

SYNOPSIS
As a class, β-lactamase inhibitors have proven successful in extending the clinical utility of β-lactam antibiotics by circumventing β-lactamase-mediated resistance. However, the rapid evolution of these β-lactamases calls for a critical re-evaluation of the relationships between susceptibility, drug exposures and bacterial response. The existing paradigm for in vitro susceptibility testing and development of β-lactam / β-lactamase inhibitor combinations may not optimally facilitate clinical use. Thus, alternative approaches for pairing these combinations and evaluating in vitro susceptibility are needed to provide better guidance to clinicians.
Keywords: Combination therapy, Pharmacokinetics/Pharmacodynamics, Susceptibility testing, Optimal dosing, Gram-negative bacteria
INTRODUCTION
Antibiotic resistance poses one of the greatest medical threats to human health. According to the Centers for Disease Control (CDC), resistant bacteria account for more than 2 million illnesses and 23,000 deaths annually in the United States alone1. In healthcare settings, infections caused by resistant Gram-negative bacteria may be especially challenging to treat because they often present with multiple mechanisms of resistance. These infections are commonly associated with inappropriate or suboptimal therapy. Several studies have shown that resistance in Gram-negative bacteria leads to additional costs and length of hospitalization, as well as high morbidity and mortality2–7. Although resistance to key antibiotics continues to rise, there is a shortage of new drug candidates in early development for the treatment of Gram-negative bacterial infections. Thus, there is a pressing need for a critical evaluation of how to optimally use existing agents and those under development.
Gram-negative bacteria are highly adaptable pathogens capable of intrinsic and acquired resistance to multiple classes of antibiotics. Resistance to these agents is mediated through a variety of mechanisms that include: decreased permeability through loss of porins, extrusion of drug through overexpression of transmembrane efflux pumps, decreased binding of drugs through target mutations, and the production of inactivating enzymes (e.g., β-lactamases or functional group transferases). Of these, the production of β-lactamases is one of the most commonly encountered resistance mechanisms.
Historically, the use of β-lactamase inhibitors in combination with β-lactams has been critical to circumventing β-lactamase-mediated resistance. However, conventional practices in the development and evaluation of these combinations may preclude optimal clinical use. This review will highlight our current practices of developing β-lactam / β-lactamase inhibitor combinations, and discuss avenues for improved testing / pairing of these combinations.
β-LACTAMASE-MEDIATED RESISTANCE
β-lactams are an important class of antibiotics used for the treatment of infections due to Gram-negative pathogens. They exert their activity by inhibiting penicillin-binding proteins (PBPs) required for bacterial cell wall synthesis. As a result of the widespread use of these β-lactams, β-lactamase-producing organisms have become a prominent concern8–12.
β-lactamases represent a heterogeneous group of enzymes capable of hydrolyzing and inactivating the core β-lactam ring required for the bactericidal activity of β-lactam antibiotics. These β-lactamases are classified either according to substrate and inhibitor profiles (Bush-Jacoby classification) or primary amino acid sequences (Ambler classification). The Ambler classification groups β-lactamases into classes A, B, C and D, as shown in Table 1. In the United States, class A enzymes are the predominant β-lactamases reported in Gram-negative species11–13. Genes encoding class A enzymes are generally located on plasmids which can be transferred between bacterial species. These enzymes include penicillinases, extended-spectrum β-lactamases (ESBLs) and carbapenemases. Penicillinases (e.g., SHV-1, TEM-1) primarily hydrolyze penicillins and early generation cephalosporins14,15. In addition to penicillins, ESBLs (e.g., CTX-M and other SHV, TEM subtypes) hydrolyze nearly all cephalosporins and monobactams14. Class A carbapenemases (e.g., Klebsiella pneumoniae carbapenemases or KPCs) are especially daunting because they can confer resistance to all currently available β-lactams. Nonetheless, the prevalence of other non-class A β-lactamases is also rising. Furthermore, many clinical isolates harbor multiple β-lactamase-encoding genes11,12. To circumvent these β-lactamases, several new β-lactamase inhibitors have been developed.
Table 1.
Major Classes of β-lactamases of Clinical Importance
| Ambler Class | Type of β-lactamase | Preferred Substrate(s) | Representative Enzymes |
|---|---|---|---|
| A | Narrow spectrum (penicillinase) | Penicillins, early cephalosporins | TEM-1/−2, SHV-1 |
| A | Extended spectrum | Narrow and extended spectrum penicillins, cephalosporins | SHV-2, CTX-M-15 |
| A | Serine carbapenemases | All β-lactams | KPC-2, KPC-3 |
| B | Metallo-β-lactamases | β-lactams except aztreonam | IMP-1, VIM-1 |
| C | Cephalosporinases | Cephalosporins | AmpC, CYM-2 |
| D | Oxacillinases | Oxacillin/cloxacillin | OXA-1, OXA-2 |
| D | Cephalosporinases | Oxacillin/cloxacillin, cephalosporins | OXA-11, OXA-15 |
| D | Carbapenemases | Oxacillin, carbapenems | OXA-48 |
PHARMACOLOGY OF β-LACTAMASE INHIBITION
The use of β-lactamase inhibitors is considered one of the most successful approaches for restoring β-lactam efficacy and continues to be of interest in drug development. Currently approved β-lactamase inhibitors generally lack significant antibacterial effect at clinically relevant concentrations; instead, they inhibit β-lactamases to preserve the efficacy of partnering β-lactams. At present, they are unavailable commercially as standalone agents and are co-formulated with a partner β-lactam. Existing combination products include piperacillin /tazobactam, ampicillin / sulbactam, amoxicillin / clavulanic acid, ceftazidime / avibactam and meropenem / vaborbactam.
