a summed branch length of 3.97653233 is shown. The tree is drawn to scale; branch lengths with the same units as the evolutionary distances are used to infer the phylogenetic tree. The evolutionary distances were computed using the Kimura 2-parameter method [28], and the units are the number of base substitutions per site. The rate variations among the sites were modeled with a gamma distribution (shape parameter = 1). The analysis included 316 reference STs from the H. pylori MLST database [http://pubmlst.org/helicobacter/]. All ambiguous positions were removed for each sequence pair. The major H. pylori populations were identified according to the assigned population available at PubMLST and the identification and positions of the 17 STs identified in this study are shown in the tree. There were 3402 positions in the final dataset. Phylogeography analyses were conducted using MEGA6 [29]
Discussion
H. pylori is a bacterium that is acquired at an early age, and mixed infections have been reported [18, 27, 29]. H. pylori recurrence after successful eradication is an infrequent event and usually involves: reinfection and recrudescence. A higher recurrence of H. pylori infection has been observed in Latin America than in other regions of the world [28, 30, 31]. In this study, H. pylori strains from two Mexican paediatric patients with recurrent infection were analysed.
Patient one, who was diagnosed with reinfection, harboured the same genotypes during both events (2006–2007), suggesting that the reinfection was attributable to recrudescence and not reinfection (Table 2). The presence of identical genotypes at different times indicates treatment failure: the bacterium was suppressed but not eradicated. Xia et al. [32] reported the unsuitability of certain antimicrobial therapies for H. pylori because they do not completely eradicate infection, thus resulting in recurrence. The presence of antibiotic-resistant strains is one of the most frequent causes of recurrent H. pylori infection; in this situation, selection pressure is exerted, resulting in the death of sensitive strains and the persistence of resistant strains. However, data shown in the Table 1 indicate that strains isolated during both events did not demonstrate resistance to antibiotic treatment (AMX and CLA). Another of the most frequent causes of recrudescence is a lack of treatment adherence; this situation conditions the strains to stimuli induced by the environment, which contains different concentrations of antibiotics.
Patient two, diagnosed with recrudescence, harboured different genotypes during the second event (2008), suggesting reinfection and not recrudescence (Table 3). Reinfection is responsible for 62.5 to 75% of H. pylori recurrence cases in the first 6 months after eradication and is primarily attributed to intrafamilial transmission [33, 34]. The predominant genotype in both paediatric patients was vacAs1m1/cagA+/cagE+/babA2- (45/80 strains); however, other genotypes with different allelic combinations for the vacA gene were identified: vacAs2m1 (vacAs2m1/cagA+/cagE+/babA2 and vacAs2m1/cagA+/cagE+/babA2+). These allelic combinations were previously reported in paediatric strains in Mexico [17]. Importantly, the babA2+ genotype was predominant, and this predominance is attributable to adherence to the gastric epithelium or to persistent or chronic infection [35, 36].
In addition, our findings show that the 80 strains of H. pylori present the EPIYA motif Western-ABC, with different repeats of EPIYA-C (41.25% ABC, 26.25% ABCC, and 26.25% ABCCC). It has been reported that in both adults and children, the prevalence of the CagA protein containing three EPIYA motifs (ABC) or four EPIYA motifs (ABCC) is the same. However, strains containing more than four EPIYA motifs (ABCCC) have also been observed in children over 10 years of age and in adults [37, 38], suggesting that CagA strains acquire more EPIYA-C repetitions over time. These results highlight the presence of EPIYA motifs in the polymorphic region of the CagA protein (Western-ABC and Eastern-ABD, the latter being more aggressive), and the number of repetitions has been associated with strains that produce greater morphological changes in gastric epithelial cells, thus resulting in an increased risk of gastric cancer [39, 40].
Strains isolated from patient one during the first event demonstrated a higher frequency of EPIYA-ABCC motifs, and strains from the second event had a higher frequency of EPIYA-ABCCC motifs (Table 2). Reyes-Leon et al. [37] reported that increases in multiple segments of the EPIYA-C motif are involved in the development of gastric diseases, and this association was observed in patient one during clinical treatment. This association was not observed in strains obtained from patient two, who presented only epigastric abdominal pain; these strains (40 isolates) presented the same frequency of the EPIYA-ABC motif during both the first and second events (Table 3). In this study, the modification of EPIYA-C motifs (DPVYA) with the ABC& (1.25%) pattern and EPIYA-B motifs (EPIYT) with the AAB&C and AB&C (1.25% each) patterns was evident. In other studies, the presence of a dipeptide (ST) was observed in the sequence upstream of the EPIYA-A motif, which has already been observed in strains isolated from Greek children (GLKN [ST] EPIYAKVNKKK) [38]. The EPIYA-B motif is highly important for IL-8 secretion and cell elongation, and modifications to this motif (EPIYT) induce lower levels of cell elongation and IL-8 secretion than those induced by isolates containing normal ABC patterns [37].
