

Abstract
Vascular inflammatory responses play an important role in several cardiovascular diseases. Of the many pro-inflammatory vasoactive factors implicated in this process, is aldosterone, an important mediator of vascular oxidative stress. Statins, such as atorvastatin, are cholesterol-lowering drugs that have pleiotropic actions, including anti-oxidant properties independently of their cholesterol-lowering effect. This study investigated whether atorvastatin prevents aldosterone-induced VSMC inflammation by reducing reactive oxygen species (ROS) production. Vascular smooth muscle cells (VSMC) from WKY rats were treated with 1 μM atorvastatin for 60 min or for 72 h prior to aldosterone (10−7 mol/L) stimulation. Atorvastatin inhibited Rac1/2 and p47phox translocation from the cytosol to the membrane, as well as reduced aldosterone-induced ROS production. Atorvastatin also attenuated aldosterone-induced vascular inflammation and macrophage adhesion to VSMC. Similarly EHT1864, a Rac1/2 inhibitor, and tiron, ROS scavenger, reduced macrophage adhesion. Through its inhibitory effects on Rac1/2 activation and ROS production, atorvastatin reduces vascular ROS generation and inhibits VSMC inflammation. Our data suggest that in conditions associated with aldosterone-induced vascular damage, statins may have vasoprotective effects by inhibiting oxidative stress and inflammation.
Keywords: Inflammation, Vascular, Statins, Mineralocorticoids, Redox signaling
1. Introduction
Aldosterone is a steroid hormone synthesized mainly in the outer layer of the adrenal cortex, the zona glomerulosa [1], although extraadrenal sources of aldosterone have been identified [2]. Aldosterone plays an important role in vascular homeostasis regulating endothelial function, vascular inflammation and remodeling, which are important processes underlying vascular injury in hypertension and atherosclerosis [1,3–5].
Statins, inhibitors of 3-hydroxy-3-methylglutaryl-coenzymeA (HMG-CoA) reductase, revolutionized the treatment of hypercholesterolemia. Statins are competitive inhibitors of HMG-CoA reductase, the rate-limiting enzyme in cholesterol synthesis, thus decreasing endogenous cholesterol synthesis [6,7]. Statins confer cardiovascular protection, which has been confirmed extensively in experimental and clinical studies [8,9]. In addition to lipid-lowering actions of statins, they exhibit a wide array of cardiovascular effects independently of their lipid-reducing properties [10], the so-called pleiotropic effects. Statins influence redox-sensitive processes through putative antioxidant properties and by inhibiting NADPH oxidase (Nox)-derived reactive oxygen species (ROS) generation [10,11]. Recently, we demonstrated that atorvastatin treatment counterbalances type 2 diabetes-associated vascular inflammation and dysfunction and kidney damage, via antioxidant-dependent mechanisms [8,12]. Since vascular cells express functionally active HMG-CoA reductase [13,14], it is also possible that vascular effects of statins may involve local inhibition of this enzyme.
Here, we investigated whether atorvastatin directly modulates aldosterone inflammatory effects in vascular smooth muscle cells (VSCM), and whether this effect is mediated via anti-oxidant properties.
2. Material and methods
2.1. VSMC culture
The study was approved by the Animal Ethics Committee of the University of Ottawa and performed according to the recommendations of the Canadian Council for Animal Care and in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. VSMCs from adult male Wistar-Kyoto rats (16 weeks-old) were euthanized with non-anesthetic gas (carbon dioxide) followed by decapitation. VSMCs derived from mesenteric arteries were isolated and characterized as previously described [15,16]. Low-passage cells (4 to 7) from 3 different batches of cell isolation were studied.
2.2. Protocols for cell stimulation
Cells were stimulated with aldosterone (10−7 mol/L). Two different times of stimulations were used: 5 and 30 min (min), to assess non-transcriptional effects, or long-term 24 h (h), to assess transcriptional effects. In some experiments cells were pre-exposed for 60 min and 72 h to atorvastatin (10−7 mol/L). Cells were also pre-treated for 30 min with EHT1864 (Rac1/2 inhibitor, 10−6 mol/L) or tiron (ROS scavenger, 10−5 mol/L).
2.3. Lucigenin-enhanced chemiluminescence
ROS generation was measured by a luminescence assay with lucigenin as the electron acceptor and NADPH as the substrate, as previously described [15,17].
