

SUMMARY
Ribosomes and a subset of cellular mRNAs are trafficked into axons of developing neurons. The axonal localization of translational machinery allows new proteins to be rapidly and locally synthesized during axonal growth and pathfinding. However, in mature neurons, axonal ribosomes are significantly reduced or even absent. The mechanism that elicits this removal is currently unknown. Here, we demonstrate that synapse formation is the trigger for ribosome reduction in mature axons. In vivo analysis shows that axonal ribosome levels decrease in rat brain at a developmental stage coincident with synapse formation. Next, we observe in vitro that different synaptogenic inducers trigger an overall decrease of ribosomal proteins and rRNA in the axons of spinal motor neurons. We further observe that this process is dependent on the ubiquitin-proteasome system but not on autophagy. Together, these data identify synaptogenesis as the long missing biological trigger that leads to ribosome disappearance during axonal maturation.
Graphical Abstract
In Brief
The mechanism behind the striking loss of ribosomes from axons during neuronal maturation is unknown. Using in vivo and in vitro models, including neuron-muscle co-cultures and combining biochemistry and imaging techniques, Costa et al. demonstrate that synapse formation triggers ribosome reduction by a mechanism involving the ubiquitin-proteasome system.
INTRODUCTION
Axonal translation is a well-regulated process that has important roles during embryogenesis. During brain development, the establishment of neuronal circuitry requires axonal ribosomes to locally synthesize proteins required for axonal growth, branching, and response to guidance cues (Yoon et al., 2009; Zhen et al., 2000). However, in the axons of adult mammalian neurons, ribosomes are more difficult to detect. Early studies of rat hippocampal neuronal development show that axons possess ribosomal RNA (rRNA) shortly after plating neurons (Kleiman et al., 1994); however, by 10 days in culture, rRNA was virtually undetectable in axons. Subsequent studies showed that axonal growth cones of rat cortical neurons contain β-actin mRNA and polyribosomes (Bassell et al., 1998). Although several studies suggest that axons maintain some protein synthesis capacity in adulthood, there is still a clear decrease in the level of translational machinery in axons as neurons adopt their final mature connectivity in the nervous system. The basis for this drop in ribosome levels is currently not known.
Understanding the mechanisms that control ribosome expression levels in axons is important for understanding how to restore the growth and regenerative capacity of axons after injury. In certain injury models, there is evidence for increased mRNA and ribosome transport into axons (Court et al., 2008; Gumy et al., 2010; Taylor et al., 2009; Verma et al., 2005). This may be essential in order for the injured axon to reacquire the growth and synaptogenic properties typically associated with axons of developing neurons.
Although the loss of ribosomes is a fundamental step during neuronal development, surprisingly little is known about the underlying mechanisms responsible for this developmentally regulated program of ribosome loss from axons. In this study, we demonstrate that synapse formation is the trigger that leads to ribosomal reduction upon axonal maturation. In vivo analysis of developing axons from rat callosal projection neurons (CPNs) shows that a decrease in ribosome levels occurs coincident with the period in which these axons begin their synaptogenic period. In primary cultures of rat motor neurons, we observe that both exogenously applied synaptogenic stimuli, or formation of neuromuscular synapses in a microfluidic co-culture assay, significantly reduces the number of ribosomes in distal axons. The decrease in axonal ribosomes is mediated by the ubiquitin-proteasome system (UPS). Overall, our studies link synapse formation to the activation of a ribosome-directed proteasomal degradation pathway.
RESULTS
Decrease in Ribosomes Correlates with Axonal Maturation
Intra-axonal protein synthesis is a well-documented phenomenon that occurs primarily during the earliest stages of neuronal development (Piper and Holt, 2004; Cioni et al., 2018). During this period, we and others found rRNA and poly(A) mRNA in distal axons and growth cones of developing axons. Local protein synthesis has also been well documented in axons, with specific roles in axon growth (Wu et al., 2005; Merianda et al., 2015), guidance (Campbell and Holt, 2001, 2003; Welshhans and Bassell, 2011) and other processes critical for the establishment of neural circuitry (Lyles et al., 2006). However, the translational capacity of axons is markedly different in the axons of mature neurons (Bassell et al., 1994). In the axons of these neurons, ribosomes are nearly undetectable, indicating that the local protein synthesis capacity has been reduced. The specific timing and trigger for the loss of ribosomes in axons during development is not known.
