

Graphical abstract
Sucrose nonfermenting AMPK-related kinase (SNARK) is a member of the AMPK family of kinases and has been implicated in the regulation of critical metabolic processes. Recent findings demonstrate that SNARK has an important role in the maintenance of muscle mass with age. Loss of skeletal muscle mass (cachexia) is a key problem for cancer patients. Thus, based on our previous findings with aging, we hypothesized that SNARK would play a role in regulating muscle mass under conditions of cancer cachexia. To test this hypothesis, Lewis Lung Carcinoma tumor cells or vehicle were injected subcutaneously in the right flank of wild type mice, muscle-specific transgenic mice expressing inactive SNARK mutant (SDN) or muscle-specific transgenic mice overexpressing wild-type SNARK (SWT). All tumor-bearing mice presented muscle wasting compared to vehicle-injected mice. However, SDN tumor-bearing mice had more pronounced atrophy compared to wild-type and SWT tumor-bearing mice. Histological analysis confirmed muscle atrophy in tumor-bearing mice, and SDN tumor-bearing mice exhibited a significantly smaller skeletal muscle cross-sectional area than wild-type and SWT tumor-bearing mice. Moreover, SDN tumor-bearing mice had increased skeletal muscle BAX protein expression, a marker of apoptosis, compared to other groups. Thus, lack of SNARK in skeletal muscle aggravates cancer-induced skeletal muscle wasting. These findings uncover a role for SNARK in the maintenance of skeletal muscle mass under cachexia conditions.
Keywords: AMPK, Muscle atrophy, Aging, Cancer cachexia
Graphical Abstract
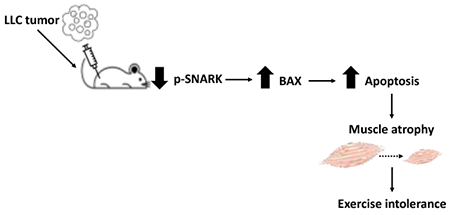
Introduction
AMP-activated protein kinase (AMPK) is a master regulator of glucose and lipid metabolism1. Sucrose nonfermenting AMPK-related kinase (SNARK) is a member of the AMPK family of kinases2 and is activated by phosphorylation on its Thr-208 residue. SNARK has been implicated in the regulation of critical metabolic processes, including contraction-stimulated glucose transport in skeletal muscle3,4. In addition, recent findings demonstrate a critical role for SNARK in myocyte survival and the maintenance of muscle mass with age in vivo5. Transgenic mice with muscle-inactive SNARK have age-accelerated muscle atrophy, activation of apoptosis in muscle, and are characterized by an approximately 40% reduction in muscle fiber cross sectional area at 58 weeks of age when compared to age-matched control mice5.
Cachexia syndrome is characterized by a progressive loss of skeletal muscle mass and is a critical issue for cancer patients. The lack of an effective treatment for cancer cachexia causes great suffering for patients and places a great burden on the healthcare system. Most patients with advanced cancer display skeletal muscle wasting associated with functional impairment and a poor prognosis6,7,8. Thus, elucidating mechanisms underlying skeletal muscle wasting that can lead to new therapies for this devastating syndrome9,10 is an important research goal. Animal models of cancer have been useful in identifying catabolic mediators of cancer cachexia, but the molecular basis of cancer cachexia is still poorly understood and the lack of therapies for cancer cachexia is problematic10–15. Given that there has been increased interest in the discovery of new targets to counteract cancer cachexia, and based on our previous findings with aging3,5, we hypothesized that SNARK plays a role in cancer-induced skeletal muscle wasting.
To test this hypothesis, we investigated the effects of Lewis Lung Carcinoma (LLC) tumor cell injection into muscle-specific SNARK transgenic mice on body mass, food intake, tumor size and muscle mass and running capacity. We find that overactivity of muscle SNARK is not protective against cancer cachexia effects, but lack of muscle SNARK activity aggravates cancer cachexia in mice. This study reveals that SNARK plays a role in mediating muscle atrophy and provides new insights into SNARK signaling as a potential target to counteract cancer cachexia.