2.4. Western blotting
Total or fractionated proteins from VSMCs were separated by electrophoresis on a polyacrylamide gel, and transferred onto a nitrocellulose membrane. Non-specific binding sites were blocked with 5% skim milk. Membranes were then incubated with specific antibodies overnight at 4 °C described in the cytosol and membrane fractionation. After incubation with secondary antibodies, signals were revealed with chemiluminescence, visualized by autoradiography and quantified densitometrically. Antibody to β-actin (Sigma Aldrich, MO-USA) was used as an internal control.
2.5. Cytosol and membrane fractionation
Cytosol to membrane translocation of p47phox, which is essential for NADPH oxidase activation, and Rac1/2 (small G protein necessary for NADPH oxidase activity) was assessed in VSMCs. Cells were lysed and fractionated to obtain cytosol- and membrane-enriched fractions. Western blotting was performed as described using anti-p47phox (Santa Cruz Biotechnology, TX-USA) and anti-Rac1/2 (Cell Signaling, MA-USA). Translocation was determined as the ratio of protein expression in membrane to cytosolic fractions.
2.6. Real time RT (reverse transcription)-PCR
Total VSMC mRNA was extracted (Trizol Plus, Invitrogen), purified with chlroform method, and eluted in 20 μL of DEPC-treated water. Complementary DNA was generated by RT-PCR with SuperScript III (Invitrogen). Reverse transcription was performed at 50 °C for 50 min; the enzyme was heat inactivated at 85 °C for 5 min, and real-time quantitative RT-PCR was performed with the SYBR Green Supermix (Bio-Rad Laboratories). The genes analyzed were: Tumor necrosis factor alpha (TNF-α): Fw: ACCACGCTCTTCTGTCTACTG; Rev.: CTTGGTGGT TTGCTACGAC, interleukine 1β (IL-1β): Fw: GCAATGGTCGGGACATA GTT; Rev.: AGACCTGACTTGGCAGAGGA and glyceraldehyde 3-phosphate dehydrogenase (GAPDH): Fw: AAGGTCATCCCAGAGCTGAA; Rev.: GTCCTCAGTGTAGCCCAGGA, which was used as house-keeping gene.
2.7. Vascular inflammatory response: macrophage adhesion
Vascular macrophage adhesion was determined according to our previously described methods [16]. Briefly, VSMCs were cultured to confluence in 6-well plates. Growth-arrested VSMCs from WKY rats were stimulated with 10−7 mol/L aldosterone for 24 h in the presence or absence of atorvastatin (60 min or 72 h) and inhibitors. Non-stimulated VSMCs served as controls. Rat-derived NR8383 monocyte/macrophage cell lines were obtained from the American Type Culture Collection (Manassas, VA). NR8383 cells, adherent and suspension, were cultured in growth medium (Ham’s F12K with 2 mmol/L L-glutamine, 1.5 g/L sodium bicarbonate and 15% heat inactivated fetal bovine serum). For cell fluorescent labeling, macrophages (105 cells/mL) were suspended in 1% bovine serum albumin (BSA)-supplemented phosphate buffered saline containing 1 μmol/L calcein-AM (Molecular Probes, Eugene, OR-USA) and incubated for 20 min at 37 °C. Labeled macrophages were washed twice with phosphate-buffered saline and suspended in Hanks’ buffered salt solution. Fluorescence labeled cells (105 cells/well) were then added to both nonstimulated and stimulated VSMCs layers and were allowed to adhere for 30 min at 37 °C in 5% CO2. After the incubation, non-adhered cells were removed by gently washing with pre-warmed Hanks’ buffered salt solution. The number of adherent cells was determined by lysing the cells with 0.1 mol/L NaOH. The cell lysate was transferred to a 96 well plate and the fluorescence intensity was measured with a fluorescence multiwell plate reader (excitation wavelength 485 nm, emission wavelength 535 nm, Cary Eclipse, Varian, CA-USA). VSMC lysate was used as a blank. Experiments were performed in duplicates.
2.8. Data analysis
Aldosterone-stimulated effects were determined as the percent increase over control, with the control normalized to 100%. Results are presented as mean ± SEM and compared by one way ANOVA. Values of P < 0.05 were considered to be significant.