First generation β-lactamase inhibitors (e.g., sulbactam, clavulanic acid and tazobactam) were designed structurally to resemble β-lactams. Their mechanism of inhibition involves irreversible binding to the β-lactamase active site to form of an acyl-enzyme complex that is hydrolyzed into enzyme and inhibitor fragments16–18. Sulbactam, clavulanic acid and tazobactam have in vitro inhibitory activity against class A penicillinases and extended-spectrum-β-lactamases19. However, they lack inhibitory activity against class A (e.g., KPC) and B carbapenemases (e.g., NDM, IMP and VIM), as well as class C (e.g., AmpC) and D β-lactamases (e.g., OXA enzymes shown in Table 1).
Recently, avibactam and vaborbactam, which belong to distinct classes of non-β-lactam inhibitors, have been approved for clinical use. Avibactam and vaborbactam possess inhibitory activity against Ambler class A and C β-lactamases, but lack activity against class B enzymes20,21. Additionally, avibactam exhibits activity against selected class D enzymes20. These novel non-β-lactam inhibitors not only differ structurally from the traditional inhibitors; mechanistically, they bind reversibly to the enzyme active site without (in most cases) being hydrolyzed. This allows for recycling of the inhibitor and potential binding to additional β-lactamase molecules.
Several other non-β-lactam inhibitors, such as relebactam, zidebactam and nacubactam, are currently under clinical development. Relebactam is being designed for use with imipenem and has shown an inhibitory spectrum similar to avibactam21. Pharmacologically, zidebactam and nacubactam perform a unique hybrid function: inactivation of important β-lactamases (i.e. KPCs, class D carbapenemases) as well as selective inhibition of penicillin-binding proteins, which may therefore also confer direct activity against metallo-b-lactamase-producing strains22, 23. As a result of their unique mechanisms of action, these newer agents have the potential to address lingering challenges in the inhibition of clinically relevant β-lactamases.
EFFICACY OF β-LACTAM / β-LACTAMASE INHIBITOR COMBINATIONS
While combinations like piperacillin / tazobactam have been widely used for decades, in recent years, there has been renewed interest in their utility against ESBL-producing Enterobacteriaceae. Traditionally, carbapenems have been considered the drug of choice for ESBL infections. However, the rapid dissemination of carbapenemases has highlighted the need to evaluate alternative treatment options. One of the first studies to explore the appropriateness of β-lactam / β-lactamase inhibitor combinations for ESBL bacteremia was a post-hoc analysis featuring data from 6 prospective bacteremia cohorts. In that study, 30-day mortality and length of hospitalization for patients with ESBL E. coli bacteremia were comparable for patients treated with β-lactam / β-lactamase inhibitor combinations (such as piperacillin / tazobactam) or a carbapenem24. Notably, the median piperacillin / tazobactam MIC in that study was relatively low (2/4 mg/L) given the current susceptibility breakpoint of ≤ 16/4 mg/L. Additionally, the bacteremia cases were mostly due to urinary and biliary infections, which are considered low to moderate inoculum infections. Following that landmark report, several observational clinical studies have further evaluated the efficacy of piperacillin / tazobactam for ESBL bacteremia.
Although some studies have since validated these findings, there is a lack of consensus regarding the efficacy of piperacillin / tazobactam for ESBL infections. The outcomes of key studies are summarized in Table 2. Variables such as the pathogen involved, severity of infection (inoculum size) and primary infection site appear to impact the clinical efficacy of piperacillin / tazobactam24–29. These studies have generally suggested that piperacillin/tazobactam might be associated with positive outcomes for isolates presenting with low MICs (< 8/4 mg/L), and for bacteremia secondary to low or moderate inoculum infections (such as urinary and biliary infections). Nonetheless, in a recent randomized clinical trial focusing on bloodstream infections due to ESBL-producing E. coli and K. pneumoniae, treatment with piperacillin/tazobactam was associated with higher mortality than meropenem30. Consistent with the observational studies, lower mortality rates (for the piperacillin/tazobactam treatment group) were observed in patients whose bacteremia was due to urinary infections. The median piperacillin MICs were 2/4 mg/L for E. coli isolates and 4/4 mg/L for K. pneumoniae, while the overall median meropenem MIC was 0.023 mg/L. However, there did not appear to be a trend towards worsening outcomes at higher piperacillin / tazobactam MICs as noted in the observational studies. Taken altogether, the data from these studies highlight discrepancies between in vitro susceptibility and observed clinical efficacy for piperacillin / tazobactam.
Table 2.
