

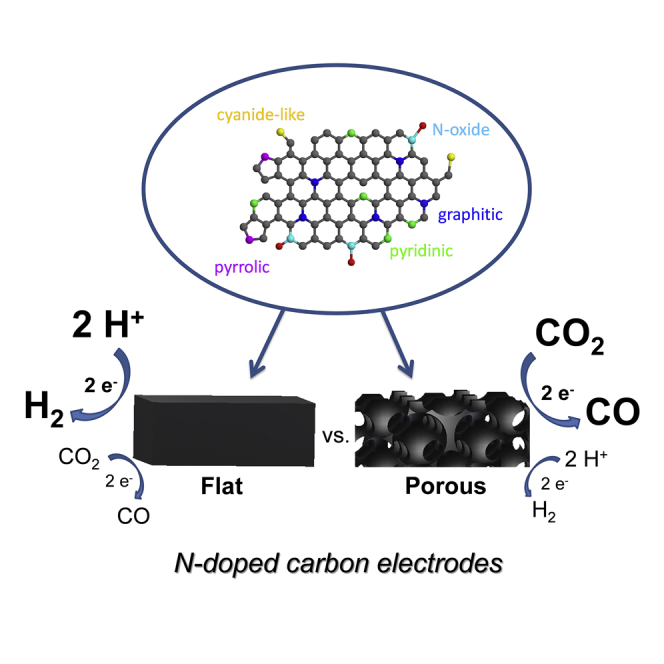
Summary
The morphology of electrode materials is often overlooked when comparing different carbon-based electrocatalysts for carbon dioxide reduction. To investigate the role of morphological attributes, we studied polymer-derived, interconnected, N-doped carbon structures with uniformly sized meso or macropores, differing only in the pore size. We found that the carbon dioxide reduction selectivity (versus the hydrogen evolution reaction) increased around three times just by introducing the porosity into the carbon structure (with an optimal pore size of 27 nm). We attribute this change to alterations in the wetting and CO2 adsorption properties of the carbon catalysts. These insights offer a new platform to advance CO2 reduction performance by only morphological engineering of the electrocatalyst.
Keywords: CO2 reduction, electrocatalyst, solar fuels, energy conversion, morphological engineering
Graphical Abstract
Highlights
-
•
The role of morphology in CO2 reduction was studied on N-doped carbon electrodes
-
•
All chemical and structural properties were set to be identical except the morphology
-
•
Both electrocatalytic activity and selectivity was tuned by changing the porosity
-
•
Wettability, adsorption strength, and geometric effects were dictating factors
Context & Scale
Producing fuels and commodity chemicals from carbon dioxide using electrochemical methods is a promising way to transform a greenhouse gas into value-added products. Scaling up this process to an industrial level, however, requires efficient, stable, and cheap electrocatalysts. One such group of materials is nitrogen-doped carbons, as demonstrated by their impressive performance improvements in recent years. Factors dictating their catalytic behavior, however, have yet to be understood to make further progress. Here, we show that both the catalytic activity and product selectivity are greatly varied by tuning the pore size of the carbon catalyst while keeping all other chemical and structural features identical. With these findings, we would like to highlight that structure-activity-stability relationships should be scrutinized before reporting on the electrocatalytic activity of different nanostructured carbons, as simple morphological factors can dictate the overall performance.
Nitrogen-doped carbon materials are attracting increasing interest as inexpensive and efficient electrocatalysts for carbon dioxide reduction. CO2 reduction selectivity (versus the hydrogen evolution reaction) was increased three times just by introducing porosity into the carbon structure (with an optimal pore size of 27 nm). This effect was quantitatively analyzed by systematically varying the porosity of the electrodes while fixing all other chemical and structural parameters. CO2 adsorption properties, wetting characteristics, and geometric effects are jointly responsible for the observed differences.
Introduction
Electrochemical reduction of CO2 (CO2R) is a promising method for converting a greenhouse gas into value-added products, utilizing renewable energy. Novel catalysts, electrode assemblies, and cell configurations are all necessary to achieve economically appealing performance.1, 2, 3 Nitrogen-doped carbon (N–C) materials are attracting increasing interest as inexpensive and efficient electrocatalysts of this process.4, 5, 6, 7, 8, 9 Despite the rapid progress, summarized in recent review articles,10, 11, 12, 13 there are still several open questions related to the mechanistic insights. The nature of active centers and the role of different heteroatoms on the catalytic performance have been studied,8, 14, 15 but the exact nature of the catalytic sites is still under debate. In addition, the composition and structure of these electrode materials are not well defined. The material can contain more than one active center and the distribution of heteroatoms and morphology can also change. Finally, several structural properties vary through these studies, such as the level of graphitization, surface functional groups, surface area, or metal impurities.8, 16, 17 This makes it very difficult (if not impossible) to compare the results of precedent art. This issue was highlighted recently since several factors, other than the inherent properties of the catalysts, can severely influence the measured activity.18
From a morphological perspective, a wide range of carbon structures such as nanofibers, nanotubes, nanospheres, nano-onions, and graphene was investigated.15, 19, 20 Furthermore, catalyst layers are prepared through different methods (e.g., dip-coating, brush painting, spray-coating, and drop-casting),5, 7, 15, 21 which usually result in a different porosity of the electrode for even the same catalyst material.22 The electrode configurations also vary from work to work, being catalysts supported on flat substrates, gas diffusion electrodes, or employed as self-supported catalysts.7, 21, 23, 24 Several mass transport limitation scenarios25, 26 can be envisioned in these structures, leading to differences in the overall activity and selectivity.27 Yet, only a few studies suspected the determinant role of catalyst morphology and porosity on the CO2R performance of carbon-based electrodes,21, 24, 28, 29 and to the best of our knowledge, no systematic studies were performed on this subject up to now.
In the case of metal electrodes, CO2R performance was shown to be affected by the catalyst morphology, particle size, and loading. Alterations in the product distribution were attributed to (1) an increased residence time of products and intermediates in the pores of the nanostructured catalyst layers,30, 31 (2) differences in mass transport requirements for hydrogen evolution (HER) and CO2R reactions,32 (3) CO2 transport enhancement induced by gas evolution,33 (4) size and morphology-dependent surface coordination effects,34, 35, 36 and (5) a field-induced reagent concentration on highly curved surfaces.37
The role of porosity was systematically studied in the oxygen reduction reaction (ORR) on N–C electrodes using well-defined porous carbon structures. Macroporous graphitic C3N4/Carbon composites outperformed its mesoporous counterpart because of the facile reactant transport in the larger pores.38 In the mesopore range, an intermediate pore size (around 22 nm in diameter) was the best,39 while the performance was further enhanced by introducing microporosity and modifying surface functionalities.40 Transport effects in ORR and CO2R, however, are different: ORR results in water formation, which can undergo condensation, while gas-phase products are common in CO2R. Furthermore, the solution chemistry of the reactants is largely different in the two cases.