Methods
Mice
All mouse experiments were approved by the Joslin Diabetes Center Institutional Animal Care and Use Committee (IACUC) and were in accordance with NIH guidelines. Ten-week-old C57BL/6 mice (Charles River Laboratories; MA, USA) were used to investigate SNARK phosphorylation in skeletal muscle during cancer cachexia progression.
Transgenic SNARK mice were generated as previously described5. Mice overexpressing wild-type SNARK (SWT) or dominant-negative SNARK (SDN) were identified using PCR, and mouse lines were established by breeding transgenic founders to wild-type C57BL/6 mice. SDN mice have reduced muscle SNARK activity due to overexpression of SNARK with a Thr-Ala mutation in its kinase domain, which reduces endogenous muscle SNARK activation. SWT mice have similar levels of SNARK overexpression compared to SDN mice, but do not display reduced SNARK activity, thus allowing us to distinguish the effects of SNARK overexpression from reduced SNARK activity. Non-transgenic littermates of both the SDN and SWT lines were pooled as wild-type controls. Ten to sixteen-week-old transgenic SNARK mice were studied to determine genotype-cancer interactions. Mice were housed in the Joslin animal facility with 12:12 hours light: dark cycle and had ad libitum access to standard diet (PharmaServ Inc., MA, USA) and water.
Cancer cachexia model
Mice were injected subcutaneously in the right flank with 105 LLC cells diluted in 100 μL of serum-free DMEM medium. Mice were killed by cervical dislocation under isoflurane anesthesia. Tumors and gastrocnemius + plantaris muscles were carefully harvested and weighed. Body mass changes (21 days post tumor cell injection) were calculated for each mouse after subtracting tumor mass from total body mass.
Running capacity test
Running capacity was assessed at 18 days post injection. Mice were acclimated to treadmill running for three consecutive days, which consisted of 10 minutes running on a treadmill at 15° inclination and moderate speed (12 m/min). After acclimation, mice completed a maximal incremental running test, which consisted of running on a graded treadmill at 15° inclination until exhaustion. The speed started at 6 m/min and was increased by 3 m/min every 3 min until mice were unable to run. The time to exhaustion was recorded.
Immunoblotting
Immunoblotting was performed as previously described5. Protein samples were run on a gradient SDS-PAGE gel and transferred to a nitrocellulose membrane. Ponceau staining or stain-free technology (Bio-Rad) were used as loading control and primary antibodies were applied overnight. SNARK phosphorylation was detected using anti-phospho SNARK (Thr-208) provided by Cell Signaling Technology, total SNARK was detected using Anti-NUAK2 antibody (ab107287), p62 (ab56416), BAX (CST2772) and GAPDH was detected using anti-GAPDH (#2118S, Cell Signaling Technology). Membranes were imaged using the ChemiDoc Touch System (Bio-Rad).
Muscle histology
Gastrocnemius muscle sections were stained with Wheat Germ Agglutinin (Alexa Fluor™ 488 Conjugate, ThermoFisher), and the cross sectional area of each fiber was measured. Histology was performed as previously described5. Because body mass, running capacity, food intake and skeletal muscle mass were similar among SNARK transgenic mice in this study, we combined wild-type, SDN and SWT non-injected mice in the same control group for the histological analysis.
Statistical analysis
Data are expressed as mean ± s.e.m. Sample sizes are indicated in the figure legends. All statistical analyses were performed using GraphPad Prism software (GraphPad Software, Inc.). Unpaired two-tailed Student’s t-test was used to compare two experimental groups. One-way analysis of variance (ANOVA), followed by Tukey’s multiple comparisons test, was applied to compare three or four groups. Statistical significance was defined as p < 0.05.