3. Results
3.1. Atorvastatin prevents ROS-generation by inhibiting Rac1/2 and p47phox assembly in VSMCs
Aldosterone (5 and 30 min) increased ROS production. Atorvastatin pre-incubation (60 min and 72 h) prevented aldosterone-induced ROS generation in VSMCs (Fig. 1). Aldosterone increased Rac1/2 and p47phox translocation from the cytosol to the membrane, which was abrogated by atorvastatin treatment (Figs. 2 and 3). In order to confirm that aldosterone produces ROS via Rac1/2 activation, we incubated VSMC with EHT1864 (Rac1/2 inhibitor), which reduced aldosterone-induced ROS generation, similar to the atorvastatin effects. Tiron (ROS scavenger) also reduced aldosterone-mediated production of ROS (Fig. 4).
Fig. 1.
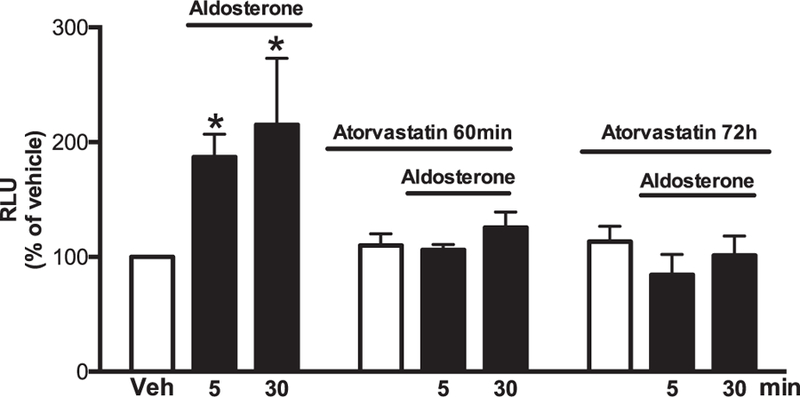
Atorvastatin prevents aldosterone-induced ROS generation in WKY VSMCs. Effects of aldosterone (10−7 mol/L) on ROS generation measured by lucigenin chemiluminescence assay in the absence and presence of 10−7 mol/L of atorvastatin (60 minutes and 72 hours pre-incubation). Results are mean ± SEM of 5–6 experiments. *P < 0.05, vs. vehicle.
Fig. 2.
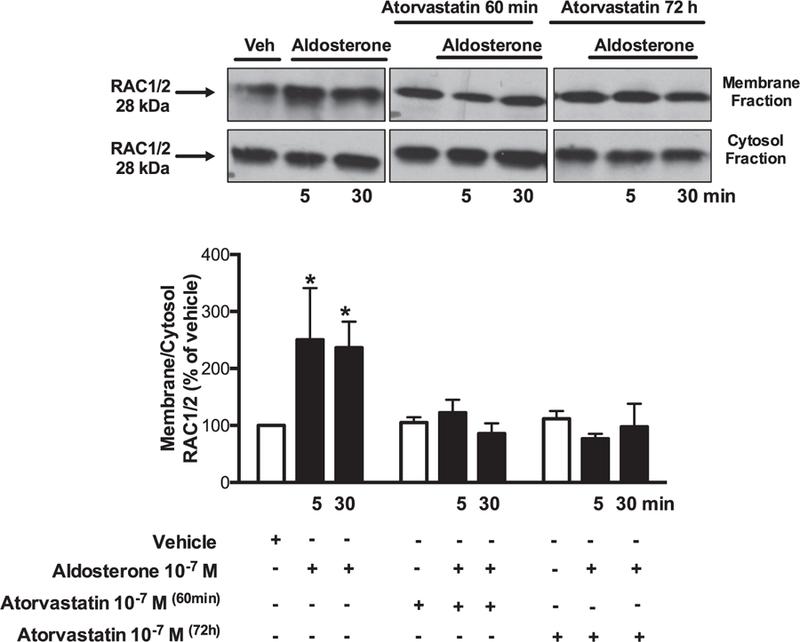
Atorvastatin prevents aldosterone-induced Rac1/2 activity in WKY VSMCs. Effects of aldosterone (10−7 mol/L) on Rac1/2 translocation from cytosol to the membrane, in the absence and presence of 10−7 mol/L of atorvastatin (60 minutes and 72 hours pre-incubation). Representative immunoblots: Rac1/2 (fractions of membrane and cytosol). Results are mean ± SEM of 5–6 experiments. *P < 0.05, vs. vehicle.