The above trends gave us the motivation of this study, where we aimed to deconvolute the effect of various structural parameters and thus focus only on the role of the porosity in the CO2R process. This study aims to shine light on the morphology-activity-stability relationships in CO2R electrocatalysis with well-defined carbon-based catalysts materials as an object. In this work, we will show that variations in the porosity of N–C electrodes alone can result in massive differences in the electrocatalytic behavior in both activity and selectivity. We will also demonstrate that altered CO2 adsorption and wetting properties, along with a curvature effect, are jointly responsible for this intriguing phenomenon.
Results and Discussion
Catalyst Synthesis and Characterization
As the first step, we have synthesized a series of N–C electrodes by a totally metal-free sacrificial support method (Scheme 1). The o-phenylenediamine (oPD) monomer was chemically polymerized in the presence of monodisperse silica nanoparticles. The resulting SiO2/PoPD composite was carbonized (pyrolyzed) at 900°C in neat N2 flow, followed by the etching of the silica template with hydrogen fluoride (HF). By this “hard templating method,” we prepared three N-C samples having pores with 13, 27, and 90 nm nominal diameters (denoted as NC-13, NC-27, and NC-90, respectively) based on the mean size of the silica template used. A nonporous catalyst (NC), without using a silica template, was also prepared for comparison. We used the same oPD/SiO2 mass ratio through the synthesis of the porous catalysts, thus fixing the total pore volume for the samples. This synthetic approach allowed us to control the morphological attributes of the electrodes while keeping all other parameters unchanged, as shown in what follows.
Scheme 1.

Scheme of the Catalyst Synthesis
Catalysts were synthesized by a sacrificial support method, starting from poly(o-phenylenediamine) polymers and using silica nanoparticles with different diameters as templates. High-temperature carbonization of the silica/polymer composites was followed by etching of the template particles with hydrogen fluoride solution.
The formation of interconnected carbon structures with uniformly sized pores was verified by transmission electron microscopy (TEM) and scanning electron microscopy (SEM) (Figure 1). The average diameter of the pores (determined from the TEM images) was 12.7 ± 2.1, 26.4 ± 3.9, and 94.8 ± 9.4 nm for the NC-13, NC-27, and NC-90 samples, respectively, mirroring the size of the template silica particles. In contrast, the NC appeared to be smooth, absent of meso- or macropores (Figure S1). The size of the template particles also affected the carbon wall thickness. Semi-quantitatively, the smaller the particles, the thinner the wall size (5–8 nm for NC-13, 10–12 nm for NC-27, and 15–20 nm for NC-90). High-resolution (HR) TEM images, however, revealed similar graphitic structures for all four samples (Figure S2).
Figure 1.

Morphology of the Studied Porous Catalysts
Transmission (A:NC-13, B: NC-27, C: NC-90) and scanning (D: NC-13, E: NC-27, F: NC-90) electron microscopy images of the porous N–C catalysts. See also Figures S1 and S2.
N2 adsorption and desorption isotherms (Figure S3) of NC-13 and NC-27 showed dominantly mesoporous features with the characteristic hysteresis loops, while the less pronounced capillary condensation for NC-90 (also shifted to higher relative pressures) confirmed its rather macroporous structure. The pore size distribution curves peaked at 13, 30, and 78 nm for NC-13, NC-27, and NC-90, respectively (Figure S4), and further confirmed that uniformly sized pores formed. Specific surface areas were 957 m2 g−1 for NC-13, 899 m2 g−1 for NC-27, 665 m2 g−1 for NC-90, and only 104 m2 g−1 for NC. The trend in the roughness factors of the electrodes (1 mg cm−2 loadings), determined form the double layer capacitance values, were in line with the BET specific surface areas (Figures S6–S8).
X-ray diffraction (XRD) and Raman spectroscopic analysis indicated very similar carbon structures for the catalysts (Figure S5). The intensity ratio of D and G bands in the Raman spectra (characteristic of the defects and the graphitic structure) was 0.89 ± 0.01 for all four samples, indicating an identical density of defect sites. The broad and very weak diffraction at 25° and 43° in the XRD pattern are typical for amorphous carbons. The surface chemical composition of the electrodes was studied by X-ray photoelectron spectroscopy (XPS) (Figure 2). The relative amounts of carbon, nitrogen, and oxygen atoms were very similar for all N–C samples: the composition was 80–85 atom % C, 6–8 atom % N, and 6–11 atom % O (Table S1). Similarly, the distribution of nitrogen moieties41, 42 in different chemical environments was almost identical (Figure 2B; Table S2) for all studied catalysts and was in good accordance with previous results on related materials.43, 44 Overall, the N–C catalysts were very similar both in terms of the surface functional groups (chemical properties) and the electronic features. Therefore, they offer a platform to systematically study the effect of morphology on the CO2R performance.
Figure 2.

X-Ray Photoelectron Spectroscopic Data for the NC-90 Sample
(A) N1s XPS pattern for the NC-90 catalyst.
(B) Distribution of the N species in different chemical environments.
Electrochemical CO2 Reduction Performance
The CO2R activity of the N–C electrodes with the same catalyst loading (1.00 ± 0.07 mg cm−2) was first tested by linear sweep voltammetry (LSV) in a CO2-saturated KHCO3 solution (Figure 3A). NC-27 and NC-13 showed the highest current densities, exceeding 25 mA cm−2 at –1.0 V (versus RHE). Only half of this current was recorded for NC-90, while currents stayed below 5 mA cm−2 for NC. The onset potential for the reduction was –0.27 ± 0.03 V (versus RHE) for all samples (Figure S9), further confirming that the chemical features of the active centers are identical. To deconvolute the evident surface area effect, currents were normalized by the roughness factors too (Figure S10). The trend in the current densities remained the same among the porous samples, implying that structural features other than the variations in the specific surface area are responsible for the differences in the catalytic performance.
Figure 3.

Electrochemical Reduction Performance of the Studied Catalysts
(A) Linear sweep voltammograms of the catalysts recorded in a CO2-saturated 0.5 M KHCO3 solution with 5 mV s−1 sweep rate.
(B) Total current densities recorded in the 40th min of the potentiostatic electrolysis in a 0.1 M KHCO3 solution, saturated with CO2. Currents are normalized by the geometrical surface area of the electrodes. Error bars reflect the average of three parallel measurements for different samples, and data are represented as mean ± SEM.
See also Figure S10.