We identified the presence of 26 new alleles and 17 new STs in H. pylori, representing the first alleles and STs reported worldwide for Mexican strains [41]. MLST analysis is a robust and consistent approach to study the ancestry and evolution of populations of H. pylori, which is an organism with a high degree of genetic diversity in housekeeping genes [2, 5, 12, 42]. When comparing the values of Pi and Theta, we observed that the value of Theta was higher than that of Pi, which suggests that the haplotypes forming the population in each patient are very divergent, thus confirming recolonization events (Table 4). It has been widely recognized that microorganisms tolerate a limited number of point mutations in their coding regions, and the mutations in H. pylori represent a possible mechanism for host adaptation [43].
Analysis of the evolutionary patterns among the H. pylori strains revealed one clonal complex with linked STs for each paediatric patient across both events as well as the presence of individual unlinked STs. Patient one had clonal complexes that comprised five and three linked STs for the first and second events, respectively (Fig. 1a, b). The second event (Fig. 1b) also included linked STs, confirming our genotypic observations (Table 2): the STs present were related, and the patient demonstrated recrudescence and not reinfection. Patient two had clonal complexes that comprised two and one linked STs for the first and second events, respectively, but one unlinked individual ST was present during the second event (Fig. 2b), confirming our genotypic observations, based on the emergence of a new unlinked ST, the patient presented reinfection. The reinfection of H. pylori is present among asymptomatic family members may facilitate the transmission within households. In addition, there is no change in the habits among the same family members (24). However, the patient two, both events recrudescence and reinfection may be involved.
The evolutionary relatedness in each paediatric patient revealed a possible recombination event between the two events (Figs. 1c and 2c). Patient one showed a greater number of recombination pathways between the two events (Fig. 1c), which explains the observed changes in the EPIYA motifs (Table 2). The presence of recombination pathways between the STs confirmed the acquisition of an EPIYA-C motif during the second event in 95% of the strains (19/20). Patient two presented several recombination pathways, based on the loss and emergence of a new ST, indicating that the strains in this patient underwent an adaptation process. Furthermore, the presence of multiple recombination pathways may contribute to the recurrence of infection (reinfection or recrudescence) observed in each patient. The presence of one or more dominant strains suggests an important natural diversification process in H. pylori strains over time, mainly via point mutations and inter-strain recombination events during mixed infections [5, 44, 45], which are regulated by natural selection favouring the presence of certain genotypes [17–19, 29, 46]. Antibiotic administration is a strong selective pressure that inhibits certain strains more than others or eradicates specific genotypes, thus changing the strain distribution in the host [46].
The phylogenetic relationships between the paediatric strains and the strains deposited in the PubMLST database for H. pylori (Fig. 3) were consistent with those observed in previous studies of H. pylori. This bacterium forms non-clonal populations because it has a high rate of mutation that generates a large number of alleles and a high rate of allelic recombination [5, 47, 48]. Our analysis confirmed the offspring patterns obtained using PHYLOViZ and the neighbour-net algorithm, indicating that all isolates in the same patient were genetically related and therefore derived from a common ancestor (Fig. 3) [49].
MLST analyses of seven concatenated housekeeping genes revealed a clear grouping of the various H. pylori strains according to different geographical regions. STs obtained in this study were clustered within the hpEurope group (76.5%) and the hspWAfrica subgroup (23.5%) (Fig. 4). The migrations of slaves from West Africa to the Americas and of European colonists to the Americas and South Africa are likely responsible for the current existence of these strains in Mexico as well as the incorporation of other Latin American strains into this group and subgroup [42, 50]. In addition, intra-genomic and inter-genomic diversity potentially play important roles in the presence of our strains within these groups. Last, our studies suggest that the strains isolated from patients one and two appear to possess the genetic diversity necessary to survive in the host, thus resulting in competition between genotypes during colonization.
Conclusions
The prevalence of H. pylori infection depends on several important elements such as host factors, environmental factors, and genetic variation of the strains [51], thus resulting in multiple infections, convergent mutations, and recombination among strains of H. pylori [52–54]. This study provides evidence of the evolutionary dynamics of the H. pylori strains in two paediatric patients during recrudescence and reinfection events. In particular, our study shows the presence of different STs that emerged before and after treatment; these changes may be due to the accumulation of mutations and recombination events during the diversification process and recolonization of the patients by different genotypes.
Methods
Patients and strains
The two paediatric patients from the Department of Gastroenterology and Nutrition of the Hospital Infantil de Mexico Federico Gómez with recurrent H. pylori infection were both diagnosed with dyspepsia and gastroesophageal reflux disease via lower panendoscopy. Patient one was diagnosed in August 2006 (First event). Treatment with amoxicillin, clarithromycin and omeprazole was indicated, and he was asymptomatic for 13 months. The second event was diagnosed in this patient in October 2007; at this time, the patient exhibited sudden bleeding in the upper digestive tract. Patient two was diagnosed in October 2007 (First event). Treatment was indicated, but it was not administered. In January 2008, treatment with amoxicillin, clarithromycin and omeprazole was again indicated, and the symptoms disappeared two months after treatment. The second event diagnosed in this patient occurred in July 2008. Clinical diagnoses of gastritis, duodenitis, and oesophagitis were determined by endoscopic evaluation.