Fig. 3.
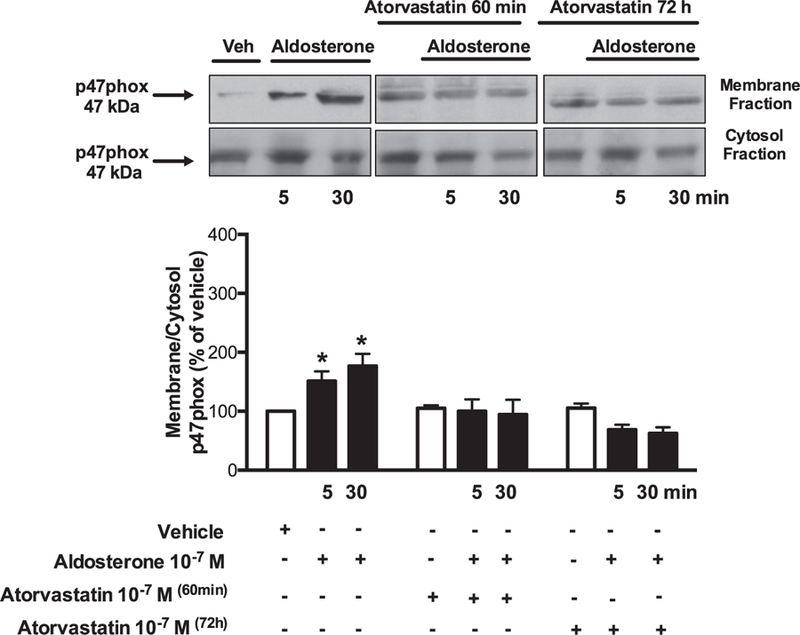
Atorvastatin prevents aldosterone-induced p47pox activity in WKY VSMCs. Effects of aldosterone (10−7 mol/L) on p47phox translocations from cytosol to the membrane, in the absence and presence of 10−7 mol/L of atorvastatin (60 minutes and 72 hours pre-incubation). Representative immunoblots: p47phox (fractions of membrane and cytosol). Results are mean ± SEM of 5–6 experiments. *P < 0.05, vs. vehicle.
Fig. 4.
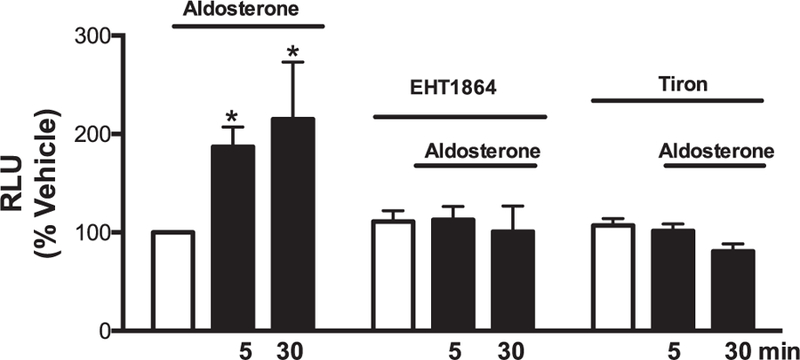
Atorvastatin prevents aldosterone-induced ROS generation via Rac1/2 in WKY VSMCs. Effects of aldosterone (10−7 mol/L) on ROS generation measured by lucigenin chemiluminescence assay in the absence and presence of EHT1864 (10−6 mol/L) or tiron (10−5 mol/L) (30 minute-incubation). Results are mean ± SEM of 5–6 experiments. *P < 0.05, vs. vehicle.
3.2. Atorvastatin blocks aldosterone-induced vascular inflammation
Aldosterone has been associated with vascular inflammation. We analyzed whether statin treatment reduces aldosterone-stimulated macrophage adhesion and inflammatory markers. Aldosterone stimulation significantly increased TNF-α expression and there was a trend to increase IL-1β genes expression. Aldosterone increased the number of adherent macrophages on VSMCs, effects that were attenuated by atorvastatin. Furthermore, Rac1/2 inhibition and ROS scavenging prevented aldosterone-induced vascular inflammatory response (Fig. 5A–C).
Fig. 5.