Product selectivity was studied by on-line gas chromatography (GC) and ex situ nuclear magnetic resonance (NMR) spectroscopic analysis of the gas and liquid-phase products during potentiostatic electrolysis. We detected the formation of CO and H2 accounting for more than 95% of the total products (in some cases, trace amounts of methane and formate were also seen). The total current density versus potential curves is plotted in Figure 3B. The order in the total currents followed the same trend as during the LSV measurements normalized either by the geometric surface areas or by the roughness factors (Figure S10).
The porosity of the N–C electrodes immensely affected the selectivity, too (Figure 4C). While the CO/H2 ratio went through a maximum peaking at –0.6 V (versus RHE) for NC-27 and NC-13; NC-90 and NC showed a continuous decrease in this ratio in the studied potential window. A general trend in decreasing CO selectivity with increasing pore size was observed at all potentials; however, differences in the product distribution were more obvious above –0.7 V (i.e., where CO2R dominated over HER). The highest CO selectivity was achieved for NC-27 at all potentials, peaking at –0.6 V with 76%.
Figure 4.

Electrochemical CO2 Reduction Selectivity of the Studied Catalysts
(A and B) Partial current densities for H2 (A) and CO (B) formation on the N–C catalysts during potentiostatic electrolysis in a CO2-saturated 0.1 M KHCO3 solution.
(C) Molar ratio of the CO and H2 products as a function of the electrode potential. Data points are measured in the 40th min of the electrolysis, and currents are normalized by the geometric surface areas. Error bars reflect the average of three parallel measurements for different samples, and data are represented as mean ± SEM.
See also Figure S11.
H2 partial current densities (IHER) exponentially increased with increasing overpotential, indicating a purely kinetic process (Figure 4A). IHER was practically the same for NC-27 and NC-13 but showed a 2- to 3-fold decrease for NC-90. In contrast, CO partial currents (ICO) increased in the order of NC-90 < NC-13 < NC-27 and showed distinct potential dependences for the various samples (Figure 4B). While ICO increased continuously with more negative potentials for NC-90 and NC, it declined at –1.0 V in case of NC-27 and NC-13. This suggests that besides the charge transfer kinetics, other complications arise in the CO2R, which are also dependent on the porosity. The superior CO production activity of NC-27 among the studied catalysts was even more pronounced when we normalized the partial currents by the roughness factors (Figure S11). Overall, the mesoporous NC-27 performed best both in terms of total activity and CO selectivity, which is in line with a recent report on similar catalysts.29 The decreased catalytic performance of NC-13 was because of the decreased CO2R, while for NC-90, it was the effect of the simultaneous attenuation of the HER and the CO2R. In this case, the CO selectivity approached the bulk-like behavior. Also, note that not the best performing NC-27 catalyst contains the highest amount of pyridinic and graphitic N-moieties, which sites are supposed to be the active sites in CO2R for such materials.13 This further supports our notion, namely that the observed trends in catalytic activity are more likely rooted in the different morphology as discussed below, and the contribution of the slightly different chemical properties is inferior.
Overall, the performance of the N-C catalyst described in this study is comparable to that of the other carbon-based CO-selective electrodes reported earlier (see an extensive compilation of precedent art in Table S4). The highest CO production activity was reached for NC-27 at ‒0.8 V with ‒1.85 mA cm−2 partial current density, which falls in the activity range of metal-free N-doped carbon catalysts measured under similar conditions (electrolyte, substrate electrode, and cell type). Preliminary experiments in a membrane-separated continuous-flow cell (data not shown) resulted in 70 mA cm−2 CO2R partial current density, which is also in the range measured for similar catalysts (see Table S4). Furthermore, the activity of our best performing catalyst is also close to many metal-nitrogen-doped carbons (M–N–Cs). The advantage of incorporating atomically dispersed metals into the carbon structure is evident from the literature comparison, especially in the case of Ni–N–C materials. The optimization of the carbon framework, however, is also crucial in the M–N–C catalysts. Taking into account these considerations, our set of catalysts is a relevant model system for the investigation of the morphological effects of the carbon structure in CO2R. Moreover, as data on rate-controlling steps in CO2R are scarce and under dispute, this work provides a baseline and a low-parameter variability study on the effect of morphology in this electrochemical process. This allows us to separate the adsorption steps in catalyst reactivity and demonstrate that a “holistic materials design,” which includes porosity at several length scales is needed for successful implementation of electrochemical processes for CO2 valorization.
Control Experiments
To prove that the above observations are not the results of the unlikely variation in the layer thicknesses for the different catalysts with the same mass loadings, we studied the product distributions for NC-27 with three different loadings (0.50, 1.00, and 1.50 mg cm−2). The total and partial current densities increased with increasing mass (Figure S12); however, the specific activity (currents normalized by the roughness factors) diminished for the thicker layers (Figure S13). This might be the result of the higher mass transfer resistance of the thicker layers. The CO/H2 selectivity was similar for the layers with 0.50 and 1.00 mg cm−2 loadings at all studied potentials. For the layers with 1.50 mg cm−2 loadings, it was around 1.3 times lower than the thinner layers above –0.7 V, while slightly higher at more negative potentials. Changes in the layer thickness influenced slightly the product distribution; however, its effect was far not as pronounced as that of the variations in the pore size.
Factors behind the Changing Selectivity
The differences in the CO2R activity and selectivity can be rationalized by taking into account three main factors: (1) wettability of the catalysts, (2) CO2 adsorption in the differently sized pores, and (3) curvature effects. The wetting properties of the NC samples were investigated by measuring the dynamic (advancing and receding) contact angles of the thin layers (Figure S14). The surface energies (Figure 5A) were calculated from the contact angle hysteresis.45 The nonporous NC catalyst was the least hydrophobic (Θ = 128.5°) and presented the highest hysteresis, resulting in the highest surface energy (γs,tot=6.35 ± 2.31 mJ m−2). Introducing pores into the carbon framework decreased the surface energy to about 2 mJ m−2 (ideal porosity) independently of the pore size. This effect is in line with previous morphology-surface energy correlations.46, 47 Importantly, this trend in the surface energies may partially account for the enhanced CO2R performance (versus HER) of the porous samples, compared to the flat NC, in the absence of flooding water.27
Figure 5.

Wettability and CO2 Adsorption Measurements to Reveal the Factors behind the Changing Selectivity
(A) Surface energies of the studied catalysts calculated from dynamic contact angle measurements. The inset shows the representative images of water drops on the surface of the catalyst layers used for the determination of contact angles for NC-27 and NC.
(B) Temperature-programmed CO2 desorption profiles of the studied catalysts.
See also Figure S14.