Isolation and identification of H. pylori
Antral gastric biopsy specimens from both patients were homogenized, inoculated and cultured as described by Mendoza-Elizalde et al. [18]. Briefly, the strains were inoculated and cultured on Casman agar plates (BD BBL, MD, USA) supplemented with 5% horse blood and antibiotics under microaerophilic conditions at 37 °C for 5–7 days. Twenty strains were isolated for each infection event, for a total of 40 strains per patient. Bacterial identification was based on colony morphology, Gram staining, and tests for urease, oxidase and catalase activity. Eighty colonies isolated from the two patients were stored at − 70 °C in 1.5 mL of Brucella broth (BD BBL) supplemented with 10% foetal bovine serum and 25% glycerol.
Susceptibility profile
The minimum inhibitory concentrations (MICs) to three different antibiotic classes, including the β-lactam amoxicillin (AMX, Sigma-Aldrich, St. Louis, MO), the macrolide clarithromycin (CLA, MP Biomedicals, Solon, OH) and the nitroimidazole metronidazole (MTZ, Sigma-Aldrich, St. Louis, MO), were performed using agar dilution methods according to the Clinical and Laboratory Standards Institute (CLSI) guidelines (2015) [55]. The reference strain used for the validation of the techniques was Helicobacter pylori ATCC® 43,504 (American Type Culture Collection, Manassas, VA, USA). The minimal inhibitory concentration interpretative criteria (μg/mL) for resistance was as follows: CLA: ≥1, AMX: ≥4, and MTZ: > 8 [56–59].
Polymerase chain reaction (PCR) detection of virulence genes
Genomic DNA was extracted from cultured H. pylori using a Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions, with slightly modified incubation times. The quantity and integrity of the DNA was analyzed as described by Mendoza-Elizalde et al. [18]. H. pylori was identified based on the presence of the glmM gene [60]. The vacA (s1, s2, m1, and m2), cagA, cagE, and babA2 genes were amplified by PCR using the conditions described by Atherton et al. [61], Mizushima et al. [62] and Kauser et al. [63]. DNA from H. pylori reference strain 26,695 was included as a positive control, and DNA from Pseudomonas aeruginosa reference strain PAO1 was included as a negative control. Amplification was performed using the conditions described by Mendoza-Elizalde et al. [18] in a T100™ Bio-Rad thermal cycler (Applied Biosystems, Foster City, CA, USA). The PCR products were separated and stained as described by Mendoza-Elizalde et al. [18].
Amplification of the 3′ variable region of cagA
The 3′ variable region of the cagA gene was amplified using the conditions described by Mendoza-Elizalde et al. [18]. The primers used were those described by Rudi et al. [64]. The PCR products were separated by electrophoresis on 1.5% agarose gels. The PCR products were purified using ExoSap IT® (Affymetrix, Cleveland, OH, USA) according to the manufacturer’s recommendations. The purified products were sequenced using a BigDye Terminator v3.1 Cycle Sequencing Kit in an ABI 3130 genetic analyser (Applied Biosystems, Foster City, CA, USA). The sequences obtained were aligned using the CAP3 Sequence Assembly program (available at: http://doua.prabi.fr/softwore/cap3). After alignment, the nucleotide sequences were translated into amino acid sequences using the Blastx program (available at http://blast.ncbi.nlm.nih.gov/Blast.cgi) and compared with sequences deposited in GenBank (http://www.ncbi.nlm.nih.gov/genbank/).
Multi-locus sequence typing (MLST)
Seven housekeeping genes of H. pylori located throughout the genome were amplified and sequenced in both directions (mutY, ureI, atpA, efp, ppa, trpC, and yphC) [8, 49]. The genes were amplified by PCR using the conditions described by Achtman et al. [14]. The PCR products were purified using ExoSAP-IT® (Affymetrix, Cleveland, OH, USA) according to the manufacturer’s recommendations. The purified products were sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit in the ABI 3130 genetic analyser (Applied Biosystems, Foster City, CA, USA). Each strain was defined based on the presence of alleles for the seven genes (the allelic profile), and every allelic profile was defined as an ST [65, 66].
The datasets obtained in this article are available in the PubMLST database for H. pylori [http://pubmlst.org/helicobacter/] [41]. The accession numbers for each allele and ST are as follows: 2144, 2218, 2227, 2237, 2252, 2280, 2289, 2303, 2315, 2333, 2336, 2338, 2340, 2341, 2371, 2388, 2412, 2413, 2452, 2378, 2385, 2386, 2590, 2591, 2592, 2593, ST288, ST313, ST813, ST2887, ST2888, ST2889, ST2890, ST2891, ST2892, ST2893, ST2894, ST2895, ST2896, ST2897, ST2898, ST2899, and ST2900.