Results and Discussion
LLC induces cancer cachexia and regulates SNARK phosphorylation in skeletal muscle
To determine effects of cancer on muscle SNARK activity, we measured SNARK phosphorylation and expression in skeletal muscle from mice bearing LLC tumors (Figure 1A). Subcutaneous LLC cell injection in mice is known to exacerbate tissue catabolism in the host13,16–20. As expected, tumor-bearing mice displayed cancer cachexia in this study, as demonstrated by lower body mass change and skeletal muscle mass compared to control mice 18 days after tumor cell injection (Figure 1B and C). Western blot analysis revealed that tumor-bearing mice had reduced skeletal muscle SNARK phosphorylation (Figure 1D and E), indicating reduced SNARK activity. Thus, the hypermetabolism and catabolism typically associated with cancer cachexia was associated with decreased skeletal muscle SNARK phosphorylation. Previous work by us and others demonstrated that decreased metabolic function as observed with high fat feeding of rodents and obesity in humans are associated with increased skeletal muscle SNARK expression4,5. This demonstrates that muscle SNARK can be differentially regulated by various pathological conditions that induce changes in metabolism.
Figure 1. SNARK phosphorylation in the skeletal muscle of wild-type mice.
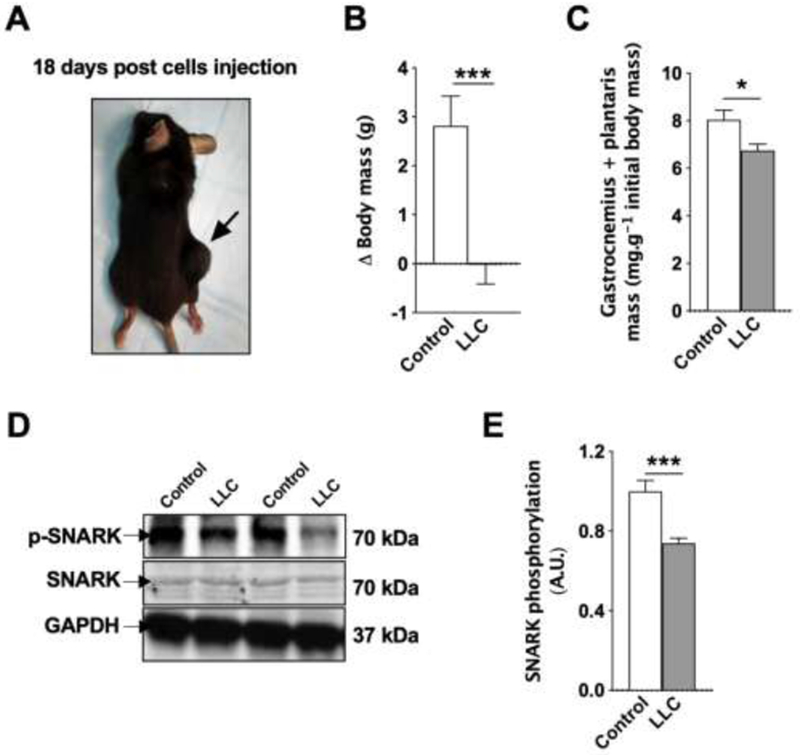
(A) C57BL/6 mice were subcutaneously injected with 105 LLC cells and analyzed 18 days post-injection, when a solid tumor was observed in all injected mice. (B) Delta changes in body mass and (C) skeletal muscle mass. n = 6 (control) and 13 (tumor-bearing mice). (D and E) SNARK phosphorylation in the skeletal muscle. n = 4 (control) and 7 (tumor-bearing mice).