As the dynamics of gas bubble formation on the electrode surface depend on the wettability and morphology (among others), the size of the bubbles was analyzed along with their residence time during potentiostatic electrolysis (Figure S15). Such analysis is important because the release of gas bubbles can enhance mass transport of the reactant by inducing convective flow, hence reducing the diffusion layer thickness, leading to higher catalytic current density.33 In addition, large bubbles, which remain on the electrode surface for a longer time, can partially block the active area resulting in lower current density.48 The departing bubbles were relatively large on all N–C catalysts (few hundred μm) because of the hydrophobic character of the samples. The increase in the overpotential (hence the current density), reduced both the departure diameters and the residence time (Figure S16A). The residence time decreased drastically for the porous samples, compared to the nonporous NC. Comparing the porous samples, both the residence times and the departure diameters decreased with the reduction of the pore size (Figure S16B) (i.e., in the order of NC-90 > NC-27 > NC-13). This finding is in agreement with previous reports, where enhanced bubble release was observed in nanostructures with smaller dimensions.33, 48 The distribution diagrams also revealed that polydispersity of the bubble size and residence time increased with increasing pore size (Figure S17) suggesting progressive nucleation. The differences in residence time can be an additional reason for the superiority of the porous catalysts compared to the flat NC and also for the better performance of NC-27 and NC-13 than NC-90. In contrast, these findings do not explain NC-27 being the best performing electrode, as the bubble-induced CO2 transport enhancement effect is expected to be the highest in the case of NC-13.
Temperature-programmed desorption was employed to study the CO2 adsorption strength of the catalysts (Figure 5B). While only one desorption peak was present in case of the NC, CO2 desorption from the porous samples happened in two distinct steps. We assume that the first step at a lower temperature (< 400 K) is connected to the adsorbed CO2 on the outer surface of the catalyst layers, and the second step is linked to the desorption of CO2 from the inner surface of the pores (which is absent for NC). The temperature of the desorption from the pores was the highest (433 K) for NC-27, implying the highest CO2 adsorption strength. As NC-27 performed best also in the CO2 conversion, we assume a link between the CO2 adsorption and reduction activity. Considering that the studied N−C catalysts are practically identical in terms of N-containing moieties, we suggest that the distinct CO2 adsorption strengths arise from the differences in the pore structure. In this case, the strain induced on the CO2 adsorption sites by the differently curved surfaces is optimal for the 27 nm pores. This curvature-dependent binding strength of CO2 is similar to what was reported for both graphene and N–C materials, based on computational methods.49, 50 Additionally, the importance of differently curved adsorption sites was highlighted in ORR for N–C materials, based on experimental evidence.51
We also note that the presented N–C structures are not ideal and present several levels of chemical and morphological defects. These are defects in the carbon structure; therefore, the contribution of edge-site-expressed defects versus in-plane sites might be of importance. The differently curved pores may indeed contribute to a different ratio of in-plane versus edge defects. For smaller pores, we may expect more edge site defects than in-plane ones, resulting in higher catalytic activity. High-resolution C1s XPS indeed revealed a higher amount of graphitic carbon in the NC than the porous samples (Figure S18; Table S3). This is probably because of the lower concentration of defects and edge sites in the case of a flat structure compared to the curved ones. Carbon speciation of all three porous catalysts, however, was identical based on the XPS (within the experimental error). Finally, we note that higher curvature (i.e., smaller pore diameter) can also lead to field-induced CO2 accumulation (condensation), which can also contribute to the enhanced CO2 reduction performance of NC-27 and NC-13, as demonstrated earlier in the case of metal electrodes.37
Activity-Selectivity-Stability Correlations
Stability of the catalytic performance is a key point in catalyst development, especially from the perspective of future application. This is a particularly intriguing issue for a nano-engineered catalyst, where the morphology might alter during operation, thus causing changes in the catalytic properties.52 To find the key parameters dictating the stability of the N–C catalysts, the decay of the catalytic currents, for both HER and CO2R, was studied as a function of potential, catalyst activity (ICO and IH2), and selectivity (CO/H2 ratio). We found that the higher the CO production rate (ICO), the more stable the CO2R activity (Figure 6A). Importantly, all data points fell on the same trendline, irrespective from the sample studied. Notably, the data points are clustered: NC-27 is the most stable, followed by NC-13, NC-90, and finally NC. Another representation of the same data for the most active and stable NC-27 sample is shown in Figure 6B. This interpretation further confirms that stability correlates with activity, and they both increase with the overpotential.
Figure 6.

CO2 Reduction Activity-Stability-Selectivity Correlations
(A) Stability of the CO partial currents during 70 min of operation as a function of the CO partial currents in the 15th min of the potentiostatic electrolysis for the different samples.
(B) CO partial currents (left) and stability of CO partial currents (right) during 70 min of electrolysis as a function of the electrode potential for NC-27.
(C) Stability of the CO partial currents during 70 min of operation as a function of the CO selectivity for NC-27. Error bars reflect the average of three parallel measurements for different samples, and data are represented as mean ± SEM.
See also Figures S19–S21.
We were also interested to see the relationship between stability and selectivity. Interestingly, a clear trend was witnessed for all porous samples, and the example of NC-27 is shown in Figure 6C. The stability rapidly decreased with the increasing CO2R selectivity (i.e., CO/H2 ratio). Although this dataset highlights the trade-off between selectivity and stability for the N–C catalyst, the underlying mechanistic insights still have to be uncovered.
Overall, we identified that higher CO formation activity and lower CO selectivity resulted in more stable CO production in time, independently of the porosity. In the HER, however, no such unambiguous correlations were found (Figure S19), and even some HER performance increase was seen, while the CO2R activity decreased. Finally, we also tested the morphological stability of NC-27. No major changes in the porosity could be observed after a 4 h long potentiostatic electrolysis at ‒0.6 V, as seen on the SEM images taken before and after CO2 reduction (Figure S20). Changes in the HER and CO2R currents followed that same pattern as discussed above (Figure S21).
In summary, we found that CO2R activity, selectivity, and stability of N–C electrodes are highly dependent on their porosity. The presence of mesopores was demonstrated to be beneficial in achieving high CO selectivity and current density, with an optimal pore size around 27 nm. Even after convoluting factors other than morphology (e.g., surface chemistry, level of graphitization, surface area), the reasons behind the observed trends are complex. CO2 adsorption properties, wetting characteristics, and geometric effects are jointly responsible for the massive difference in the CO2R performance. All these properties must be taken into consideration when we aim to understand the reduction mechanism on different catalysts and while improving the performance further to a technologically relevant level (as alternatives to precious metal catalysts).
Experimental Procedures
Catalyst Synthesis and Electrode Preparation
All chemicals were used as received, without further purification. Aqueous solutions were prepared using deionized water (Millipore Direct Q3-UV, 18.2 MΩ cm).