Studies Comparing the Efficacy of Piperacillin/Tazobactam versus Carbapenems for ESBL Bacteremia
| Author (Year) | Study Design | Primary Organism(s) | Primary Infection Source | 30 Day Mortality |
|||
|---|---|---|---|---|---|---|---|
| PTZa | CBPb | P-value | Interpretation | ||||
| Rodriguez-Baño (2004) | Prospective | E. coli | Urinary/biliary | 9%c | 17% | > 0.05 | Comparabled |
| Harris (2015) | Retrospective |
E. coli K. pneumoniae |
Urinary/biliary | 8% | 17% | > 0.05 | Comparable |
| Tamma (2015) | Retrospective |
E. coli K. pneumoniae |
Catheter | 26% | 11% | < 0.05 | Inferior |
| Ofer-Friedman (2015) | Retrospective |
E. coli K. pneumoniae |
Pneumonia | 60% | 34% | = 0.1e | Comparabled |
| Ng (2016) | Retrospective |
E. coli K. pneumoniae |
Urinary | 31% | 30% | > 0.05 | Comparable |
| Gutiérrez-Gutiérrez (2016) | Retrospective |
E. coli K. pneumoniae |
Urinary | 10% | 14% | > 0.05 | Comparable |
| Harris (2018) | Prospective |
E. coli K. pneumoniae |
Urinary | 12.3% | 3.7% | 0.90f | Inferior |
PTZ = piperacillin/tazobactam
CBP = carbapenem
Composite mortality associated with amoxicillin/clavulanic acid and piperacillin/tazobactam
Comparable within power of study to detect differences
P-value shown for 30-day mortality; for 90-day mortality, p < 0.05
P-value for non-inferiority
Given the broader inhibitory spectra of the new inhibitors, combinations such as ceftazidime / avibactam and meropenem / vaborbactam have been evaluated primarily against carbapenemase-producing organisms. Although both combinations have shown efficacy against KPCs, there have already been reports of resistance to ceftazidime / avibactam. Clinical resistance was first reported for a K. pneumoniae isolate harboring KPC-3 obtained from a patient who had no previous exposure to ceftazidime / avibactam31. Since that report, there have been clinical cases of resistance development following treatment with ceftazidime / avibactam in isolates harboring KPC-2 and KPC-332,33. The rapid emergence of resistance soon after the commercial availability of ceftazidime / avibactam may further illustrate shortcomings in our assessments and dosing of these combinations.
LIMITATIONS IN CURRENT PRACTICE WITH β-LACTAM / β-LACTAMASE INHIBITOR COMBINATIONS
Fixed Agent Pairings
The utility of β-lactams / β-lactamase inhibitors may be fundamentally limited by current practices in the design of these combinations. Currently, β-lactams and β-lactamase inhibitors are paired most commonly based on shared pharmacokinetics (e.g., similar elimination half-lives, bio-distribution and metabolic pathways). While matching the pharmacokinetics of the two agents is key to ensuring the presence of both agents at the site of infection and protecting the integrity of the β-lactam antibiotic, these considerations alone may not ensure optimal efficacy against all clinical isolates. Furthermore, when more than 1 β-lactamase is present, individual enzymes may display different affinities and susceptibilities (to different β-lactamase inhibitors), thus a fixed agent combination may not always be optimal.
Fixed Dose Ratio Pairings
β-lactam / β-lactamase inhibitor pairs are generally available only as a fixed dose ratio combination. For instance, commercial piperacillin / tazobactam formulations are all in 8:1 ratio (piperacillin to tazobactam). Yet, the rationale for this fixed ratio remains unclear. In some clinical scenarios [e.g., enzyme hyperproduction or severe (high inoculum) infections] modification of this ratio may be necessary to ensure adequate inhibitor exposures. The exception to this practice is with oral formulations of amoxicillin / clavulanic acid, where compositions of 2:1, 4:1, and 7:1 of β-lactam to β-lactamase inhibitor are available. This would allow some flexibility for clinicians to customize dosing to different clinical scenarios.
Current Approaches to Susceptibility Testing
Since the advent of β-lactamase inhibitors, there has been much debate regarding the most appropriate approach to assess in vitro susceptibility for β-lactam / β-lactamase inhibitor combinations. For some combinations, susceptibility is evaluated using a fixed inhibitor concentration (e.g., 4 mg/L of tazobactam) with a range of concentrations for the β-lactam (reflective of dynamic concentrations observed in vivo). The resulting MIC is designated as susceptible, intermediate or resistant based on established breakpoints for efficacy. This scheme is predicated on the assumption that the magnitude of enhanced susceptibility remains constant in the presence of an inhibitor, grossly neglecting the contribution that varying concentrations of inhibitor may have on susceptibility (Figure 1).
Figure 1.