Muscle-specific SNARK-inactive transgenic mice are more susceptible to cancer cachexia
To test whether skeletal muscle SNARK plays a role in the progression of cancer cachexia, we performed subcutaneous injections of vehicle or LLC tumor cells in muscle-specific SNARK transgenic mice5, expressing either dominant-negative inactive mutant SNARK (SDN), or overexpressing wild-type SNARK (SWT), as well as wild-type controls. Body mass, running capacity, food intake and tumor size were assessed (Figure 2A). As expected, tumor-bearing mice had lower body mass compared to control mice (Figure 2B). However, the loss of body mass was more pronounced in SDN tumor-bearing mice than the other groups (Figure 2B), suggesting that the lack of active muscle SNARK aggravated the cachexia condition. To assess functional impairment in response to cachexia, running capacity was measured. Consistent with body weight findings, tumor-bearing mice had lower running capacity than control mice, and SDN tumor-bearing mice displayed significantly lower running capacity compared to wild-type or SWT tumor-bearing mice (Figure 2C). Importantly, these differences in SDN tumor-bearing mice were not due to differences in food intake or tumor size (Figure 2D,E). Thus, our data demonstrate that the progression of cachexia was similar in control and SWT mice, indicating that worsening cachexia in SDN was due to loss of SNARK activity, and not SNARK overexpression, per se. These results suggest that overexpression of SNARK protein in SWT does not protect from the effects of cancer cachexia, but that loss of SNARK activity exacerbates cancer cachexia, as evidenced by greater loss of body mass and exercise capacity in SDN mice. As SDN and SWT models are muscle-specific, our data demonstrate a clear role for SNARK in the response of muscle to cancer cachexia. Future studies should examine whether SNARK also contributes to the cachexia response in other tissues, such as adipose and liver.
Figure 2. Phenotype of SNARK transgenic mice bearing LLC tumors.
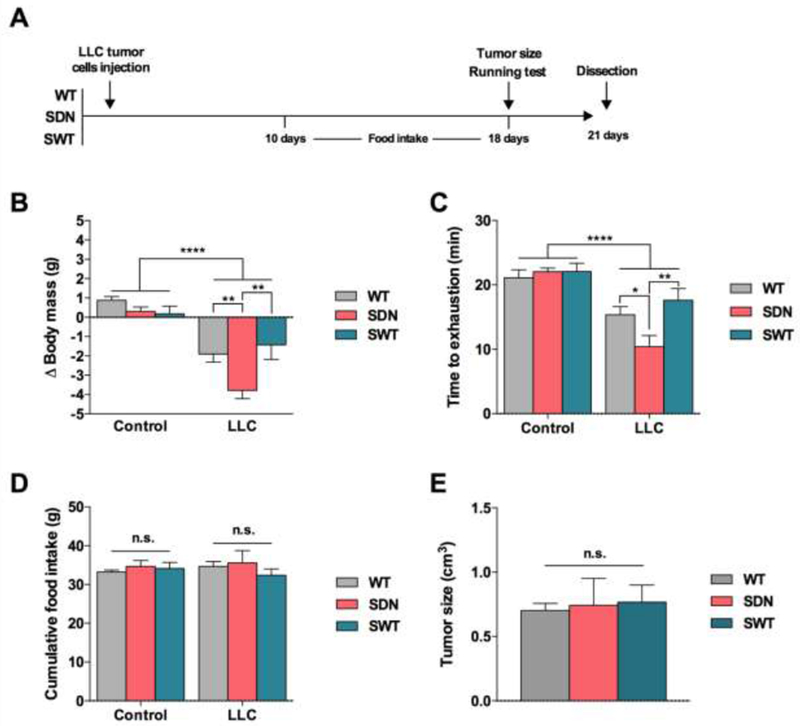
(A) Wild type mice (WT), transgenic mice expressing muscle-specific dominant-negative inactive SNARK mutant (SDN), and transgenic mice overexpressing muscle-specific wild-type SNARK (SWT) were subcutaneously injected with LLC cells and assessed at 18 (running capacity) or 21 days (body mass, food intake and tumor size) after tumor cell injection. (B) Delta changes in body mass. (C) Time to exhaustion in a maximal incremental running test. (D) Cumulative food intake. (E) Tumor size. Data are presented as mean ± s.e.m. n = 7-18. *p < 0.05; **P < 0.01; ***p < 0.001, ****P < 0.0001 between groups.