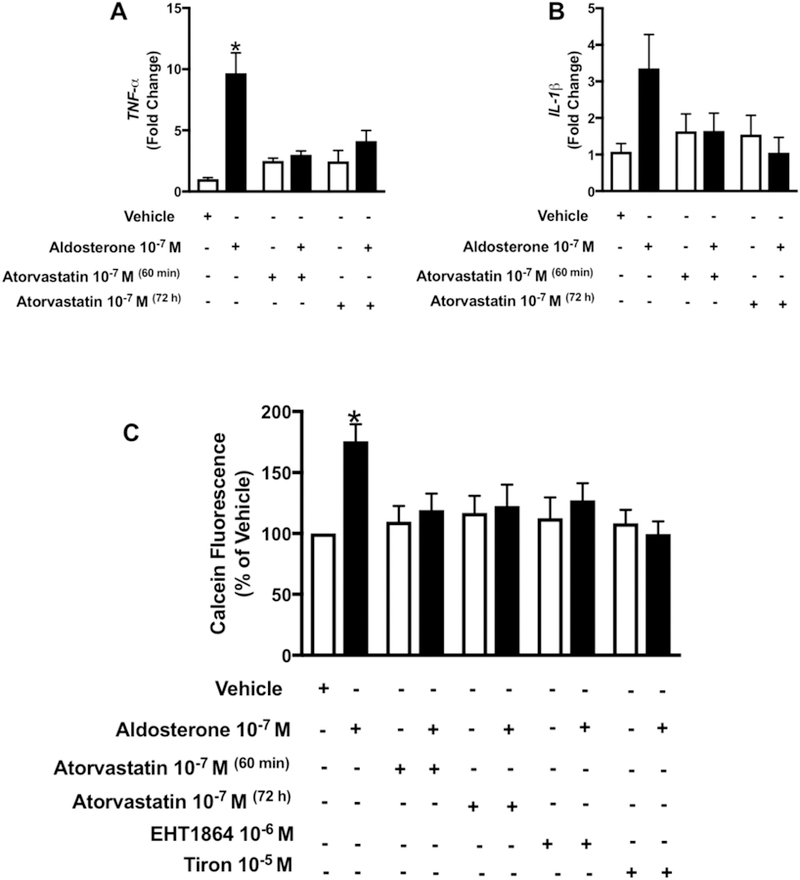
Atorvastatin prevents aldosterone-induced inflammation and macrophages adhesion to WKY VSMCs by Rac1/2 and ROS-sensitive mechanisms. Effects of aldosterone (10−7 mol/L) on TNF-α (A) and IL-1β (B) gene expression macrophages adhesion (C) in the absence and presence of 10−7 mol/L of atorvastatin (60 minutes or 72 h pre-incubation), EHT1864 (10−6 mol/L) or tiron (10−5 mol/L) (30 min pre-incubation). Results are mean ± SEM of 4–6 experiments. *P < 0.05, vs. vehicle.
4. Discussion
Major findings in the present study demonstrate that atorvastatin attenuates inflammatory effects induced by aldosterone in VSMCs by inhibiting Rac1/2 and reducing ROS production. Statins as an adjuvant therapy in the management of cardiovascular diseases such as hypertension and atherosclerosis may have beneficial vascular effects beyond their lipid-lowering effects.
Aldosterone has potent inflammatory and pro-fibrotic actions mediated by mineralocorticoid receptor (MR) activation [16,18,19]. Here, we have shown that aldosterone increases inflammatory markers and macrophage adhesion to VSMCs, supporting a pro-inflammatory vascular phenotype typically observed in atherosclerosis, hypertension and other cardiovascular diseases [21–25]. In the present study, we showed that aldosterone-induced VSMC inflammation is blunted by atorvastatin incubation and that both acute and long-term effects of aldosterone are modulated by atorvastatin. Statins possess broad immunomodulatory and anti-inflammatory properties, e.g. in endothelial cells statins increase endothelial nitric oxide synthase (eNOS) mRNA expression and nitric oxide bioavailability, decreases chemokines receptors, adhesion protein and cytokines production [20]. In addition, statin treatment reduces vascular remodeling and oxidative stress in angiotensin II treated mice, as well as attenuates type I collagen formation in isolated VSMC under angiotensin II stimulus via ROS production [21].