The catalysts were synthesized by a sacrificial support method using oPD as the carbon and nitrogen precursors. A solution containing 0.58 M oPD (C6H8O2, Merck, 108.14 g/mol), 0.72 M hydrochloric acid (HCl; VWR International, 37%, 36.46 g/mol), 0.19 g/cm3 silica nanoparticle (see Table 1) was vigorously stirred for ∼15 min to adsorb the monomer molecules on the silica surface. A solution of 2.7 M ammonium persulfate (APS; (NH4)2S2O8, Acros Organics, 228.19 g/mol) in 1 M HCl was dropwise added to the oPD solution (n(oPD) / n(APS) = 0.8) at 0°C (ice bath) and stirred for 24 h to complete the polymerization process. The obtained PoPD / SiO2 composites were dry-freezed, then pyrolyzed at 900°C in a tube furnace in N2 flow (110 cm3/min). Heating program: room temperature (RT)–5°C/min–180°C (1 h)–5°C/min–900°C (2 h). The silica nanoparticles were etched out overnight with an excess amount of 15 wt % HF (VWR, 40 wt %, 20.01 g/mol) solution. Finally, the nitrogen-doped carbon catalysts were washed thoroughly with ultrapure water, until close to neutral pH (>5) was reached. The nonporous (NC) sample was prepared by the same procedure but without the use of silica colloid.
Table 1.
Silica Colloids Used as Templates in the Synthesis of the N–C Catalysts
| Silica colloid | Mean diameter/nm | wt % |
|---|---|---|
| LUDOX-HS40 (Aldrich) | 13 | 40% |
| LUDOX-TM50 (Aldrich) | 27 | 50% |
| LEVASIL CT3-PL (AkzoNobel) | 90 | 50% |
For the electrochemical measurements, the catalysts were spray-coated onto the surface of glassy carbon plates. The substrates were polished with 0.05 μm MicroPolish Alumina (Buehler), rinsed, and sonicated in acetone (C3H6O, 58.08 g/mol, VWR), ethanol (C2H6O, 46.07 g/mol, 99%, VWR), and ultrapure water before spray-coating. The catalyst ink consisted of 5 mg/mL catalyst dispersed in 10 mL ethanol-ultrapure water mixture (50 V/V%) containing 100 μL Nafion dispersion (FuelCell Store, 10%). Spray-coating was performed with a homemade automated spray-coating equipment at 110°C until the desired catalyst loading was achieved. The exact amount of catalyst coated was always measured with a microbalance.
Electrochemical Measurements
Electrochemical measurements were carried out using an Autolab PGSTAT 204 instrument. Potentials were measured against an Ag/AgCl/3 M NaCl reference electrode, but they are given versus the reversible hydrogen electrode (RHE) in the text (ERHE = EAg/AgCl + 0.20 V + 0.059 V × pH).
Cyclic voltammograms (for the determination of the electrochemically active surface areas of the electrodes) were recorded in a one-compartment three-electrode setup using the catalyst-coated glassy carbon substrates as the working electrodes, a Pt foil as the counter electrode and the Ag/AgCl/3 M NaCl electrode as the reference. These measurements were carried out in an Ar-saturated (Messer, 99.995 %) 1 M sodium sulfate solution (Na2SO4 anhydrous, 142.04 g/mol, 99% Alfa Aesar).
Linear sweep voltammetric and chronoamperometric electrolysis measurements were performed in a two-compartment sealed electrochemical cell with the cathode and anode compartments separated by a Nafion-117 membrane. For the analysis of gas-phase products, the cathode compartment of the cell was directly connected to the inlet of the gas chromatograph via a six-port valve. In the CO2 reduction experiments, CO2-saturated (Messer; 99.995%) potassium hydrogen carbonate (KHCO3, 100.12 g/mol, VWR) solutions were used. The error bars on the figures reflect the average of three parallel measurements for different catalyst layers.
Roughness factors of the N–C electrodes were calculated from the electrochemical double layer capacitance (Qdl) values determined by cyclic voltammetry. Cyclic voltammograms were recorded in an Ar-saturated 1 M Na2SO4 solution between −0.21 and 0.61 V (versus RHE) applying different sweep rates. The double layer current (Idl) was determined at 0.41 V as the difference between the anodic and cathodic currents (Idl = Ia−Ic). Idl was plotted as a function of the sweep rate (v) and Qdl was calculated from the slope (s) of this curve (Qdl = s/2). The Qdl values obtained for the N–C electrodes were compared to that of a bare, smooth glassy carbon electrode (assuming surface roughness of 1). Roughness factors were given by the ratio of these two values.
CO2 Reduction Products Analysis
Gas-phase CO2 reduction products were analyzed by on-line GC, using a Shimadzu-2010 Plus GC equipped with a barrier ionization discharge detector. A Shincarbon ST column was used for the separation. During electrolysis, 0.5 mL of the headspace gas was injected into the GC at around 15, 40, and 68 min. Analysis parameters were the following: carrier gas: helium; oven program: Tstart = 35°C (2.5 min), ΔTramp = 20°C min−1, Tend = 270°C (3 min); injection temperature: T = 150°C; linear velocity was controlled by the pressure pstart = 250 kPa (2.5 min) Δpramp = 15 kPa min−1 pend = 400 kPa (7.5 min); and split ratio: 10.
Liquid-phase products were analyzed by NMR spectroscopy acquired on a Bruker NMR Avance 500 MHz instrument. Water suppression was used to eliminate the peak of the water. 450 μL of the electrolyte sample was mixed with 50 μL D2O (Sigma Aldrich, 99.9 atom% D) containing dimethyl-sulfoxide (DMSO, C2H6SO, 78.13 g/mol, Alfa Aesar) and phenol (C6H6O, 94.11 g/mol 99.5% Sigma Aldrich) as the internal standards. The ratio of peak areas of the products and the internal standards were used for the calibration. Peak areas of the products right to the water peak were compared to the peak area of the DMSO, while peak areas of products left to that were compared to the peak area of the phenol.
Error bars were determined from at least three parallel measurements for separate electrodes.
Materials Characterization Techniques
XPS spectra were acquired on a Kratos Axis Ultra DLD spectrometer. A monochromatic Al Kα source operated at 300 W. No charge neutralization was needed. CasaXPS was used for the data analysis (three regions were analyzed per sample). The survey patterns were acquired at 80 eV pass energy. The high-resolution patterns for C 1s and N 1s were acquired at 20 eV pass energy and fitted with a 70% Gaussian-30% Lorentzian line shape with fixed full-width half maximum of 1.0–1.2 eV for C and of 1.3–1.5 eV for N. For consistency, the positions and width of peaks used in the curve-fit was adapted from previously established protocol.43, 53
XRD patterns were recorded between 2θ = 10°–80° at 1° per min scan rate on a Rigaku Miniflex II instrument, operating with a Cu Kα,1 radiation source (λ = 0.1541 nm)
For the determination of the BET surface areas, the samples were outgassed under N2 flow at 120°C overnight using a Micromeritics flow prep system. The surface area was then measured using N2 adsorption at 77 K in a Micromeritics Gemini 2360 multipoint BET analyzer.