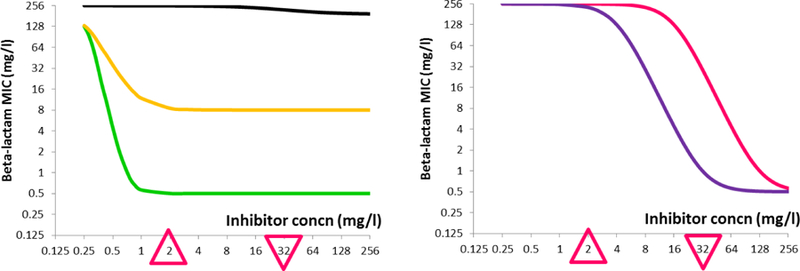
Different Hypothetical Pharmacodynamic Profiles of a β-lactamase Inhibitor
A hypothetical β-lactamase inhibitor is known to have the following therapeutic concentration range with a standard dosing regimen: Cmax = 32 mg/L (inverted triangle) and Cmin = 2 mg/L (upright triangle). When used in combination with a β-lactam, various response profiles (i.e., MIC reduction) can be anticipated for bacteria expressing different β-lactamase(s):
Black - minimal change in susceptibility (inactive inhibitor)
Green - dramatic reduction in susceptibility below Cmin, minimal change over the therapeutic range (ideal active inhibitor)
Gold - moderate reduction in susceptibility below Cmin, minimal change over the therapeutic range (active inhibitor rendered ineffective by other non-enzymatic resistance mechanisms)
Purple - gradual reduction in susceptibility over the therapeutic range (typical active inhibitor)
Pink - minimal change in susceptibility over the therapeutic range but gradual reduction above the therapeutic range (potentially active inhibitor with more aggressive dosing)
Hence, only a partial assessment of efficacy may be achieved with a single inhibitor concentration, and the resulting susceptibility data may not always correlate with in vivo efficacy. For combinations like ampicillin / sulbactam and amoxicillin / clavulanic acid, a fixed 2:1 ratio of β-lactam to inhibitor is used in susceptibility testing. For ampicillin / sulbactam, this latter approach reflects the 2:1 dose ratio used in all commercial formulations. Hence, it may be argued that this could better reflect the in vivo concentration ratio achieved for the combination and provide better insights of in vivo efficacy34,35. However, for amoxicillin / clavulanic acid where parenteral formulations use a fixed ratio of 5:1 while oral formulations use varied dose ratios, the correlation between susceptibility testing ratios and anticipated in vivo ratios is less clear.
Conventional PK/PD Characterization of Combinations
Pharmacokinetic/pharmacodynamic (PK/PD) indices such as the maximum concentration divided by the MIC (Cmax/MIC), the area under the 24 h concentration-time curve divided by the MIC (AUC/MIC) and the percentage of free-time above MIC (%fT>MIC) are commonly used to characterize killing profiles for various antibiotics (Figure 2). For β-lactams such as piperacillin and ceftazidime, the PK/PD index that best correlates with efficacy is the fT>MIC, which represents the duration of the dosing interval that the β-lactam concentration exceeds the MIC.
Figure 2.
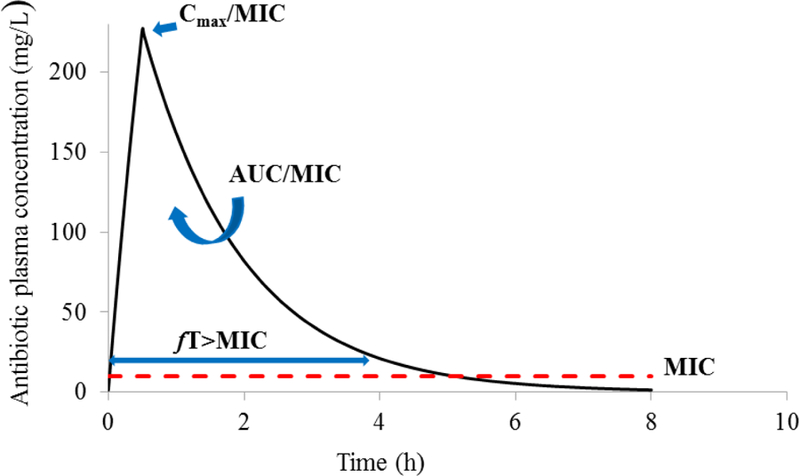
Pharmacokinetic/Pharmacodynamic Indices for Killing Activity of Antibiotics
PK/PD indices used to characterize the killing activity of various antibiotics:
Cmax/MIC – peak concentration divided by MIC
AUC/MIC - the area under the concentration-time curve divided by the MIC
%fT>MIC - the percentage of free-time above MIC
In all cases, the MIC (red dashed line) is expected to remain unchanged over time
Relatively speaking, our understanding of the PK/PD of the β-lactam in combination with a β-lactamase inhibitor is more limited. When an inhibitor is co-administered, it is commonly assumed to have a fixed effect over the entire concentration range observed during a dosing interval (Figure 1). While traditional inhibitors lack appreciable intrinsic killing, they are more likely to impact susceptibility in a concentration-dependent manner, as illustrated in Figure 1. Thus, the assumption of a fixed (all-or-nothing) inhibitory effect may not always be appropriate and may hamper efforts to optimally dose these combinations.
ALTERNATIVE STRAGETIES FOR PAIRING AND ASSESSING β-LACTAM / β-LACTAMASE INHIBITOR COMBINATIONS
Evaluating the complex interplay between a β-lactam, β-lactamase inhibitor and an infectious organism is not a trivial task. There is a critical need to address limitations in the current paradigm to guide rational pairing and dosing of β-lactam / β-lactamase inhibitor combinations. Several approaches have been proposed to improve our understanding of the dynamics of these combinations and to better inform dosing.