Besides being considered a marker for inflammation, ROS have been considered as a seconder messenger for the inflammatory response [4,15,19,22–24]. The deleterious effects produced by aldosterone have been mainly associated with ROS production [3,4,25,26]. Here, we show that aldosterone elevates ROS production in VSMC, which is abrogated by atorvastatin. Small GTPases such as Rac1/2 are essential for NADPH oxidase activation. Our findings suggest that atorvastatin might reduce aldosterone-induced ROS generation by inhibiting Rac1/2 and consequently Nox-activation, since atorvastatin pre-incubation blocked aldosterone-induced Rac1/2 and p47phox activation. Nox1 and Nox2 are constitutively associated with p22phox, and the full activation of these complexes requires interaction with other cytosolic subunits, including p47phox [8,23,27]. Further supporting a role for Rac1/2, NADPH oxidase and ROS in aldosterone-induced inflammation are the findings that tiron, the ROS scavenger, and EHT1864, Rac1/2 inhibitor, prevented macrophage adhesion to VSMC induced by aldosterone. This was associated with reduced ROS production and suggests that atorvastatin reduces VSMCs inflammation by inhibiting Rac1/2 and reducing ROS production, possibly mediated by Nox enzymes.
Statins are considered potent inhibitors of cholesterol biosynthesis. However, the overall benefits observed with statins also include effects beyond cholesterol lowering effects, e.g.: blocking Small GTPase such as Rac1/2 [10,13,20,21,28]. Here, 60 min of pre-exposure to atorvastatin generated vascular beneficial effects most likely by directly inhibiting Rac1/2 activity, whereas 72 h of pre-treatment might produce beneficial effects via dual actions: by lowering cholesterol content and by inhibiting Rac1/2 activity. The cell membrane contains lipid rafts, which have high concentrations of cholesterol and sphingolipid and which are responsible for stabilization of several proteins including the NADPH oxidases [29,30]. Although we did not check the lipid rafts content in the present study, 72 h of statin incubation might be inhibiting ROS production and vascular inflammation by disrupting lipid rafts content. Some studies have shown that statin long-term incubation reduces the lipid rafts content [31,32].
We have not analyzed the effects of statin treatment in experiments in vivo in the present study, but it is worth mentioning that statins can reduce aldosterone plasma levels in hypertensive and diabetic patients, as well as diminish Ang II-induced aldosterone secretion in mouse zona glomerulosa cells [33]. These findings reinforce the notion that atorvastatin may protect the vasculature against pro-inflammatory effects of aldosterone, as well as reduce aldosterone production.
In conclusion, our findings indicate that atorvastatin prevents aldosterone-induced vascular inflammation associated with Nox–mediated ROS generation. Our findings indicate that atorvastatin may protect the vasculature in diseases that are associated with elevated levels of aldosterone. In addition, we propose that statins might have dual beneficial effects in cardiovascular disease through its lipid-lowering actions and direct pleiotropic vascular effects.
Acknowledgments
The authors gratefully acknowledge Ms. Ying He for her help in the VSMC isolation procedure.
Funding
This work was supported by the São Paulo Research Foundation [grant numbers 2010/52214-6 (to R.C.T); 2011/01785-6; 2011/22035-5 (to T.B.-N.)]; Coordination for the Improvement of Higher Education Personnel (CAPES) [grant number: 88887.092495/2015-00 (to T.B.-N)]; the Agence Universitaire de la Francophonie [grant number 58145FT103]; the Juvenile Diabetes Research Foundation [grant number 4-2010-528]; the Canadian Institutes of Health Research [grant number 44018]; and the British Heart Foundation [grant number CH/12/429762].
Footnotes
Conflict of interest
None declared.