N2 adsorption/desorption isotherms were recorded at 77.4 K on a Quantachrome Nova 3000e instrument. Samples were outgassed at 200°C for 2 h before measurement. Pore size distribution curves were calculated using the Barrett-Joyner-Halenda method excluding points below 0.35 relative pressure.
Raman spectroscopy was measured with a Senterra II Compact Raman microscope (Bruker), using 532 nm laser excitation wavelength, operating at a power of ≤ 2.5 mW, and a 50× objective.
Transmission electron microscopic (TEM) images were recorded by using a FEI Tecnai G2 20 X-Twin type instrument, operating at an acceleration voltage of 200 kV. A Hitachi S4700 field emission scanning electron microscope (SEM) was operated at an accelerating voltage of 10 kV.
The dynamic advancing (Θadv) and receding (Θrec) contact angles of the layers were measured applying an EasyDrop drop shape analysis system (Krüss GmbH, Hamburg, Germany) controlled with the DSA100 software, equipped with a Peltier temperature chamber (T = 25.0°C ± 0.5°C, under atmospheric pressure and constant humidity) and a steel syringe needle of 0.5 mm diameter. During the measurements, distilled water was used as a test liquid. The total apparent surface free energies (γs,tot) of the layers were calculated from these dynamic contact angle data, knowing the surface tension of the distilled water, (γl, 72.1 mN m−1 at 25°C) and the contact angle hysteresis, which is defined as the difference between the Θadv and Θrec as shown by Equation 1.45
| (Equation 1) |
In-situ bubble formation during potentiostatic electrolysis was recorded with a digital microscope (500 × magnification) at different potentials in a CO2-saturated 0.1 M KHCO3-solution. The size of the bubbles—right before their departure from the electrode surface—was determined with the ImageJ image processing and analysis software.
Temperature-programmed CO2 desorption (TPD) profiles were recorded with a BELCAT-A instrument. Samples were heated to 500°C for 1 h in N2 atmosphere prior to the measurements to remove all the adsorbed species from the surface. The catalysts were cooled in He atmosphere to 50°C, and CO2 was adsorbed at this temperature for 30 min. This was followed by a He rinsing step (15 min), when the excess (not adsorbed) CO2 was removed. In these steps, the flow rate of the gases was 50 mL min−1. For the CO2, desorption step samples were heated in He with 10°C min−1 heating rate (30 mL min−1 flow rate). The desorbed CO2 was analyzed with a TCD detector.
Acknowledgments
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 716539). This work was supported by the GINOP-2.3.2-15-2016-00013 project. Ministry of Human Capacities, Hungary grant 20391-3/2018/FEKUSTRAT is also acknowledged. K.A., T.A., and P.A. would like to thank the Army Research Office MURI grant for financial support (#W911NF1410263). D.H. acknowledges the support of the New National Excellence Program of the Ministry of Human Capacities (grant UNKP-18-3). The authors are thankful to AkzoNobel for the donation of the LEVASIL silica colloid. The authors also thank Kornélia Ábrahámné Baán for performing the temperature programmed desorption measurements, László Nagy for the help with the N2 adsorption and desorption measurements, Egon Kecsenovity for taking the TEM, Dr. Attila Kormányos for taking the SEM images, and Dr. Bíborka Janáky-Bohner for her help with the manuscript preparation.
Author Contributions
P.A. and C.J. conceived the research and designed the experiments. D.H. and A.S. synthesized the samples and performed all electrochemical measurements. K.A. supervised, designed, and analyzed XPS experiments. T.A. performed the XPS and some BET measurements. L.J. carried out and analyzed the contact angle measurements. All authors discussed the results and assisted during manuscript preparation, which was completed by D.H. and C.J.
Declaration of Interests
The authors declare no competing interests.
Published: May 30, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.joule.2019.05.007.
Supplemental Information
References
- 1.Endrődi B., Bencsik G., Darvas F., Jones R., Rajeshwar K., Janáky C. Continuous-flow electroreduction of carbon dioxide. Prog. Energy Combust. Sci. 2017;62:133–154. [Google Scholar]
- 2.Seh Z.W., Kibsgaard J., Dickens C.F., Chorkendorff I., Nørskov J.K., Jaramillo T.F. Combining theory and experiment in electrocatalysis: insights into materials design. Science. 2017;355:eead4998. doi: 10.1126/science.aad4998. [DOI] [PubMed] [Google Scholar]
- 3.Bushuyev O.S., De Luna P., Dinh C.T., Tao L., Saur G., van de Lagemaat J., Kelley S.O., Sargent E.H. What should we make with CO2 and how can we make it? Joule. 2018;2:825–832. [Google Scholar]
- 4.Hursán D., Janáky C. Electrochemical reduction of carbon dioxide on nitrogen-doped carbons: insights from isotopic labeling studies. ACS Energy Lett. 2018;3:722–723. doi: 10.1021/acsenergylett.8b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roy A., Hursán D., Artyushkova K., Atanassov P., Janáky C., Serov A. Nanostructured metal-N-C electrocatalysts for CO2 reduction and hydrogen evolution reactions. Appl. Catal. B. 2018;232:512–520. [Google Scholar]
- 6.Ju W., Bagger A., Hao G.P., Varela A.S., Sinev I., Bon V., Roldan Cuenya B., Kaskel S., Rossmeisl J., Strasser P. Understanding activity and selectivity of metal-nitrogen-doped carbon catalysts for electrochemical reduction of CO2. Nat. Commun. 2017;8:944. doi: 10.1038/s41467-017-01035-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leonard N., Ju W., Sinev I., Steinberg J., Luo F., Varela A.S., Roldan Cuenya B., Strasser P. The chemical identity, state and structure of catalytically active centers during the electrochemical CO2 reduction on porous Fe–nitrogen–carbon (Fe–N–C) materials. Chem. Sci. 2018;9:5064–5073. doi: 10.1039/c8sc00491a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu J., Liu M., Sharma P.P., Yadav R.M., Ma L., Yang Y., Zou X., Zhou X.D., Vajtai R., Yakobson B.I. Incorporation of nitrogen defects for efficient reduction of CO2 via two-electron pathway on three-dimensional graphene foam. Nano Lett. 2016;16:466–470. doi: 10.1021/acs.nanolett.5b04123. [DOI] [PubMed] [Google Scholar]