β-lactamase Inhibitors as Standalone Agents
There are no standard regulatory pathways with respect to the development of β-lactamase inhibitors alone. The availability of standalone inhibitors would allow much needed flexibility in these pairings to suit unique clinical needs. Additionally, it would allow the tailoring of inhibitor dose to accommodate clinical isolates that express different degrees of enzyme activity. Experimental data suggests that a flexible pairing / dosing scheme could be instrumental to inhibiting isolates that do not respond to a conventional fixed ratio or fixed β-lactam / β-lactamase inhibitor pair36,37. In this scheme, pharmacokinetic considerations would remain relevant as dosing agents with dissimilar half-lives in tandem could be implemented by asymmetric dosing frequency (i.e., dosing the two agents independently at different intervals) to reduce accumulation of the longer half-life drug. Furthermore, other approaches, such as staggered dosing (i.e., dosing the β-lactam first and the β-lactamase inhibitor later after a lag time) could also maximize the effect of the agent combination in selected cases. Clinical microbiology testing could be used to guide selection of combinations and optimize ratios of the components.
PK/PD Index to Characterize the Effect of β-lactamase Inhibitor
Using tazobactam as a reference inhibitor, Nicasio et al. evaluated the PK/PD determinant that best predicts efficacy within the context of a fixed β-lactam (piperacillin) exposure38. This study also evaluated the impact of β-lactamase gene transcription (low, moderate and high) on the magnitude of PK/PD associated with efficacy. Using data from MIC studies, the percentage of time above a threshold inhibitor concentration (%Time > threshold) was identified as the index that best correlated to tazobactam efficacy. This threshold value signified a critical concentration (dependent on enzyme transcription levels) at which enzyme inhibition was maximal. These findings suggested that tazobactam exposures may need to be customized for individual isolates (based on differences in enzyme expression) to meet efficacy targets. From a dosing perspective, this approach is more informative than the current scheme, in which a fixed concentration of inhibitor (irrespective of β-lactamase activity) is used to demonstrate in vitro efficacy. However, it may still overlook inhibitor effects below and above the threshold concentration, and thus provides an incomplete overview of inhibitor pharmacodynamics.
Characterization of a Unique PK/PD Index Accounting for the Effect of both the Inhibitor and the β-lactam
To address similar issues, Bhagunde et al. used relebactam (previously MK7655) in combination with imipenem against KPC-producing K. pneumoniae to illustrate the combined effects of a β-lactam and β-lactamase inhibitor39. In that study, a full factorial design was used to explore susceptibility to the combination. MICs were determined using a range of inhibitor concentrations (as opposed to using a single fixed concentration) to better reflect the fluctuations in inhibitor concentration observed in vivo, and adapted to a modified inhibitory sigmoid Emax model (as shown in Figure 3):
In this model, MIC = MIC in the presence of inhibitor; MIC0 = MIC in the absence of inhibitor; Imax = maximum inhibitor effect; H = sigmoidicity coefficient; I = inhibitor concentration and IC50 = inhibitor concentration required for 50% maximal inhibition. The model was used to characterize a theoretical concept known as the instantaneous MIC (MICi), which reflected changing pathogen susceptibility as inhibitor concentrations oscillated over a typical dosing interval. Bhagunde et al. elaborated on this concept to define the percentage of free-time above instantaneous MIC (fT>MICi) as the PK/PD index that best correlated with the efficacy of imipenem / relebactam. Since MICi (a surrogate for susceptibility) is dependent on the relationship between individual β-lactamase producing isolates and the inhibitor, this framework could be used to evaluate the efficacy of various β-lactamase inhibitor exposures against individual isolates. This approach is illustrated in Figure 3 using piperacillin / tazobactam and an ESBL-producing clinical K. pneumoniae isolate. In a small collection of ESBL-producing isolates, exposures of piperacillin / tazobactam yielding fT>MICi ≥ 55.1% were observed to suppress bacterial growth36. Although the model is relatively simple, additional operational elements (such as automated computation) would be needed in clinical settings to generate the more robust susceptibility profiling data required for generating optimal dosing recommendations.
Figure 3.
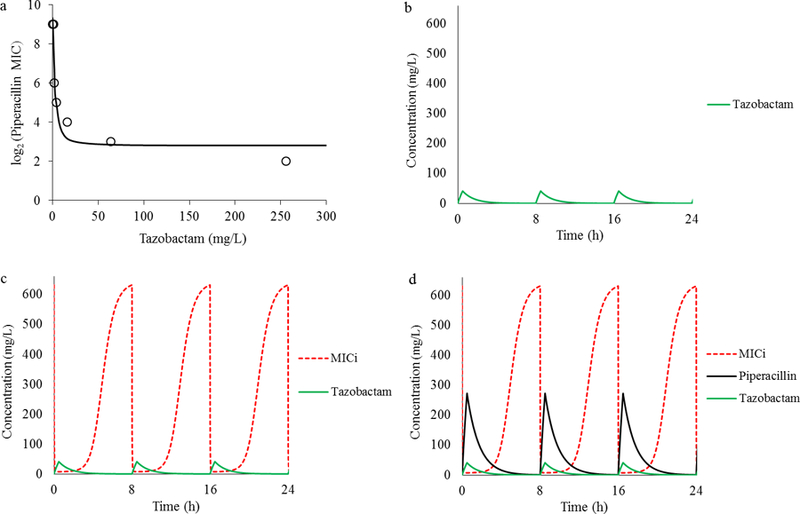
Depiction of MIC as Function of Inhibitor Concentration and Estimation of %fT>MICi
Piperacillin MICs for a clinical isolate were determined in the presence of tazobactam concentrations ranging from 0–256 mg/L and modeled using the sigmoid inhibitory Emax model (Figure 3a) to generate isolate-specific model paramater estimates. A free (unbound) tazobactam pharmacokinetic profile associated with a 0.5 g dose delivered every 8 h was then simulated (Figure 3b) and integrated with the Emax model parameter estimates to simulate a theoretical instantaneous MIC (MICi) profile (Figure 3c). In contrast to the common approch, the MICi profile (red dashed line) reflected changing pathogen susceptibility as the inhibitor concentration fluctuated over time. Finally, a simulated unbound piperacillin pharmacokinetic profile associated with a 4 g dose every 8 h was superimposed on the theoretical MICi profile (Figure 3d). The %fT>MICi was then estimated as the duration of the dosing interval over which the piperacillin concentration exceed the MICi (fT>MICi = 39.6% in Figure 3d).