The loss of axonal ribosomes is seen by day 10 in cultured rat embryonic hippocampal neurons (Kleiman et al., 1994). This time point is associated with the initiation of synapse formation. Therefore, we investigated whether ribosomal disappearance also occurs at a similar time point in vivo. To test this, we monitored axonal ribosomes during the development of rat CPNs. These neurons extend long axons that connect the two hemispheres of the cerebral cortex via the corpus callosum (Kumar and Huguenard, 2001). To target and visualize CPN, we performed in utero electroporation of progenitors of neurons committed to layers II/III of the somatosensory cortex (Kumar and Huguenard, 2001; Fame et al., 2011) with pCAGGS-tdTomato (Figure 1A). We also electroporated pCAGGS-EGFP-L10a plasmid to visualize ribosomes. EGFP-L10a encodes a ribosome subunit protein and is efficiently incorporated into ribosomes in neurons (Heiman et al., 2008, 2014; Gong et al., 2010; Doyle et al., 2008; Walker et al., 2012). Previous studies using heterologous expression of EGFP-L10a show that essentially all EGFP-L10a is present in ribosomes, with essentially no unincorporated EGFP-L10a in neurons (Heiman et al., 2008). Furthermore, EGFP-L10a-containing ribosomes were shown to be functional based on the presence of EGFP-L10a in polysomes as measured by immunoelectron microscopy (Heiman et al., 2008). Last, it was demonstrated that EGFP-L10a can be expressed via viral transduction of the axons themselves, resulting in the localization of EGFP-L10a to ribosomes in axons (Walker et al., 2012). Thus, EGFP-L10 can be used as a selective tag for imaging ribosome localization in vivo (Heiman et al., 2014; Heiman et al., 2008; Doyle et al., 2008).
Figure 1. Axonal Ribosome Reduction Correlates with Synaptogenesis In Vivo.

(A) In utero electroporation experimental design for rat embryonic somatosensory cortex progenitor cells.
(B) Development of CPN in vivo at different post-natal days (PND). tdTomato (red) was used as a reporter protein to allow corpus callosum visualization and DAPI (blue) as nuclear marker. Inset scale bar is 250 μm.
(C) EGFP-tagged ribosomal protein (RibP) L10a and tdTomato are co-expressed at somatosensory cortex. Co-expression of tdTomato (red) and RibP L10a (green) at somatosensory cortex cell bodies (C) is shown. Nucleolus position is indicated by the white arrow in the top inset (C). Scale bar is 25 μm.
(D–G) RibP L10a levels are reduced in CPN axons as they reach the contralateral target area but do not decrease in callosal CPN cell bodies. Electroporated brains, at different PND after perfusion and fixation (D; Scale bar is 1 cm), are shown. Corpus callosum (C.C.) midline area and cell bodies (C.B.) regions were used for protein extraction (E). Representative image of CPN cell bodies region (C; tdTomato; red) or corpus callosum midline area containing CPN axons (tdTomato; red) and RibP L10a (green) (E′; scale bar is 15 μm) is shown. Western blot analysis of EGFP and tdTomato expression (F and G) is shown. Bars represent the mean ± SEM of at least 2 (F) or 4 (G) independent experiments. For each independent experiment, results were normalized to tdTomato/β-actin ratio. Statistical significance by one-way ANOVA followed by multiple-comparison Tukey’s post hoc test is shown.
Coronal brain slices (80 μm thick) were prepared from animals at different postnatal ages (postnatal day [PND] 7, PND 12, PND 21). The CPN axonal tracts were readily detected as emanating from the transfected somatosensory cortex to the contralateral hemisphere (Figure 1B; Figure S1, for further details on CPN axonal development). At PND 7, CPN axons (red) have already crossed the corpus callosum midline. At this developmental stage, axons have just reached the contralateral hemisphere and start to ramify. These ramifications only become readily visible at PND 12 and completely spread at PND 21 (Figure 1B, insets).