- 9.Zheng T., Jiang K., Ta N., Hu Y., Zeng J., Liu J., Wang H. Large-scale and highly selective CO2 electrocatalytic reduction on nickel single-atom catalyst. Joule. 2019;3:265–278. [Google Scholar]
- 10.Varela A.S., Ju W., Strasser P. Molecular nitrogen–carbon catalysts, solid metal organic framework catalysts, and solid metal/nitrogen-doped carbon (MNC) catalysts for the electrochemical CO2 reduction. Adv. Energy Mater. 2018;8:1703614. [Google Scholar]
- 11.Vasileff A., Zheng Y., Qiao S.Z. Carbon solving carbon’s problems: recent progress of nanostructured carbon-based catalysts for the electrochemical reduction of CO2. Adv. Energy Mater. 2017;7:1700759. [Google Scholar]
- 12.Lai J., Nsabimana A., Luque R., Xu G. 3D porous carbonaceous electrodes for electrocatalytic applications. Joule. 2018;2:76–93. [Google Scholar]
- 13.Duan X., Xu J., Wei Z., Ma J., Guo S., Wang S., Liu H., Dou S. Metal-free carbon materials for CO2 electrochemical reduction. Adv. Mater. 2017;29:1701784. doi: 10.1002/adma.201701784. [DOI] [PubMed] [Google Scholar]
- 14.Daiyan R., Tan X., Chen R., Saputera W.H., Tahini H.A., Lovell E., Ng Y.H., Smith S.C., Dai L., Lu X. Electroreduction of CO2 to CO on a mesoporous carbon catalyst with progressively removed nitrogen moieties. ACS Energy Lett. 2018;3:2292–2298. [Google Scholar]
- 15.Liu S., Yang H., Huang X., Liu L., Cai W., Gao J., Li X., Zhang T., Huang Y., Liu B. Identifying active sites of nitrogen-doped carbon materials for the CO2 reduction reaction. Adv. Funct. Mater. 2018;28:1800499. [Google Scholar]
- 16.Lum Y., Kwon Y., Lobaccaro P., Chen L., Clark E.L., Bell A.T., Ager J.W. Trace levels of copper in carbon materials show significant electrochemical CO2 reduction activity. ACS Catal. 2016;6:202–209. [Google Scholar]
- 17.Zhao Y., Liang J., Wang C., Ma J., Wallace G.G. Tunable and efficient tin modified nitrogen-doped carbon nanofibers for electrochemical reduction of aqueous carbon dioxide. Adv. Energy Mater. 2018;8:1702524. [Google Scholar]
- 18.Clark E.L., Resasco J., Landers A., Lin J., Chung L.-T., Walton A., Hahn C., Jaramillo T.F., Bell A.T. Standards and protocols for data acquisition and reporting for studies of the electrochemical reduction of carbon dioxide. ACS Catal. 2018;8:6560–6570. [Google Scholar]
- 19.Zhang S., Kang P., Ubnoske S., Brennaman M.K., Song N., House R.L., Glass J.T., Meyer T.J. Polyethylenimine-enhanced electrocatalytic reduction of CO2 to formate at nitrogen-doped carbon nanomaterials. J. Am. Chem. Soc. 2014;136:7845–7848. doi: 10.1021/ja5031529. [DOI] [PubMed] [Google Scholar]
- 20.Liu T., Ali S., Lian Z., Si C., Su D.S., Li B. Phosphorus-doped onion-like carbon for CO2 electrochemical reduction: the decisive role of the bonding configuration of phosphorus. J. Mater. Chem. A. 2018;6:19998–20004. [Google Scholar]
- 21.Song Y., Chen W., Zhao C., Li S., Wei W., Sun Y. Metal-free nitrogen-doped mesoporous carbon for electroreduction of CO2 to ethanol. Angew. Chem. Int. Ed. 2017;56:10840–10844. doi: 10.1002/anie.201706777. [DOI] [PubMed] [Google Scholar]
- 22.Jhong H.R., Brushett F.R., Kenis P.J.A. The effects of catalyst layer deposition methodology on electrode performance. Adv. Energy Mater. 2013;3:589–599. [Google Scholar]
- 23.Wu J., Ma S., Sun J., Gold J.I., Tiwary C., Kim B., Zhu L., Chopra N., Odeh I.N., Vajtai R. A metal-free electrocatalyst for carbon dioxide reduction to multi-carbon hydrocarbons and oxygenates. Nat. Commun. 2016;7:13869. doi: 10.1038/ncomms13869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang H., Wu Y., Lin Q., Fan L., Chai X., Zhang Q., Liu J., He C., Lin Z. Composition tailoring via N & S co-doping and structure tuning by constructing hierarchical pores: metal-free catalysts for high-performance electrochemical reduction of CO2. Angew. Chem. Int. Ed. 2018;130:15702–15706. doi: 10.1002/anie.201809255. [DOI] [PubMed] [Google Scholar]
- 25.Jaouen F., Lindbergh G. Transient techniques for ivestigating mass-transport limitations in gas diffusion electrodes: I. Modeling the PEFC cathode. J. Electrochem. Soc. 2003;150:A1699–A1717. [Google Scholar]
- 26.Kong C.S., Kim D.-Y., Lee H.-K., Shul Y.-G., Lee T.-H. Influence of pore-size distribution of diffusion layer on mass-transport problems of proton exchange membrane fuel cells. J. Power Sources. 2002;108:185–191. [Google Scholar]
- 27.Weng L.C., Bell A.T., Weber A.Z. Modeling gas-diffusion electrodes for CO2 reduction. Phys. Chem. Chem. Phys. 2018;20:16973–16984. doi: 10.1039/c8cp01319e. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto T., Tryk D.A., Hashimoto K., Fujishima A., Okawa M. Electrochemical reduction of CO2 in the micropores of activated carbon fibers. J. Electrochem. Soc. 2000;147:3393–3400. [Google Scholar]
- 29.Wang H., Jia J., Song P., Wang Q., Li D., Min S., Qian C., Wang L., Li Y.F., Ma C. Efficient electrocatalytic reduction of CO2 by nitrogen-doped nanoporous carbon/carbon nanotube membranes: a step towards the electrochemical CO2 refinery. Angew. Chem. Int. Ed. 2017;56:7847–7852. doi: 10.1002/anie.201703720. [DOI] [PubMed] [Google Scholar]
- 30.Dutta A., Rahaman M., Luedi N.C., Mohos M., Broekmann P. Morphology matters: tuning the product distribution of CO2 electroreduction on oxide-derived Cu foam catalysts. ACS Catal. 2016;6:3804–3814. [Google Scholar]