SDN tumor-bearing mice display more pronounced skeletal muscle wasting
Based on the data described above, we hypothesized that SNARK plays a role in the maintenance of skeletal muscle mass during cancer cachexia progression. To test this hypothesis, we evaluated skeletal muscle morphology in wild-type, SDN and SWT tumor-bearing mice. Skeletal muscle mass was significantly decreased in all tumor-bearing mice, an effect that was greatest in the SDN tumor-bearing mice. The SDN mice had a 30% decreases in muscle mass compared to control mice. This degree of muscle atrophy in the tumor-bearing SDN mice was 2-fold greater than the atropy of the SWT or wild-type tumor-bearing mice (Figure 3A). Histological analysis confirmed muscle atrophy in tumor-bearing mice, showing smaller cross sectional area of gastrocnemius muscle fibers compared to control mice (Figure 3B and C). SDN tumor-bearing mice exhibited significantly reduced cross sectional area compared to wild-type and SWT tumor-bearing mice (Figure 3B and C). Taken together, these findings demonstrate that loss of SNARK activity aggravates LLC-induced skeletal muscle wasting.
Figure 3. Muscle morphology in SNARK transgenic mice bearing LLC tumors.
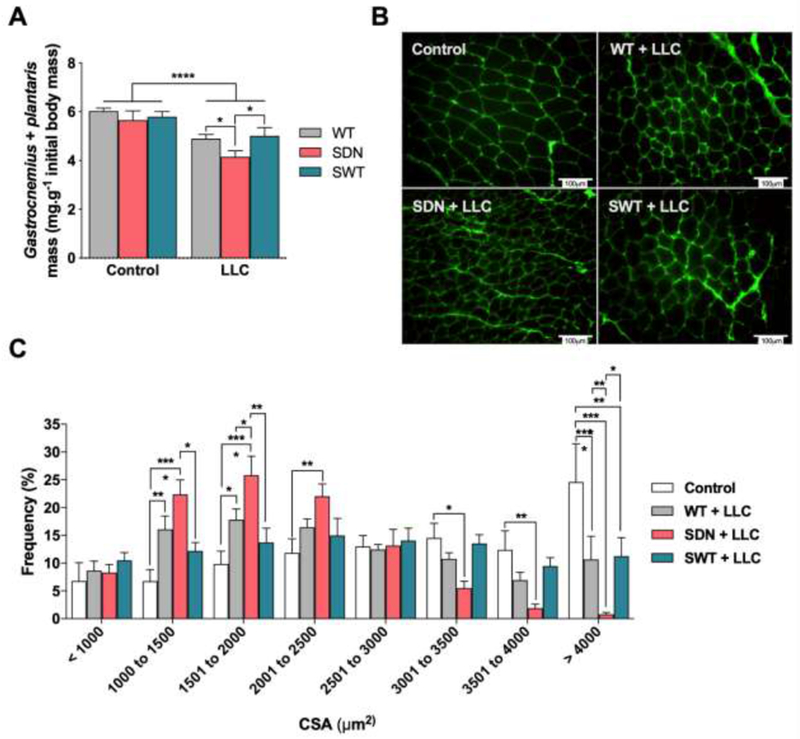
Wild type mice (WT), transgenic mice with muscle-specific dominant-negative inactive SNARK mutant (SDN), and transgenic mice with muscle-specific overexpression of wild-type SNARK (SWT) were subcutaneously injected with LLC cells and assessed 21 days after tumor cell injection. (A) Skeletal muscle mass. (B) Representative image of cross sections taken from gastrocnemius muscle. Muscle sections were stained with Wheat Germ Agglutinin, and the cross-sectional area (CSA) of each fiber was measured. (C) Quantification of fiber distribution. Data are presented as mean ± s.e.m. n = 7-12 mice/group. *p < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 between groups.