Future Application in the Clinical Microbiology Laboratory: Susceptibility Profiling Based on a Concentration Ratio (Truncated Factorial Design)
When a β-lactam and a β-lactamase inhibitor have very similar pharmacokinetics, the concentrations of the agents are expected to remain at a fixed proportion at the site of infection over a dosing interval. In this context, the concept of susceptibility profiling can be abbreviated to testing a fixed ratio of the agents, based on the dose ratio of the formulation administered. For example, meropenem / vaborbactam is administered clinically as a 1:1 ratio. A strategy for susceptibility testing would be to use meropenem / vaborbactam in a similar ratio as 64/64 mg/L, 32/32 mg/L, 16/16 mg/L, 8/8 mg/L, 4/4 mg/L, etc. Extending the fixed ratio method to various β-lactam / β-lactamase inhibitor combinations would provide a simple, easily implemented approach for automated in vitro susceptibility testing that could better reflect PK/PD knowledge for these agents. This should be an area of increased investigation by organizations such as CLSI that provide guidance on antimicrobial susceptibility testing constituents. If further evidence for strain-dependent threshold effects is obtained based on β-lactamase expression and inhibitor engagement, then testing of fixed critical breakpoint concentrations of β-lactam with different concentrations of inhibitor may also provide data for individualized dosing strategies against specific pathogens.
WHAT RESEARCH IS NEEDED IN THE FUTURE?
In clinical settings, the ideal method for evaluating these combinations would have to provide informative insights and a quick turnaround time for bedside decisions to be made. In the long run, increasing involvement of robotics and artificial intelligence in automated platforms seem inevitable. Moreover, innovations in microfluidic design and liquid handling could promote the development of point-of-care devices to expedite identification and antimicrobial susceptibility profiling.
In the foreseeable future (next 5–10 years), advancements in whole genome sequencing could lead the path forward by facilitating timely identification of enzyme-encoding genes in clinical isolates. This could be leveraged to rule out β-lactamase inhibitor pairings (e.g., selection of avibactam in the presence of metallo-β-lactamases). However, detection of β-lactamase genes alone lends no additional information on the degree of functional gene expression, thus potentially limiting the ability to individualize therapy. Given the potential differences in β-lactamase present, levels of gene expression and substrate affinity among different clinical isolates, customizable β-lactam / β-lactamase inhibitor regimens could ensure the appropriate selection and dosing of inhibitors. Allowing some flexibility in the selection and dose of inhibitor could also improve clinical efficacy and/or delay the development of resistance against these combinations. Nonetheless, regulatory hurdles and concerns about medication errors could hinder the commercialization of standalone inhibitors. With the continued discovery of more sophisticated inhibitors, improved methods for the in vitro evaluation of β-lactam / β-lactamase inhibitor combinations in the clinical microbiology laboratory are warranted.
CONCLUSION
The continued dissemination of multidrug-resistant bacteria is an inevitable consequence of antibiotic use. β-lactam / β-lactamase inhibitor combinations remain viable options for the treatment of infections due to β-lactamase producing bacteria and could help address worsening resistance. However, current susceptibility testing practices do not provide the best guidance for β-lactam / β-lactamase inhibitor therapy. New approaches based on evolving understanding of PK/PD for combination therapy are needed to guide clinicians in the optimal selection and rational dosing of these combinations.
KEY POINTS.
The continued dissemination of β-lactamases among Gram-negative bacteria presents a major challenge to the treatment of nosocomial infections
β-lactamase inhibitors have been important to preserving the activity of β-lactam antibiotics against β-lactamase-producing Gram-negative bacteria
Currently, there are technical gaps in the development and evaluation of β-lactam / β-lactamase inhibitor combinations
Improved understanding of the joint action of these combinations will better inform dosing strategies and preserve their efficacy in the face of evolving β-lactamase resistance
Acknowledgments
Funding
V.H.T. is supported by the National Institutes of Health (R01AI140287-01).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement
V.H.T. has received honorarium from Merck. The other authors have nothing to disclose.