To quantify the ribosomal levels during CPN axonal development in the same animals, the corpus callosum was dissected at the midline region to assess ribosome content in these axons (Figures 1D and 1E). After this tissue was harvested, total protein was extracted for western blot analysis (Figures 1F and 1G). Any EGFP-L10a in this fraction must derive from axonal projections from the somatosensory cortex since the plasmid was selectively incorporated into these neurons. An anti-GFP antibody was used to detect the ribosomal protein (RibP) L10a fused to enhanced green fluorescent protein (EGFP). tdTomato was used both as an axonal marker and, together with β-actin, was used as a loading control. Western blotting shows a decrease in ribosomal levels at PND 12 CPN axons (35%, p < 0.05), which become more significant at PND 21 (28%, p < 0.01), in comparison to PND 7 (Figure 1G; Figure S2). These results demonstrate that during brain development ribosomal levels decrease in vivo, and this occurs soon after axons reach the contralateral side and extend their ramifications.
We next investigated whether the ribosomal decrease was a generalized event that occurs throughout the entire neuron or whether it was limited to axons. EGFP-L10a and tdTomato expression levels in CPN cell body were evaluated by fluorescence microscopy. This analysis showed co-expression of both proteins (Figure 1C) including the EGFP-L10a fusion protein in the nucleolus of CPN cell bodies (Figure 1C, white arrow). Importantly, EGFP-L10a expression levels in the cell body were not reduced at any time point, as evaluated by western blot (Figure 1F). Together, these data indicate that there is an axon-selective loss of ribosomes shortly after CPN axons reach their targets on the contralateral hemisphere in the brain.
Synaptogenic Stimuli Induce a Decrease in Axonal Ribosomes
Synapse formation is characterized by the differentiation of the pre-and post-synaptic terminals. The former is regulated by postsynaptic stimuli, which can either be a target-derived molecule or a transmembrane protein present in the postsynaptic cell (Phillips et al., 2001). In the presence of these stimuli, neurotransmitter-containing synaptic vesicles (SVs) form clusters along the axons. These are sites where new synapses will be formed (Südhof, 2000).
To test the hypothesis that synapse formation is the trigger that leads to the decrease of ribosome levels in axons, we sought to induce presynaptic differentiation in a spatial and temporally controlled manner. Thus, we used fibroblast growth factor (FGF22), a target-derived presynaptic organizing molecule, capable of inducing the formation of functional presynaptic terminals in vitro and in vivo (Umemori et al., 2004; Terauchi et al., 2010). In order to confine FGF22 treatment to isolated axons we cultured rat motor neurons in a microfluidic chamber device (Figures 2A and 2B). This culturing device allows fluidic isolation of the distal axons from the cell bodies and creates a physical separation of axons (axonal compartment) from cell bodies and dendrites (somal compartment) (Taylor et al., 2005; Pinto et al., 2016b; Cristovão et al., 2014). In these devices, the axons of the motor neurons elongate through the microgrooves into the axonal compartment and establish a complex network of axonal processes. Axons stochastically cross the 450 μm long microgrooves and then cover the entire surface of the axonal compartment. Dendrites, due to their length, do not reach the axonal compartment (Figure 2B).
Figure 2. Axonal Application of FGF22 Induces Synapse Formation and RibPs and rRNA 5.8S Decrease.

(A) Schematic representation of microfluidic chambers.
(B) Representative image of spinal motor neurons (MNs) cultured in microfluidic chambers. At DIV 4, neurons were immunostained against MAP2 (somatodendritic marker; green) and Tau (axonal marker; red).
(C and E) Presynaptic clusters increase upon FGF22 treatment. At DIV 3/4, MN axons were stimulated with either BSA (Ctr) or FGF22 (2 nM). At DIV 4, neurons were immunostained against synapsin (C) or SV2 (E) (green) and neurofilament (NF; red). Scale bar is 2.5 μm.
(G, I, and K) Rib P and rRNA 5.8S decrease upon FGF22 treatment. At DIV 3/4, MN axons were stimulated with either BSA (Ctr) or FGF22 (2 nM). At DIV 4, neurons were immunostained against RibP P0 (G), RibP S6 (I), or rRNA 5.8S (K; green) and neurofilament (NF; red). Scale bar is 2.5 μm.
(D, F, H, J, and L) Quantitative data of the number, area, and integrated density of synapsin (D) and SV2 (F) and number and area of RibP P0 (H) RibP S6 (J) and rRNA 5.8S (L) clusters per axonal length. Bars represent the mean ± SEM. Statistical significance by unpaired Student’s t test.
(M) Exogenous RibP EGFP-L10a decrease upon FGF22 treatment. MNs were transduced at DIV 1 with an EGFP-RibP L10a lentivirus and stimulated at DIV 3/4, with either BSA (Ctr) or FGF22 (2 nM). Neurons were immunostained against GFP (green) and neurofilament (NF; red). Scale bar is 2.5 μm.