References
- [1].Bollag WB, Regulation of aldosterone synthesis and secretion, Compr. Physiol 4 (2014) 1017–1055. [DOI] [PubMed] [Google Scholar]
- [2].Briones AM, Nguyen Dinh Cat A, Callera GE, Yogi A, Burger D, He Y, Correa JW, Gagnon AM, Gomez-Sanchez CE, Gomez-Sanchez EP, Sorisky A, Ooi TC, Ruzicka M, Burns KD, Touyz RM, Adipocytes produce aldosterone through calcineurin-dependent signaling pathways: implications in diabetes mellitus-associated obesity and vascular dysfunction, Hypertension 59 (2012) 1069–1078. [DOI] [PubMed] [Google Scholar]
- [3].Briet M, Schiffrin EL, Vascular actions of aldosterone, J. Vasc. Res 50 (2013) 89–99. [DOI] [PubMed] [Google Scholar]
- [4].Briet M, Schiffrin EL, Aldosterone: effects on the kidney and cardiovascular system, Nat. Rev. Nephrol 6 (2010) 261–273. [DOI] [PubMed] [Google Scholar]
- [5].Fritsch Neves M, Schiffrin EL, Aldosterone: a risk factor for vascular disease, Curr. Hypertens. Rep 5 (2003) 59–65. [DOI] [PubMed] [Google Scholar]
- [6].Stancu C, Sima A, Statins: mechanism of action and effects, J. Cell. Mol. Med 5 (2001) 378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Martin G, Duez H, Blanquart C, Berezowski V, Poulain P, Fruchart JC, Najib-Fruchart J, Glineur C, Staels B, Statin-induced inhibition of the Rho-signaling pathway activates PPARalpha and induces HDL apoA-I, J. Clin. Invest 107 (2001) 1423–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bruder-Nascimento T, Callera GE, Montezano AC, He Y, Antunes TT, Cat AN, Tostes RC, Touyz RM, Vascular injury in diabetic db/db mice is ameliorated by atorvastatin: role of Rac1/2-sensitive Nox-dependent pathways, Clin. Sci. (Lond.) 128 (2015) 411–423. [DOI] [PubMed] [Google Scholar]
- [9].Sahebkar A, Ponziani MC, Goitre I, Bo S, Does statin therapy reduce plasma VEGF levels in humans? A systematic review and meta-analysis of randomized controlled trials, Metabolism 64 (2015) 1466–1476. [DOI] [PubMed] [Google Scholar]
- [10].Gotto AM Jr., Farmer JA, Pleiotropic effects of statins: do they matter? Curr. Opin. Lipidol 12 (2001) 391–394. [DOI] [PubMed] [Google Scholar]
- [11].Antonopoulos AS, Margaritis M, Shirodaria C, Antoniades C, Translating the effects of statins: from redox regulation to suppression of vascular wall inflammation, Thromb. Haemost 108 (2012) 840–848. [DOI] [PubMed] [Google Scholar]
- [12].Bruder-Nascimento T, Callera G, Montezano AC, Antunes TT, He Y, Cat AN, Ferreira NS, Barreto PA, Olivon VC, Tostes RC, Touyz RM, Renoprotective effects of atorvastatin in diabetic mice: downregulation of RhoA and upregulation of Akt/GSK3, PLoS One 11 (2016) e0162731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Geng Y, Hsu JJ, Lu J, Ting TC, Miyazaki M, Demer LL, Tintut Y, Role of cellular cholesterol metabolism in vascular cell calcification, J. Biol. Chem 286 (2011) 33701–33706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen GP, Zhang XQ, Wu T, Li L, Han J, Du CQ, Alteration of mevalonate pathway in proliferated vascular smooth muscle from diabetic mice: possible role in high-glucose-induced atherogenic process, J. Diabetes Res 2015 (2015) 379287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Callera GE, Touyz RM, Tostes RC, Yogi A, He Y, Malkinson S, Schiffrin EL, Aldosterone activates vascular p38MAP kinase and NADPH oxidase via c-Src, Hypertension 45 (2005) 773–779. [DOI] [PubMed] [Google Scholar]
- [16].Callera GE, Yogi A, Briones AM, Montezano AC, He Y, Tostes RC, Schiffrin EL, Touyz RM, Vascular proinflammatory responses by aldosterone are mediated via c-Src trafficking to cholesterol-rich microdomains: role of PDGFR, Cardiovasc. Res 91 (2011) 720–731. [DOI] [PubMed] [Google Scholar]
- [17].Montezano AC, Callera GE, Yogi A, He Y, Tostes RC, He G, Schiffrin EL, Touyz RM, Aldosterone and angiotensin II synergistically stimulate migration in vascular smooth muscle cells through c-Src-regulated redox-sensitive RhoA pathways, Arterioscler. Thromb. Vasc. Biol 28 (2008) 1511–1518. [DOI] [PubMed] [Google Scholar]