Phylogenetic and genealogic analyses, genetic diversity analysis, and recombination
The sequences of the seven loci were aligned using ClustalX v2 [67], manually edited with Seaview v4.2.5 [68] and FinchTV V.1.4.0 software (Geospiza, Inc.), and compared with all known alleles of H. pylori deposited in the PubMLST database [http://pubmlst.org/helicobacter/]. To establish the open reading frame of the protein, the nucleotide sequences of different STs from each housekeeping gene were translated into amino acid sequences using the translate tool in ExPASy [http://www.expasy.org]. We used DnaSP v5.10 [69] to assess the nucleotide diversity, including the average nucleotide diversity per site (π) and the expected variation per site under the assumption of neutral evolution (θ), for each housekeeping gene. For phylogenetic analysis, the seven housekeeping genes from each strain were manually concatenated after their independent alignment.
The genealogic relationships among the H. pylori strains was inferred with the PHYLOViZ (http://www.phyloviz.net/) platform. PHYLOViZ infers evolutionary descent patterns among allelic profiles using the goeBURST algorithm and a full minimal spanning tree (MST)-like approach that uses a heuristic local optimization procedure [70]. The possible recombination events among the studied strains were explored with the neighbour-net algorithm [71] implemented in the SplitsTree4 program [72] using uncorrected P distances. The reliability of this network was confirmed with a non-parametric bootstrap analysis after 1000 pseudoreplications. Furthermore, the relatedness among the strains in this study and those deposited in the MLST database [http://pubmlst.org/helicobacter/] was determined using eBURST V3.0 [http://www.phyloviz.net/goeburst/]. This algorithm subdivides large MLST datasets into non-overlapping groups of related STs or clonal complexes to discern the location of the most parsimonious isolates within groups or clonal complexes based on the predicted founder. In addition, eBURST allows the observation of a “snapshot” population with a general view of the clonal complexes; the central part shows the main clonal complexes, the triple (TLV) and double (DLV) linkages, and the individual unlinked STs [65].
To determine the geographical type of H. pylori to which the strains analysed in this study belonged, a phylogenetic tree was generated with MEGA V6.0 software [26] using the neighbour-joining method and the Kimura 2-parameter model of nucleotide substitution [25, 73]. The concatenated nucleotide sequences of the seven housekeeping genes in the studied strains (17 ST), and reference strains (299) [http://pubmlst.org/helicobacter/], which were representative of different geographical groups, were aligned in Muscle software [74]. The reliability of clustering was evaluated with a non-parametric bootstrap test after 1000 pseudoreplications. The reference sequences of the geographical groups were as follows: hpEurope: 75 sequences, hpsNEAfrica: 14 sequences, hspWAfrica: 40 sequences, hspSAfrica: 50 sequences, hpAfrica2: 21 sequences, hspAmerind: 18 sequences, hspEastAsia: 30 sequences, hspMaori: 45 sequences, and hpAsia2: 6 sequences.
Additional files
Table S1. EPIYA motifs identified in the 80 paediatric strains of Helicobacter pylori. Nucleotide sequences of the EPIYA motifs identified in this study (XLSX 11 kb)
Table S2. PubMLST accession numbers for the alleles and STs of Helicobacter pylori strains obtained from paediatric patients described in this study. *Alleles that are present in isolates from other global sources are indicated in red. List of PubMLST accession numbers for the alleles and STs identified in this study as well as alleles previously reported worldwide. (XLSX 11 kb)
Acknowledgements
The authors gratefully acknowledge the Consejo Nacional de Ciencia y Tecnología (CONACyT). This work was part of SME’s PhD dissertation, and she was a CONACyT (216177) fellow.
Abbreviations
- AMX
Amoxicillin
- ATCC
American Type Culture Collection
- atpA
gene encoding an ATP synthase alpha chain
- babA
gene encoding an outer membrane protein that binds to fucosylated Lewis b blood group antigen
- cagA
gene encoding a cytotoxin-associated gene A
- CLA
Clarithromycin
- CLSI
Clinical and Laboratory Standards Institute
- efp
gene encoding an elongation factor P
- EPIYA
Glu-Pro-Ile-Tyr-Ala
- MIC
Minimal Inhibitory Concentration
- MLST
Multilocus Sequence Typing
- MTZ
Metronidazole
- mutY
gene encoding a DNA glycosylase
- PCR
Polymerase Chain Reaction
- ppa
gene encoding an inorganic pyrophosphatase
- ST
Sequence Type
- trpC
gene encoding an anthranilate isomerase
- ureI
gene encoding a urease subunit I
- vacA
gene encoding a vacuolating cytotoxin A
- yphC
gene encoding a GTPase
Authors’ contributions
SME, and NVG designed and planned the research. SME, GZ, RC, HOC and NVG analysed and interpreted the data and wrote the manuscript. SME and ACCM performed the research and generated the database. PVM and ACS collected the specimens and performed the clinical diagnoses. All authors revised and agreed on the manuscript.
Funding
This study was supported by federal resources from the Ministry of Health and Assistance (SSA), México HIM/2011/080-SSA 1005. The funders had no role in the study design, data collection and analysis, decision to publish, or manuscript preparation.