To explore the mechanisms underlying LLC-induced skeletal muscle wasting, we measured the protein expression of the autophagy substrate p62 and the apoptosis regulator BAX. p62 is a surrogate marker of the intracellular degradation system that links autophagy and proteasome pathways21. LLC tumor-bearing mice had higher p62 protein expression in the skeletal muscle compared to control mice, and there was no significant difference between wild-type and transgenic tumor-bearing mice (Figure 4A and B). BAX is a member of the proapoptotic BCL-2 family22. Remarkably, only SDN tumor-bearing mice had increased BAX protein expression compared to other groups (Figure 4A and C). These data suggest that lack of SNARK activity may regulate apoptosis and that could be the mechanism behind the more pronounced muscle wasting in SDN tumor-bearing mice. Thus, regulation of apoptosis through the apoptotic activator BAX is a potential mechanism underlying the exacerbated muscle atrophy during both aging5 and cancer cachexia progression, and in future studies will be important to elucidate the precise mechanism linking SNARK activity and apoptosis.
Figure 4. Markers of autophagy and apoptosis in skeletal muscle of SNARK transgenic mice bearing LLC tumors.
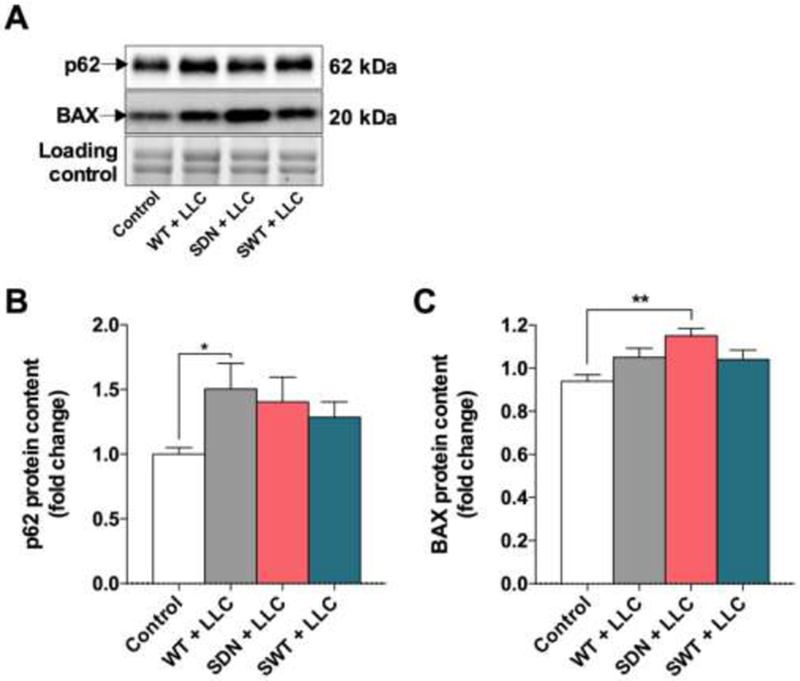
Wild type mice (WT), transgenic mice with muscle-specific dominant-negative inactive SNARK mutant (SDN), and transgenic mice with muscle-specific overexpression of wild-type SNARK (SWT) were subcutaneously injected with LLC cells and assessed 21 days after tumor cell injection. (A) Representative blots for p62 and BAX protein expression. (B) Quantification of p62 and (C) BAX protein content. Data are presented as mean ± s.e.m. n = 5-9 mice/group. *p < 0.05; **P < 0.01 between groups.
In summary, SNARK is implicated in the regulation of critical metabolic processes, including the maintenance of muscle mass with age, and our novel data now demonstrate that reduced SNARK activity aggravates cancer cachexia. By conducting experiments with LLC tumor-bearing mice, which is a well-characterized model of cancer cachexia13,17,18,23, we demonstrate for the first time that muscle SNARK activity is reduced in cancer cachexia and plays a role in cachexia progression. These novel findings provide the new insight that preventing reductions in SNARK activation could provide a therapeutic strategy to counteract cancer cachexia.
Highlights.