- [18].Callera GE, Montezano AC, Yogi A, Tostes RC, He Y, Schiffrin EL, Touyz RM, c-Src-dependent nongenomic signaling responses to aldosterone are increased in vascular myocytes from spontaneously hypertensive rats, Hypertension 46 (2005) 1032–1038. [DOI] [PubMed] [Google Scholar]
- [19].Bruder-Nascimento T, Ferreira NS, Zanotto CZ, Ramalho FN, Pequeno IO, Olivon VC, Neves KB, Lopes RA, Campos E, Aguiar Silva CA, Fazan R, Carlos D, Mestriner FL, Prado D, Pereira FV, Braga TT, Luiz JP, Cau SB, Elias PL, Moreira AC, Camara NO, Zamboni DS, Alves-Filho JC, Tostes RC, NLRP3 inflammasome mediates aldosterone-induced vascular damage, Circulation 134 (23) (2016) 1866–1880. [DOI] [PubMed] [Google Scholar]
- [20].Greenwood J, Mason JC, Statins and the vascular endothelial inflammatory response, Trends Immunol 28 (2007) 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Briones AM, Rodriguez-Criado N, Hernanz R, Garcia-Redondo AB, Rodrigues-Diez RR, Alonso MJ, Egido J, Ruiz-Ortega M, Salaices M, Atorvastatin prevents angiotensin II-induced vascular remodeling and oxidative stress, Hypertension 54 (2009) 142–149. [DOI] [PubMed] [Google Scholar]
- [22].Touyz RM, Berry C, Recent advances in angiotensin II signaling, Braz. J. Med. Biol. Res 35 (2002) 1001–1015. [DOI] [PubMed] [Google Scholar]
- [23].Nguyen Dinh Cat A, Montezano AC, Burger D, Touyz RM, Angiotensin NADPH II oxidase, and redox signaling in the vasculature, Antioxid. Redox Signal 19 (2013) 1110–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bruder-Nascimento T, da Silva MA, Tostes RC, The involvement of aldosterone on vascular insulin resistance: implications in obesity and type 2 diabetes, Diabetol. Metab. Syndr 6 (2014) 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Silva MA, Bruder-Nascimento T, Cau SB, Lopes RA, Mestriner FL, Fais RS, Touyz RM, Tostes RC, Spironolactone treatment attenuates vascular dysfunction in type 2 diabetic mice by decreasing oxidative stress and restoring NO/GC signaling, Front. Physiol 6 (2015) 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Silva MA, Cau SB, Lopes RA, Manzato CP, Neves KB, Bruder-Nascimento T, Mestriner FL, Montezano AC, Nguyen Dinh Cat A, Touyz RM, Tostes RC, Mineralocorticoid receptor blockade prevents vascular remodelling in a rodent model of type 2 diabetes mellitus, Clin. Sci. (Lond.) 129 (2015) 533–545. [DOI] [PubMed] [Google Scholar]
- [27].Montezano AC, Touyz RM, Oxidative stress, Noxs, and hypertension: experimental evidence and clinical controversies, Ann. Med 44 (Suppl. 1) (2012) S2–16. [DOI] [PubMed] [Google Scholar]
- [28].Forstermann U, Sessa WC, Nitric oxide synthases: regulation and function, Eur. Heart J 33 (829–837) (2012) 837a–837d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Catalgol B, Kartal Ozer N, Lipid rafts and redox regulation of cellular signaling in cholesterol induced atherosclerosis, Curr. Cardiol. Rev 6 (2010) 309–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nordzieke DE, Medrano-Fernandez I, The plasma membrane: a platform for intra-and intercellular redox signaling, Antioxid. Basel 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ghittoni R, Patrussi L, Pirozzi K, Pellegrini M, Lazzerini PE, Capecchi PL, Pasini FL, Baldari CT, Simvastatin inhibits T-cell activation by selectively impairing the function of Ras superfamily GTPases, FASEB J 19 (2005) 605. [DOI] [PubMed] [Google Scholar]
- [32].Liu Y, Chen L, Gong Z, Shen L, Kao C, Hock JM, Sun L, Li X, Lovastatin enhances adenovirus-mediated TRAIL induced apoptosis by depleting cholesterol of lipid rafts and affecting CAR and death receptor expression of prostate cancer cells, Oncotarget 6 (2015) 3055–3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Baudrand R, Pojoga LH, Vaidya A, Garza AE, Vohringer PA, Jeunemaitre X, Hopkins PN, Yao TM, Williams J, Adler GK, Williams GH, Statin use and adrenal aldosterone production in hypertensive and diabetic subjects, Circulation 132 (2015) 1825–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]