Availability of data and materials
The datasets generated and/or analysed in the current study are available in the PubMLST database for H. pylori [http://pubmlst.org/helicobacter/]. Furthermore, the datasets supporting the conclusions of this article are included within the article and its supplementary tables.
Ethics approval and consent to participate
The ethics (Dr. Luis Jasso Hernández: Chairman of the Committee), biosafety (Dra. Herlinda Vera Hermosillo: Chairman of the Committee) and scientific (Dr. Onofre Muñoz Hernández: Chairman of the Committee) committees at the Health Institute (HIMFG) approved the study protocol in advance (HIM/2011/080-SSA 1005).
All parents were informed about the nature of the study and provided written consent (HIM-LC-RC-PR. 01). Our study was conducted according to the principles established by the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Sandra Mendoza-Elizalde, Email: smendoza@himfg.edu.mx.
Ana Caren Cortés-Márquez, Email: yurico_cts@hotmail.com.
Gerardo Zuñiga, Email: capotezu@hotmail.com.
René Cerritos, Email: renecerritos@gmail.com.
Pedro Valencia-Mayoral, Email: pvalencia@himfg.edu.mx.
Alejandra Consuelo Sánchez, Email: draalejandraconsuelo@yahoo.com.mx.
Hector Olivares-Clavijo, Email: hectorolivaresclavijo@gmail.com.
Norma Velázquez-Guadarrama, Phone: +52 (55) 52289917, Email: normave@himfg.edu.mx.
References
- 1.Algood HM, Cover TL. Helicobacter pylori persistence: an overview of interactions between H. pylori and host immune defenses. Clin Microbiol Rev. 2006;19:597–613. doi: 10.1128/CMR.00006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fischer W, Breithaupt U, Kern B, Smith SI, Spicher C, Haas R. A comprehensive analysis of Helicobacter pylori plasticity zones reveals that they are integrating conjugative elements with intermediate integration specificity. BMC Genomics. 2014;15:2–17. doi: 10.1186/1471-2164-15-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergman M, Del Prete G, Van Kooyk Y, Appelmelk B. Helicobacter pylori phase variation, immune modulation and gastric autoimmunity. Nat Rev Microbiol. 2006;4:151–159. doi: 10.1038/nrmicro1344. [DOI] [PubMed] [Google Scholar]
- 4.Isaacson PG, Du MQ. Gastrointestinal lymphoma: where morphology meets molecular biology. J Pathol. 2005;205:255–274. doi: 10.1002/path.1703. [DOI] [PubMed] [Google Scholar]
- 5.Suerbaum S, Smith JM, Bapumia K, Morelli G, Smith NH, Kunstmann E, et al. Free recombination within Helicobacter pylori. Proc Natl Acad Sci U S A. 1998;95:12619–12624. doi: 10.1073/pnas.95.21.12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farrell S, Doherty GM, Milliken I, Shield MD, McCallion WA. Risk factors for Helicobacter pylori infection in children: an examination of the role played by intrafamilial bed sharing. Pediatr Infect Dis J. 2005;24:149–152. doi: 10.1097/01.inf.0000151104.14058.70. [DOI] [PubMed] [Google Scholar]
- 7.Raymond J, Thiberge JM, Chevalier C, Kalach N, Bergeret M, Labigne A, et al. Genetic and transmission analysis of Helicobacter pylori strains within a family. Emerg Infect Dis. 2004;10:1816–1821. doi: 10.3201/eid1010.040042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kivi M, Rodin S, Kupershmidt I, Lundin A, Tindberg Y, Granstrom M, et al. Helicobacter pylori genome variability in a framework of familial transmission. BMC Microbiol. 2007;7:1–8. doi: 10.1186/1471-2180-7-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwarz S, Morelli G, Kusecek B, Manica A, Balloux F, Owen RJ, et al. Horizontal versus familial transmission of Helicobacter pylori. PLoS Pathog. 2008;4:e1000180. doi: 10.1371/journal.ppat.1000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Atherton JC, Blaser MJ. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest. 2009;119:2475–2487. doi: 10.1172/JCI38605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suerbaum S, Achtman M. Helicobacter pylori: recombination, population structure and human migrations. Int J Med Microbiol. 2004;294:133–139. doi: 10.1016/j.ijmm.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 12.Linz B, Balloux F, Moodley Y, Manica A, Liu H, Roumagnac P, et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007;445:915–918. doi: 10.1038/nature05562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suerbaum S, Josenhans C. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat Rev Microbiol. 2007;5:441–452. doi: 10.1038/nrmicro1658. [DOI] [PubMed] [Google Scholar]
- 14.Achtman M, Azuma T, Berg DE, Ito Y, Morelli G, Pan ZJ, et al. Recombination and clonal groupings within Helicobacter pylori from different geographical regions. Mol Microbiol. 1999;32:459–470. doi: 10.1046/j.1365-2958.1999.01382.x. [DOI] [PubMed] [Google Scholar]