(N) Quantitative data of the number of RibP L10a clusters per axonal length. Bars represent the mean ± SEM of at least 2 independent experiments. Statistical significance by unpaired Student’s t test.
(O) Ribosomal levels are constant in developing axons. Unstimulated MNs were maintained in culture for increasing time periods (DIV 1–15). At the indicated points, neurons were fixed and immunostained against RibP S6 and neurofilament. Bars represent the mean ± SEM of at least 1–3 independent experiments. No significant differences were found among the groups, using one-way ANOVA followed by Dunnett’s post hoc statistic test.
We applied FGF22 (2 nM) to the axonal compartment and immunostained motor neurons to detect synapsin and SV2, two widely used presynaptic markers (early steps in synaptogenesis) (Pinto and Almeida, 2016). Neurofilament immunostaining was used as an axonal marker. FGF22 treatment increased the number of synapsin puncta per axon length (148.66%, p < 0.0026), synapsin puncta area per axon length (173.17%, p < 0.0004), and also synapsin puncta integrated intensity per axon length (157.96%, p < 0.0065; Figures 2C and 2D).
The synaptogenic effect of FGF22 was also confirmed with SV2 staining (Figures 2E and 2F). FGF22 treatment resulted in a statistically significant increase in the number of SV2 puncta per axon length (188.07%, p < 0.0001), in SV2 puncta area per length (191.54%, p < 0.0001), and in SV2 puncta intensity per axon length (194.16%, p < 0.0001; Figures 2E and 2F). Taken together, these data show that FGF22, when added exclusively to the axonal compartment, induces an increase in synapsin and SV2 clustering along the axonal shaft, indicating robust presynaptic differentiation. Similar results were observed when motor neurons were globally stimulated (i.e., bath application of FGF22 to neurons grown in standard cultures, rather than microfluidic devices) with FGF22 (Figures S3A–S3D).
We next asked whether axonal application of FGF22 triggers the reduction of axonal ribosomes by analyzing their components, namely, RibPs and rRNA. We cultured rat spinal motor neurons in microfluidic chambers, and the axonal compartment was stimulated at days in vitro (DIV) 4 with FGF22. To quantify ribosomes, we measured levels of two endogenous RibPs P0 and RibP S6, which are markers of the 60S and 40S ribosome subunits, respectively. We also measured the 5.8S rRNA by immunofluorescence using an antibody specific for this RNA (Y10b antibody). RibPs and rRNA were distributed in a punctuated pattern along distal axons, which was in accordance with previous observations (Kun et al., 2007). Neurofilament staining was used as an axonal marker. FGF22 triggered a significant decrease in the number of RibP P0 puncta and RibP P0 puncta area per axon length (78.21%, p < 0.0092; 77.94%, p < 0.0275, respectively) (Figures 2G and 2H). RibP S6 and rRNA 5.8S puncta number and area were also reduced upon FGF22 stimulation (Figures 2I–2L). RibP S6 puncta number and area per axon length were reduced (80.32%, p < 0.0001; 65.14%, p < 0.0121, respectively; Figures 2I and 2J) as well as rRNA 5.8S puncta number and area per axon length (57.16%, p < 0.0001; 33.47%, p < 0.0001, respectively; Figures 2K and 2L).
We next investigated whether the observed reduction of RibPs and rRNA 5.8S from axons is accompanied by a reduction in intra-axonal translation. We used the experimental design described above (Figure 2) and analyzed the levels of p-4E-BP1 in axons, which is commonly used to assess translational activation (Cox et al., 2008; Pfeiffer and Huber, 2006). FGF22 triggered a decrease in p-4E-BP1 intensity levels (Figures 3A and 3B; 54.88%, p < 0.0428). To measure translation using another method, we labeled nascent peptides using the O-propargyl-puromycin (OPP) method, followed by Click chemistry to visualize puromycinylated peptides (Liu et al., 2012; Slomnicki et al., 2016). FGF22 induced a reduction in intra-axonal nascent peptides levels (Figures 3C and 3D; 74.44%, p < 0.0395), confirming an overall drop in intra-axonal translation. These data indicate that FGF22 induces loss of ribosomes from distal axons and a consequent reduction in overall axonal translation levels. Similar results were observed when motor neurons were globally stimulated by bath application of FGF22 (Figures S3E–S3J).