Contributor Information
Henrietta Abodakpi, Department of Pharmacological and Pharmaceutical Sciences, University of Houston College of Pharmacy, Houston, TX
Audrey Wanger, Department of Pathology and Laboratory Medicine, University of Texas Health Science Center at Houston, Houston, TX
Vincent H. Tam, Department of Pharmacy Practice and Translational Research, Department of Pharmacological and Pharmaceutical Sciences, University of Houston College of Pharmacy, Houston, TX
REFERENCES
- 1.Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States, 2013 2014. Available at: https://www.cdc.gov/drugresistance/threat-report-2013/index.html. Accessed October 24, 2018.
- 2.Slama TG. Gram-negative antibiotic resistance: there is a price to pay. Crit Care 2008;12 (Suppl 4):S4. doi: 10.1186/cc6820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cerceo E, Deitelzweig SB, Sherman BM, Amin AN. Multidrug-Resistant Gram-Negative Bacterial Infections in the Hospital Setting: Overview, Implications for Clinical Practice, and Emerging Treatment Options. Microb Drug Resist 2016;22(5):412–431. [DOI] [PubMed] [Google Scholar]
- 4.Tam VH, Rogers CA, Chang K-T, Weston JS, Caeiro J-P, Garey KW. Impact of multidrug-resistant Pseudomonas aeruginosa bacteremia on patient outcomes. Antimicrob Agents Chemother 2010;54(9):3717–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mauldin PD, Salgado CD, Hansen IS, Durup DT, Bosso JA. Attributable hospital cost and length of stay associated with health care-associated infections caused by antibiotic-resistant gram-negative bacteria. Antimicrob Agents Chemother January 2010;54(1):109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ibrahim EH, Sherman G, Ward S, Fraser VJ, Kollef MH. The influence of inadequate antimicrobial treatment of bloodstream infections on patient outcomes in the ICU setting. Chest July 2000;118(1):146–155. [DOI] [PubMed] [Google Scholar]
- 7.Kollef MH, Sherman G, Ward S, Fraser VJ. Inadequate antimicrobial treatment of infections: a risk factor for hospital mortality among critically ill patients. Chest February 1999;115(2):462–474. [DOI] [PubMed] [Google Scholar]
- 8.Adler A, Katz DE, Marchaim D. The Continuing Plague of Extended-spectrum beta-lactamase-producing Enterobacteriaceae Infections. Infect Dis Clin North Am June 2016;30(2):347–375. [DOI] [PubMed] [Google Scholar]
- 9.Paterson DL. Resistance in gram-negative bacteria: enterobacteriaceae. Am J Med June 2006;119(6 Suppl 1):S20–28; discussion S62–70. [DOI] [PubMed] [Google Scholar]
- 10.Ruppe E, Woerther PL, Barbier F. Mechanisms of antimicrobial resistance in Gram-negative bacilli. Ann Intensive Care December 2015;5(1):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castanheira M, Farrell SE, Deshpande LM, Mendes RE, Jones RN. Prevalence of β-lactamase-encoding genes among Enterobacteriaceae bacteremia isolates collected in 26 U.S. hospitals: report from the SENTRY Antimicrobial Surveillance Program (2010). Antimicrob Agents Chemother 2013;57(7):3012–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castanheira M, Farrell SE, Krause KM, Jones RN, Sader HS. Contemporary Diversity of β-Lactamases among Enterobacteriaceae in the Nine U.S. Census Regions and Ceftazidime-Avibactam Activity Tested against Isolates Producing the Most Prevalent β-Lactamase Groups. Antimicrob Agents Chemother 2014;58(2):833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nordmann P, Naas T, Poirel L. Global spread of Carbapenemase-producing Enterobacteriaceae. Emerg Infect Dis October 2011;17(10):1791–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bush K Bench-to-bedside review: The role of β-lactamases in antibiotic-resistant Gram-negative infections. Crit Care June/29 2010;14(3):224–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bush K, Jacoby GA. Updated Functional Classification of β-Lactamases. Antimicrob Agents Chemother December/07 2010;54(3):969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bush K Beta-lactamase inhibitors from laboratory to clinic. Clin Microbiol Rev 1988;1(1):109–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wright AJ. The penicillins. Mayo Clin Proc March 1999;74(3):290–307. [DOI] [PubMed] [Google Scholar]
- 18.Drawz SM, Bonomo RA. Three decades of beta-lactamase inhibitors. Clin Microbiol Rev January 2010;23(1):160–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bush K A resurgence of beta-lactamase inhibitor combinations effective against multidrug-resistant Gram-negative pathogens. Int J Antimicrob Agents November 2015;46(5):483–493. [DOI] [PubMed] [Google Scholar]