- 15.Kraft C, Stack A, Josenhans C, Niehus E, Dietrich G, Correa P, et al. Genomic changes during chronic Helicobacter pylori infection. J Bacteriol. 2006;188:249–254. doi: 10.1128/JB.188.1.249-254.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzuki R, Shiota S, Yamaoka Y. Molecular epidemiology, population genetics, and pathogenic role of Helicobacter pylori. Infect Genet Evol. 2012;12:203–213. doi: 10.1016/j.meegid.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mendoza-Elizalde S, Arteaga-Resendiz NK, Valencia-Mayoral P, Luna RC, Moreno-Espinosa S, Arenas-Huertero F, et al. Diversification of the vacAs1m1 and vacAs2m2 strains of Helicobacter pylori in Meriones unguiculatus. Front Microbiol. 2016;7:1–11. doi: 10.3389/fmicb.2016.01758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mendoza-Elizalde S, Cortes-Marquez AC, Giono-Cerezo S, Zuniga G, Consuelo-Sanchez A, Valencia-Mayoral P, et al. Analysis of the genotypic diversity of strains of Helicobacter pylori isolated from pediatric patients in Mexico. Infect Genet Evol. 2015;29:68–74. doi: 10.1016/j.meegid.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 19.Didelot X, Nell S, Yang I, Woltemate S, van der Merwe S, Suerbaum S. Genomic evolution and transmission of Helicobacter pylori in two south African families. Proc Natl Acad Sci U S A. 2013;110:13880–13885. doi: 10.1073/pnas.1304681110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Secka O, Moodley Y, Antonio M, Berg DE, Tapgun M, Walton R, et al. Population genetic analyses of Helicobacter pylori isolates from Gambian adults and children. PLoS One. 2014;9:e109466. doi: 10.1371/journal.pone.0109466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soto G, Bautista CT, Roth DE, Gilman RH, Velapatino B, Ogura M, et al. Helicobacter pylori reinfection is common in Peruvian adults after antibiotic eradication therapy. J Infect Dis. 2003;188:1263–1275. doi: 10.1086/379046. [DOI] [PubMed] [Google Scholar]
- 22.McMahon BJ, Bruce MG, Hennessy TW, Bruden DL, Sacco F, Peters H, et al. Reinfection after successful eradication of Helicobacter pylori: a 2-year prospective study in Alaska natives. Aliment Pharmacol Ther. 2006;23:1215–1223. doi: 10.1111/j.1365-2036.2006.02880.x. [DOI] [PubMed] [Google Scholar]
- 23.Niv Y. Helicobacter pylori recurrence after successful eradication. World J Gastroenterol. 2008;14:1477–1478. doi: 10.3748/wjg.14.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryu KH, Yi SY, Na YJ, Baik SJ, Yoon SJ, Jung HS, et al. Reinfection rate and endoscopic changes after successful eradication of Helicobacter pylori. World J Gastroenterol. 2010;16:251–255. doi: 10.3748/wjg.v16.i2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Francisco AP, Bugalho M, Ramirez M, Carrico JA. Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinformatics. 2009;10:152–156. doi: 10.1186/1471-2105-10-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bryant D, Moulton V. Neighbor-net: an agglomerative method for the construction of phylogenetic networks. Mol Biol Evol. 2004;21:255–265. doi: 10.1093/molbev/msh018. [DOI] [PubMed] [Google Scholar]
- 27.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 28.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 29.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Secka O, Antonio M, Berg DE, Tapgun M, Bottomley C, Thomas V, et al. Mixed infection with cagA positive and cagA negative strains of Helicobacter pylori lowers disease burden in the Gambia. PLoS One. 2011;6:e27954. doi: 10.1371/journal.pone.0027954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao Q, Didelot X, Wu Z, Li Z, He L, Li Y, et al. Progressive genomic convergence of two Helicobacter pylori strains during mixed infection of a patient with chronic gastritis. Gut. 2015;64:554–561. doi: 10.1136/gutjnl-2014-307345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Corral JE, Mera R, Dye CW, Morgan DR. Helicobacter pylori recurrence after eradication in Latin America: implications for gastric cancer prevention. World J Gastrointest Oncol. 2017;9:184–193. doi: 10.4251/wjgo.v9.i4.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rollan A, Arab JP, Camargo MC, Candia R, Harris P, Ferreccio C, et al. Management of Helicobacter pylori infection in Latin America: a Delphi technique-based consensus. World J Gastroenterol. 2014;20:10969–10983. doi: 10.3748/wjg.v20.i31.10969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duque X, Vilchis J, Mera R, Trejo-Valdivia B, Goodman KJ, Mendoza ME, et al. Natural history of Helicobacter pylori infection in Mexican schoolchildren: incidence and spontaneous clearance. J Pediatr Gastroenterol Nutr. 2012;55:209–216. doi: 10.1097/MPG.0b013e318248877f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia HX, Windle HJ, Marshall DG, Smyth CJ, Keane CT, O'Morain CA. Recrudescence of Helicobacter pylori after apparently successful eradication: novel application of randomly amplified polymorphic DNA fingerprinting. Gut. 1995;37:30–34. doi: 10.1136/gut.37.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kayali S, Manfredi M, Gaiani F, Bianchi L, Bizzarri B, Leandro G, et al. Helicobacter pylori, transmission routes and recurrence of infection: state of the art. Acta Biomed. 2018;89:72–76. doi: 10.23750/abm.v89i8-S.7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okimoto T, Murakami K, Sato R, Miyajima H, Nasu M, Kagawa J, et al. Is the recurrence of Helicobacter pylori infection after eradication therapy resultant from recrudescence or reinfection, in Japan. Helicobacter. 2003;8:186–191. doi: 10.1046/j.1523-5378.2003.00143.x. [DOI] [PubMed] [Google Scholar]