Lewis Lung Carcinoma regulates SNARK phosphorylation in skeletal muscle
Muscle-specific SNARK-inactive transgenic mice are more susceptible to cachexia
Lack of SNARK activity in skeletal muscle aggravates cancer-induced skeletal muscle wasting
Lack of SNARK activity increases BAX protein expression, a marker of apoptosis
Acknowledgements
This work was supported by an American Heart Association grant to S.J. Lessard (Award #15SDG25560057) and NIH grant R01DK099511 and R01DK101043 to Dr. Laurie J. Goodyear, and the Joslin Diabetes Center DRC (P30 DK36836). We thank Elsa Cesari and Miracle Onyeoziri for technical support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing financial interests
The authors have declared that no conflicts of interest exist.
References
- 1.Witczak CA, Sharoff CG & Goodyear LJ AMP-activated protein kinase in skeletal muscle: from structure and localization to its role as a master regulator of cellular metabolism. Cell. Mol. Life Sci 65, 3737–55 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lefebvre DL et al. Identification and characterization of a novel sucrose-non-fermenting protein kinase/AMP-activated protein kinase-related protein kinase, SNARK. Biochem. J 355, 297–305 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koh H-J et al. Sucrose nonfermenting AMPK-related kinase (SNARK) mediates contraction-stimulated glucose transport in mouse skeletal muscle. Proc. Natl. Acad. Sci 107, 15541–15546 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rune A, Osler ME, Fritz T & Zierath JR Regulation of skeletal muscle sucrose, non-fermenting 1/AMP-activated protein kinase-related kinase (SNARK) by metabolic stress and diabetes. Diabetologia 52, 2182–2189 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lessard SJ et al. The AMPK-related kinase SNARK regulates muscle mass and myocyte survival. J. Clin. Invest 1–11 (2015). doi: 10.1172/JCI79197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.K. F Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncology. Nat. Rev. Clin. Oncol 10: 90–9 (2013). [DOI] [PubMed] [Google Scholar]
- 7.K. F Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 12: 489–495 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Kimura M et al. Prognostic impact of cancer cachexia in patients with advanced non-small cell lung cancer. Support. Care Cancer (2014). doi: 10.1007/s00520-014-2534-3 [DOI] [PubMed] [Google Scholar]
- 9.Fearon K, Arends J & Baracos V Understanding the mechanisms and treatment options in cancer cachexia. Nat. Rev. Clin. Oncol 10, 90–9 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Fearon KCH, Glass DJ & Guttridge DC Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 16, 153–166 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Argilés JM, Busquets S, Stemmler B & López-Soriano FJ Cancer cachexia: understanding the molecular basis. Nat. Rev. Cancer 14, 754–62 (2014). [DOI] [PubMed] [Google Scholar]
- 12.He W a et al. Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proc. Natl. Acad. Sci. U. S. A 111, 4525–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kir S et al. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature 1, 1–19 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo Y et al. Cancer usurps skeletal muscle as an energy repository. Cancer Res. 74, 330–340 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Schäfer M et al. Ataxin-10 is part of a cachexokine cocktail triggering cardiac metabolic dysfunction in cancer cachexia. Mol. Metab 5, 67–78 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kir S et al. PTH/PTHrP receptor mediates cachexia in models of kidney failure and cancer. Cell Metab. 23, 315–323 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puppa MJ, Gao S, Narsale AA & Carson JA Skeletal muscle glycoprotein 130’s role in Lewis lung carcinoma-induced cachexia. FASEB J. 28, 998–1009 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruas JL et al. A PGC-1α isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 151, 1319–1331 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pin F et al. Combination of exercise training and erythropoietin prevents cancer-induced muscle alterations. Oncotarget 6, 43202–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Penna F et al. Combined approach to counteract experimental cancer cachexia: Eicosapentaenoic acid and training exercise. J. Cachexia. Sarcopenia Muscle 2, 95–104 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu WJ et al. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cellular and Molecular Biology Letters (2016). doi: 10.1186/s11658-016-0031-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oltval ZN, Milliman CL & Korsmeyer SJ Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programed cell death. Cell (1993). doi: 10.1016/0092-8674(93)90509-O [DOI] [PubMed] [Google Scholar]
- 23.Das SK et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 333, 233–238 (2011). [DOI] [PubMed] [Google Scholar]