- 20.Drawz SM, Papp-Wallace KM, Bonomo RA. New beta-lactamase inhibitors: a therapeutic renaissance in an MDR world. Antimicrob Agents Chemother 2014;58(4):1835–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong D, van Duin D. Novel Beta-Lactamase Inhibitors: Unlocking Their Potential in Therapy. Drugs 2017;77(6):615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sader HS, Castanheira M, Huband M, Jones RN, Flamm RK. WCK 5222 (Cefepime-Zidebactam) Antimicrobial Activity against Clinical Isolates of Gram-Negative Bacteria Collected Worldwide in 2015. Antimicrob Agents Chemother 2017;61(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moya B, Barcelo IM, Bhagwat S, et al. Potent β-Lactam Enhancer Activity of Zidebactam and WCK 5153 against Acinetobacter baumannii, Including Carbapenemase-Producing Clinical Isolates. Antimicrob Agents Chemother 2017;61(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodriguez-Bano J, Navarro MD, Retamar P, Picon E, Pascual A, Extended-Spectrum Beta-Lactamases-Red Espanola de Investigacion en Patologia Infecciosa/Grupo de Estudio de Infeccion Hospitalaria G. beta-Lactam/beta-lactam inhibitor combinations for the treatment of bacteremia due to extended-spectrum beta-lactamase-producing Escherichia coli: a post hoc analysis of prospective cohorts. Clin Infect Dis January 15 2012;54(2):167–174. [DOI] [PubMed] [Google Scholar]
- 25.Harris PN, Yin M, Jureen R, et al. Comparable outcomes for beta-lactam/beta-lactamase inhibitor combinations and carbapenems in definitive treatment of bloodstream infections caused by cefotaxime-resistant Escherichia coli or Klebsiella pneumoniae. Antimicrob Resist Infect Control 2015;4:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tamma PD, Han JH, Rock C, et al. Carbapenem therapy is associated with improved survival compared with piperacillin-tazobactam for patients with extended-spectrum beta-lactamase bacteremia. Clin Infect Dis May 1 2015;60(9):1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gutierrez-Gutierrez B, Perez-Galera S, Salamanca E, et al. A Multinational, Preregistered Cohort Study of beta-Lactam/beta-Lactamase Inhibitor Combinations for Treatment of Bloodstream Infections Due to Extended-Spectrum-beta-Lactamase-Producing Enterobacteriaceae. Antimicrob Agents Chemother July 2016;60(7):4159–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ofer-Friedman H, Shefler C, Sharma S, et al. Carbapenems Versus Piperacillin-Tazobactam for Bloodstream Infections of Nonurinary Source Caused by Extended-Spectrum Beta-Lactamase-Producing Enterobacteriaceae. Infect Control Hosp Epidemiol August 2015;36(8):981–985. [DOI] [PubMed] [Google Scholar]
- 29.Ng TM, Khong WX, Harris PNA, et al. Empiric Piperacillin-Tazobactam versus Carbapenems in the Treatment of Bacteraemia Due to Extended-Spectrum Beta-Lactamase-Producing Enterobacteriaceae. PLoS One 2016;11(4):e0153696–e0153696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harris PNA, Tambyah PA, Lye DC, et al. Effect of Piperacillin-Tazobactam vs Meropenem on 30-Day Mortality for Patients With E coli or Klebsiella pneumoniae Bloodstream Infection and Ceftriaxone Resistance: A Randomized Clinical Trial. JAMA September 11 2018;320(10):984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Humphries RM, Yang S, Hemarajata P, et al. First Report of Ceftazidime-Avibactam Resistance in a KPC-3-Expressing Klebsiella pneumoniae Isolate. Antimicrob Agents Chemother 2015;59(10):6605–6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shields RK, Chen L, Cheng S, et al. Emergence of Ceftazidime-Avibactam Resistance Due to Plasmid-Borne blaKPC-3 Mutations during Treatment of Carbapenem-Resistant Klebsiella pneumoniae Infections. Antimicrob Agents Chemother 2017;61(3):e02097–02016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giddins MJ, Macesic N, Annavajhala MK, et al. Successive emergence of ceftazidime-avibactam resistance through distinct genomic adaptations in blaKPC-2-harboring Klebsiella pneumoniae ST307. Antimicrob Agents Chemother 2017. [DOI] [PMC free article] [PubMed]
- 34.Thomson CJ, Miles RS, Amyes SG. Susceptibility testing with clavulanic acid: fixed concentration versus fixed ratio. Antimicrob Agents Chemother 1995;39(11):2591–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pfaller MA, Barry AL, Fuchs PC, Gerlach EH, Hardy DJ, McLaughlin JC. Comparison of fixed concentration and fixed ratio options for dilution susceptibility testing of gram-negative bacilli to ampicillin and ampicillin/sulbactam. Eur J Clin Microbiol Infect Dis May 1993;12(5):356–362. [DOI] [PubMed] [Google Scholar]
- 36.Abodakpi H, Chang K-T, Gao S, Sánchez-Díaz AM, Cantón R, Tam VH. Optimal Piperacillin-Tazobactam Dosing Strategies against Extended-Spectrum-β-Lactamase-Producing Enterobacteriaceae. Antimicrob Agents Chemother 2019;63(2):e01906–01918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abodakpi H, Chang KT, Zhou J, Byerly C, Tam VH. A novel framework to compare the effectiveness of beta-lactamase inhibitors against extended-spectrum beta-lactamase-producing Enterobacteriaceae. Clin Microbiol Infect 2019. [DOI] [PMC free article] [PubMed]
- 38.Nicasio AM, VanScoy BD, Mendes RE, et al. Pharmacokinetics-Pharmacodynamics of Tazobactam in Combination with Piperacillin in an In Vitro Infection Model. Antimicrob Agents Chemother April 2016;60(4):2075–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhagunde P, Chang KT, Hirsch EB, Ledesma KR, Nikolaou M, Tam VH. Novel modeling framework to guide design of optimal dosing strategies for beta-lactamase inhibitors. Antimicrob Agents Chemother 2012;56(5):2237–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]