- 38.Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, et al. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279:373–377. doi: 10.1126/science.279.5349.373. [DOI] [PubMed] [Google Scholar]
- 39.Gerhard M, Lehn N, Neumayer N, Boren T, Rad R, Schepp W, et al. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc Natl Acad Sci U S A. 1999;96:12778–12783. doi: 10.1073/pnas.96.22.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reyes-Leon A, Atherton JC, Argent RH, Puente JL, Torres J. Heterogeneity in the activity of Mexican Helicobacter pylori strains in gastric epithelial cells and its association with diversity in the cagA gene. Infect Immun. 2007;75:3445–3454. doi: 10.1128/IAI.01951-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sgouras DN, Panayotopoulou EG, Papadakos K, Martinez-Gonzalez B, Roumbani A, Panayiotou J, et al. CagA and VacA polymorphisms do not correlate with severity of histopathological lesions in Helicobacter pylori-infected Greek children. J Clin Microbiol. 2009;47:2426–2434. doi: 10.1128/JCM.00159-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer. 2004;4:688–694. doi: 10.1038/nrc1433. [DOI] [PubMed] [Google Scholar]
- 43.Naito M, Yamazaki T, Tsutsumi R, Higashi H, Onoe K, Yamazaki S, et al. Influence of EPIYA-repeat polymorphism on the phosphorylation-dependent biological activity of Helicobacter pylori CagA. Gastroenterology. 2006;130:1181–1190. doi: 10.1053/j.gastro.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 44.Jolley KA, Chan MS, Maiden MC. mlstdbNet - distributed multi-locus sequence typing (MLST) databases. BMC Bioinformatics. 2004;5:1–8. doi: 10.1186/1471-2105-5-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, Kidd M, et al. Traces of human migrations in Helicobacter pylori populations. Science. 2003;299:1582–1585. doi: 10.1126/science.1080857. [DOI] [PubMed] [Google Scholar]
- 46.Kawai M, Furuta Y, Yahara K, Tsuru T, Oshima K, Handa N, et al. Evolution in an oncogenic bacterial species with extreme genome plasticity: Helicobacter pylori east Asian genomes. BMC Microbiol. 2011;11:104–131. doi: 10.1186/1471-2180-11-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stewart AD, Logsdon JM, Kelley SE. An empirical study of the evolution of virulence under both horizontal and vertical transmission. Evolution. 2005;59:730–739. doi: 10.1111/j.0014-3820.2005.tb01749.x. [DOI] [PubMed] [Google Scholar]
- 48.Morelli G, Didelot X, Kusecek B, Schwarz S, Bahlawane C, Falush D, et al. Microevolution of Helicobacter pylori during prolonged infection of single hosts and within families. PLoS Genet. 2010;6:e1001036. doi: 10.1371/journal.pgen.1001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taylor NS, Fox JG, Akopyants NS, Berg DE, Thompson N, Shames B, et al. Long-term colonization with single and multiple strains of Helicobacter pylori assessed by DNA fingerprinting. J Clin Microbiol. 1995;33:918–923. doi: 10.1128/jcm.33.4.918-923.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feil EJ, Spratt BG. Recombination and the population structures of bacterial pathogens. Annu Rev Microbiol. 2001;55:561–590. doi: 10.1146/annurev.micro.55.1.561. [DOI] [PubMed] [Google Scholar]
- 51.Turner KM, Hanage WP, Fraser C, Connor TR, Spratt BG. Assessing the reliability of eBURST using simulated populations with known ancestry. BMC Microbiol. 2007;7:1–14. doi: 10.1186/1471-2180-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lundin A, Björkholm B, Kupershmidt I, Unemo M, Nilsson P, Andersson DI, et al. Slow genetic divergence of Helicobacter pylori strains during long-term colonization. Infect Immun. 2005;73:4818–4822. doi: 10.1128/IAI.73.8.4818-4822.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Camorlinga-Ponce M, Perez-Perez G, Gonzalez-Valencia G, Mendoza I, Penaloza-Espinosa R, Ramos I, et al. Helicobacter pylori genotyping from American indigenous groups shows novel Amerindian vacA and cagA alleles and Asian African and European admixture. PLoS One. 2011;6:e27212. doi: 10.1371/journal.pone.0027212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin D, Koskella B. Friend and foe: factors influencing the movement of the bacterium Helicobacter pylori along the parasitism-mutualism continuum. Evol Appl. 2015;8:9–22. doi: 10.1111/eva.12231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones N, Chiba N, Fallone C, Thompson A, Hunt R, Jacobson K, et al. Helicobacter pylori in first nations and recent immigrant populations in Canada. Can J Gastroenterol. 