Figure 3. Axonal Translation Is Reduced after Presynaptic Differentiation.

(A) FGF22 stimulation decreases p-4E-BP1 intra-axonal levels. Spinal MNs axons were stimulated, at DIV 3/4, with either BSA (Ctr) or FGF22 (2 nM). At DIV 4, neurons were immunostained against p-4E-BP1 (firescale) and neurofilament (NF; red). Scale bar is 2.5 μm.
(B) Quantitative data of the levels of p-4E-BP1. Bars represent the mean ± SEM of 4 independent experiments. Bars represent the mean ± SEM. Statistical significance by unpaired Student’s t test is shown.
(C) FGF22 treatment reduces the levels of intra-axonal nascent peptides. MN axons were stimulated, at DIV 3/4, with either BSA (Ctr) or FGF22 (2 nM). At DIV 4, intra-axonal nascent peptides were labeled using the O-propargyl-puromycin (OPP) method followed by Click chemistry to label peptide-incorporated puromycin (green). Motor neuron axons were immunostained against neurofilament (NF; red). Scale bar is 2.5 μm.
(D) Quantitative data of the levels of intra-axonal nascent peptides. Bars represent the mean ± SEM. Statistical significance by unpaired Student’s t test.
To further extend our observations, we next investigated whether EGFP-L10a would behave in a similar manner. We transduced rat spinal motor neurons at DIV1 with EGFP-L10a (Figures 2M and 2N). Consistent with previous studies by Heiman and colleagues that demonstrated the assembly of EGFP-L10a into intact ribosomes (Heiman et al., 2008), EGFP-L10a co-localizes with the endogenous RibP S6 and rRNA 5.8S and can also be found in nucleoli (Figures S4A and S4B). These results confirm that EGFP-L10a incorporates into ribosomes in rat spinal motor neurons. Axons were then stimulated at DIV 4 with FGF22, and EGFP-L10a levels were measured by immunocytochemistry. This analysis showed a decrease in EGFP-L10a puncta number per axon length upon FGF22 stimulation (51.79%, p < 0.432) (Figures 2M and 2N). Notably, FGF22 treatment did not reduce the presence of EGFP-L10a in the nucleolus (Figure S4A), suggesting that FGF22 does not affect ribosome biogenesis.
We next evaluated whether ribosome loss would occur spontaneously without FGF22. To test this, we assessed RibP levels in rat spinal motor neurons at different time points (DIV 1–15) in non-stimulated cultures. Immunofluorescence staining shows that there was no significant differences in RibP S6 puncta number per axon length up to DIV 15 (Figure 2O), indicating that ribosomal reduction does not occur on its own during this in vitro culturing protocol.
Neuromuscular Synapse Formation Triggers Ribosomal Decrease in Axons
In order to corroborate the results obtained with FGF22, we developed an approach to elicit synapse formation using a target cell. In these experiments, we prepared a neuron-muscle co-culture in microfluidic chambers (Figure 4A). This approach simulates the spatial separation of neurons and muscles, along with the physiological events that occur in vivo. Spinal motor neurons were plated in the somal compartment, while C2C12 or HEK293 cells were plated in the axonal compartment, according to the indicated timeline (Figure 4A). C2C12 cells are a mouse myoblast cell line that differentiates rapidly to form contractile myotubes, produce characteristic muscle proteins, and secrete several trophic factors (e.g., FGFs, brain derived neurotrophic factor [BDNF], etc.) (Madison et al., 2014). These cells also establish synapses with axons of motor neurons (Figure S5), resulting in neuromuscular junction-like synapses (Martin et al., 2015). In contrast, HEK293 cells are not able to establish synapses with neurons (Biederer et al., 2002; Scheiffele et al., 2000). Thus, we used HEK293 cells as a control to exclude the possibility that non-synaptic axonal contacts could mediate the observed effects on ribosomal levels.
Figure 4. Neuromuscular Synapse Formation Induces Ribosomal Decrease in Distal Axons.

(A) Schematic representation of the co-culture system in microfluidic chambers. Spinal motor neurons (MNs), C2C12 muscle fibers, or HEK293 cells (control) were cultured in microfluidic chambers compartments according to the timeline. At DIV 4, MNs were transduced with a Sindbis virus expressing EGFP-L10a for 24 h.