- 31.Sen S., Liu D., Palmore G.T.R. Electrochemical reduction of CO2 at copper nanofoams. ACS Catal. 2014;4:3091–3095. [Google Scholar]
- 32.Hall A.S., Yoon Y., Wuttig A., Surendranath Y. Mesostructure-induced selectivity in CO2 reduction catalysis. J. Am. Chem. Soc. 2015;137:14834–14837. doi: 10.1021/jacs.5b08259. [DOI] [PubMed] [Google Scholar]
- 33.Burdyny T., Graham P.J., Pang Y., Dinh C.-T., Liu M., Sargent E.H., Sinton D. Nanomorphology-enhanced gas-evolution intensifies CO2 reduction electrochemistry. ACS Sustain. Chem. Eng. 2017;5:4031–4040. [Google Scholar]
- 34.Reske R., Mistry H., Behafarid F., Roldan Cuenya B., Strasser P. Particle size effects in the catalytic electroreduction of CO2 on Cu nanoparticles. J. Am. Chem. Soc. 2014;136:6978–6986. doi: 10.1021/ja500328k. [DOI] [PubMed] [Google Scholar]
- 35.Tang W., Peterson A.A., Varela A.S., Jovanov Z.P., Bech L., Durand W.J., Dahl S., Nørskov J.K., Chorkendorff I. The importance of surface morphology in controlling the selectivity of polycrystalline copper for CO2 electroreduction. Phys. Chem. Chem. Phys. 2012;14:76–81. doi: 10.1039/c1cp22700a. [DOI] [PubMed] [Google Scholar]
- 36.Lu Q., Rosen J., Zhou Y., Hutchings G.S., Kimmel Y.C., Chen J.G., Jiao F. A selective and efficient electrocatalyst for carbon dioxide reduction. Nat. Commun. 2014;5:3242. doi: 10.1038/ncomms4242. [DOI] [PubMed] [Google Scholar]
- 37.Liu M., Pang Y., Zhang B., De Luna P., Voznyy O., Xu J., Zheng X., Dinh C.T., Fan F., Cao C. Enhanced electrocatalytic CO2 reduction via field-induced reagent concentration. Nature. 2016;537:382–386. doi: 10.1038/nature19060. [DOI] [PubMed] [Google Scholar]
- 38.Liang J., Zheng Y., Chen J., Liu J., Hulicova-Jurcakova D., Jaroniec M., Qiao S.Z. Facile oxygen reduction on a three-dimensionally ordered macroporous graphitic C3N4/carbon composite electrocatalyst. Angew. Chem. Int. Ed. 2012;51:3892–3896. doi: 10.1002/anie.201107981. [DOI] [PubMed] [Google Scholar]
- 39.Wei W., Liang H., Parvez K., Zhuang X., Feng X., Müllen K. Nitrogen-doped carbon nanosheets with size-defined mesopores as highly efficient metal-free catalyst for the oxygen reduction reaction. Angew. Chem. Int. Ed. 2014;53:1570–1574. doi: 10.1002/anie.201307319. [DOI] [PubMed] [Google Scholar]
- 40.Liang H.W., Zhuang X., Brüller S., Feng X., Müllen K. Hierarchically porous carbons with optimized nitrogen doping as highly active electrocatalysts for oxygen reduction. Nat. Commun. 2014;5:4973. doi: 10.1038/ncomms5973. [DOI] [PubMed] [Google Scholar]
- 41.Chen Y., Artyushkova K., Rojas-Carbonell S., Serov A., Matanovic I., Santoro C., Asset T., Atanassov P. Inhibition of surface chemical moieties by tris(hydroxymethyl)aminomethane: a key to understanding oxygen reduction on iron–nitrogen–carbon catalysts. ACS Appl. Energy Mater. 2018;1:1942–1949. [Google Scholar]
- 42.Matanovic I., Artyushkova K., Atanassov P. Understanding PGM-free catalysts by linking density functional theory calculations and structural analysis: perspectives and challenges. Curr. Opin. Electrochem. 2018;9:137–144. [Google Scholar]
- 43.Kabir S., Artyushkova K., Serov A., Atanassov P. Role of nitrogen moieties in N-doped 3D-graphene nanosheets for oxygen electroreduction in acidic and alkaline media. ACS Appl. Mater. Interfaces. 2018;10:11623–11632. doi: 10.1021/acsami.7b18651. [DOI] [PubMed] [Google Scholar]
- 44.Kabir S., Artyushkova K., Serov A., Kiefer B., Atanassov P. Binding energy shifts for nitrogen-containing graphene-based electrocatalysts – Experiments and DFT calculations. Surf. Interface Anal. 2016;48:293–300. [Google Scholar]
- 45.Chibowski E. Surface free energy of a solid from contact angle hysteresis. Adv. Colloid Interface Sci. 2003;103:149–172. doi: 10.1016/S0001-8686(02)00093-3. [DOI] [PubMed] [Google Scholar]
- 46.Wenzel R.N. Surface roughness and contact angle. J. Phys. Chem. 1949;53:1466–1467. [Google Scholar]
- 47.Bico J., Thiele U., Quéré D. Wetting of textured surfaces. Colloids Surf. A Physicochem. Eng. Asp. 2002;206:41–46. [Google Scholar]
- 48.Faber M.S., Dziedzic R., Lukowski M.A., Kaiser N.S., Ding Q., Jin S. High-Performance Electrocatalysis using metallic cobalt pyrite (CoS2) micro- and nanostructures. J. Am. Chem. Soc. 2014;136:10053–10061. doi: 10.1021/ja504099w. [DOI] [PubMed] [Google Scholar]
- 49.Dutta D., Wood B.C., Bhide S.Y., Ayappa K.G., Narasimhan S. Enhanced gas adsorption on graphitic substrates via defects and local curvature: a density functional theory study. J. Phys. Chem. C. 2014;118:7741–7750. [Google Scholar]
- 50.Chai G.L., Guo Z.X. Highly effective sites and selectivity of nitrogen-doped graphene/CNT catalysts for CO2 electrochemical reduction. Chem. Sci. 2016;7:1268–1275. doi: 10.1039/c5sc03695j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yadav R.M., Wu J., Kochandra R., Ma L., Tiwary C.S., Ge L., Ye G., Vajtai R., Lou J., Ajayan P.M. Carbon nitrogen nanotubes as efficient bifunctional electrocatalysts for oxygen reduction and evolution reactions. ACS Appl. Mater. Interfaces. 2015;7:11991–12000. doi: 10.1021/acsami.5b02032. [DOI] [PubMed] [Google Scholar]
- 52.Grosse P., Gao D., Scholten F., Sinev I., Mistry H., Roldan Cuenya B. Dynamic changes in the structure, chemical state and catalytic selectivity of Cu nanocubes during CO2 electroreduction: size and support effects. Angew. Chem. 2018;57:6192–6197. doi: 10.1002/anie.201802083. [DOI] [PubMed] [Google Scholar]
- 53.Workman M.J., Dzara M., Ngo C., Pylypenko S., Serov A., McKinney S., Gordon J., Atanassov P., Artyushkova K. Platinum group metal-free electrocatalysts: effects of synthesis on structure and performance in proton-exchange membrane fuel cell cathodes. J. Power Sources. 2017;348:30–39. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.