2012;26:97–103. doi: 10.1155/2012/174529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kodaman N, Pazos A, Schneider BG, Piazuelo MB, Mera R, Sobota RS, et al. Human and Helicobacter pylori coevolution shapes the risk of gastric disease. Proc Natl Acad Sci U S A. 2014;111:1455–1460. doi: 10.1073/pnas.1318093111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chattopadhyay S, Chi PB, Minin VN, Berg DE, Sokurenko EV. Recombination-independent rapid convergent evolution of the gastric pathogen Helicobacter pylori. BMC Genomics. 2018;19:835–846. doi: 10.1186/s12864-018-5231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.CLSI . Methods for antimicrobial dilution and disk susceptibility testing of infrequently isolated or fastidious bacteria. CLSI guideline M45. 3rd ed. Clinical and Laboratory Standards Institute: Wayne, PA; 2015. [Google Scholar]
- 59.Kobayashi I, Muraoka H, Saika T, Nishida M, Fujioka T, Nasu M. Antimicrobial susceptibilities of Helicobacter pylori isolates under microaerophilic atmospheres established by two different methods. J Clin Microbiol. 2001;39:2646–2647. doi: 10.1128/JCM.39.7.2646-2647.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Torres J, Camorlinga-Ponce M, Perez-Perez G, Garza AMD, Dehesa M, Gonzalez-Valencia G, et al. Increasing multidrug resistance in Helicobacter pylori strains isolated from children and adults in Mexico. J Clin Microbiol. 2001;39:2677–2680. doi: 10.1128/JCM.39.7.2677-2680.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mégraud F, Lehours P. Helicobacter pylori detection and antimicrobial susceptibility testing. Clin Microbiol Rev. 2007;20:280–322. doi: 10.1128/CMR.00033-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gosciniak G, Biernat M, Grabinska J, Binkowska A, Poniewierka E, Iwanczak B. The antimicrobial susceptibility of Helicobacter pylori strains isolated from children and adults with primary infection in the lower Silesia region. Poland Pol J Microbiol. 2014;63:57–61. [PubMed] [Google Scholar]
- 63.Smith SI, Oyedeji KS, Arigbabu AO, Cantet F, Megraud F, Ojo OO, et al. Comparison of three PCR methods for detection of Helicobacter pylori DNA and detection of cagA gene in gastric biopsy specimens. World J Gastroenterol. 2004;10:1958–1960. doi: 10.3748/wjg.v10.i13.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Atherton JC, Cao P, Peek RM, Jr, Tummuru MK, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem. 1995;270:17771–17777. doi: 10.1074/jbc.270.30.17771. [DOI] [PubMed] [Google Scholar]
- 65.Mizushima T, Sugiyama T, Komatsu Y, Ishizuka J, Kato M, Asaka M. Clinical relevance of the babA2 genotype of Helicobacter pylori in Japanese clinical isolates. J Clin Microbiol. 2001;39:2463–2465. doi: 10.1128/JCM.39.7.2463-2465.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kauser F, Khan AA, Hussain MA, Carroll IM, Ahmad N, Tiwari S, et al. The cag pathogenicity island of Helicobacter pylori is disrupted in the majority of patient isolates from different human populations. J Clin Microbiol. 2004;42:5302–5308. doi: 10.1128/JCM.42.11.5302-5308.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rudi J, Kolb C, Maiwald M, Kuck D, Sieg A, Galle PR, et al. Diversity of Helicobacter pylori vacA and cagA genes and relationship to VacA and CagA protein expression, cytotoxin production, and associated diseases. J Clin Microbiol. 1998;36:944–948. doi: 10.1128/jcm.36.4.944-948.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004;186:1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vazquez JA, Berron S. Multilocus sequence typing: the molecular marker of the internet era. Enferm Infecc Microbiol Clin. 2004;22:113–120. doi: 10.1016/S0213-005X(04)73045-1. [DOI] [PubMed] [Google Scholar]
- 70.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 71.Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 72.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 73.Huson DH. SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics. 1998;14:68–73. doi: 10.1093/bioinformatics/14.1.68. [DOI] [PubMed] [Google Scholar]
- 74.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. EPIYA motifs identified in the 80 paediatric strains of Helicobacter pylori. Nucleotide sequences of the EPIYA motifs identified in this study (XLSX 11 kb)
Table S2. PubMLST accession numbers for the alleles and STs of Helicobacter pylori strains obtained from paediatric patients described in this study. *Alleles that are present in isolates from other global sources are indicated in red. List of PubMLST accession numbers for the alleles and STs identified in this study as well as alleles previously reported worldwide. (XLSX 11 kb)
Data Availability Statement
The datasets generated and/or analysed in the current study are available in the PubMLST database for H. pylori [http://pubmlst.org/helicobacter/]. Furthermore, the datasets supporting the conclusions of this article are included within the article and its supplementary tables.