(B) Nerve-muscle interaction causes SV accumulation and exogenous RibP L10a decrease. At DIV 5, co-cultures were immunostained against synapsin (red) and EGFP (green). Scale bar is 100 μm.
(C and D) Quantitative data of the puncta number of synapsin (C) and RibP L10a (D) clusters per axonal length. Bars represent the mean ± SEM. Statistical significance by unpaired Student’s t test.
In these experiments, spinal motor neurons were cultured in the microfluidic devices and axons were allowed to enter the microgrooves and extend into the axonal compartment. C2C12 cells were previously induced to form myotubes prior to plating in the axonal compartment by switching them to culture media containing 2% horse serum. The C2C12 cells were then transferred to the axonal compartment. The axons are readily detectable in the axonal compartment after DIV 2 and were allowed to encounter the C2C12 muscle fibers or HEK293 cells for up to 3 days in culture. Synapsin levels were then evaluated along the axonal segments that were in contact with either cell type. To specifically evaluate neuronal ribosomal levels, motor neurons were transduced with a virus expressing EGFP-L10a. Axonal RibP L10a levels were then evaluated by immunofluorescence in the axonal compartment. Neurofilament was used as an axonal marker. As expected, axonal contact with C2C12 cells triggered a significant increase in the number of synapsin puncta per axon length (142.83%, p < 0.0386; Figures 4B and 4C). Notably, axonal contact with C2C12 cells triggered a significant decrease in the number L10a ribosomal puncta per axon length (62.65%, p < 0.0127) when compared with HEK293 cells (Figures 4B and 4D). These results demonstrate that synapse formation induces a robust decrease in the levels of ribosomes present in distal axons.
Ribosomes Are Degraded by the Ubiquitin Proteasome System
We next sought to understand the mechanism of ribosome reduction in axons after synaptogenic stimuli. We first asked whether the loss of ribosomes is due to the activation of an autophagy pathway. This mechanism has been previously associated with ribosomal degradation in yeast (Kraft et al., 2008). To test this hypothesis, we used 3-methyladenine (3-MA), an autophagy inhibitor that blocks autophagosome formation via inhibition of type III phosphatidylinositol 3-kinases (PI-3K) (Wu et al., 2010). We also used leupeptin, an autophagy inhibitor that inhibits lysosomal proteases (Kovács et al., 1982; Goo et al., 2017). At DIV 4, the axonal compartment was treated with FGF22, and RibP P0 levels were assessed by immunocytochemistry (Figure 5A). Tau was used as an axonal marker (Figures 5B and 5C). As shown previously, FGF22 induced a significant decrease of P0 levels from axons (Figure 5D, 76.25%, p < 0,05; Figure 5F, 71%, p < 0.05). However, 3-MA and leupeptin were unable to prevent this effect (72.02% and 63.6%, respectively; non-statistically significant [n.s.], when compared to FGF22-treated condition) (Figures 5B–5D and 5F). Importantly, when 3-MA or leupeptin was added to axons without adding FGF22, we did not observe any alterations in ribosomal levels (Figures 5B–5D and 5F), and no neuronal degradation was observed due to the application of 3-MA or leupeptin. These results indicate that autophagy is not a major mechanism used by axons to eliminate ribosomes upon exposure to synaptogenic stimuli.
Figure 5. Synapse Formation Induces a Mechanism of Ribosomal Loss from Axons.

(A) Schematic representation of the experimental design.
(B and C) Ribosomal decrease induced by FGF22 is dependent on ubiquitin proteasome system (UPS) but not dependent on autophagy. Spinal MNs axons were stimulated, at DIV 3/4, with either BSA (Ctr) or FGF22 (2 nM). Inhibitors of autophagy 3-MA (10 mM; B) and leupeptin (200 μM; C) or UPS inhibitors MG132 (1 μM; B) and epoxomicin (1 μM; C) were applied 6 h before the end of FGF22 stimulus. Neurons were immunostained against RibP P0 (green) and Tau (red). Scale bar is 2.5 μm.
(D–G) Quantitative data of the number of RibP P0 clusters per axonal length using different autophagy and UPS inhibitors. 3-MA (D), MG132 (E), leupeptin (F), and epoxomicin (G). Bars represent the mean ± SEM. Statistical significance by one-way ANOVA followed by Tukey’s post hoc test. (n.s., non-statistically significant.)
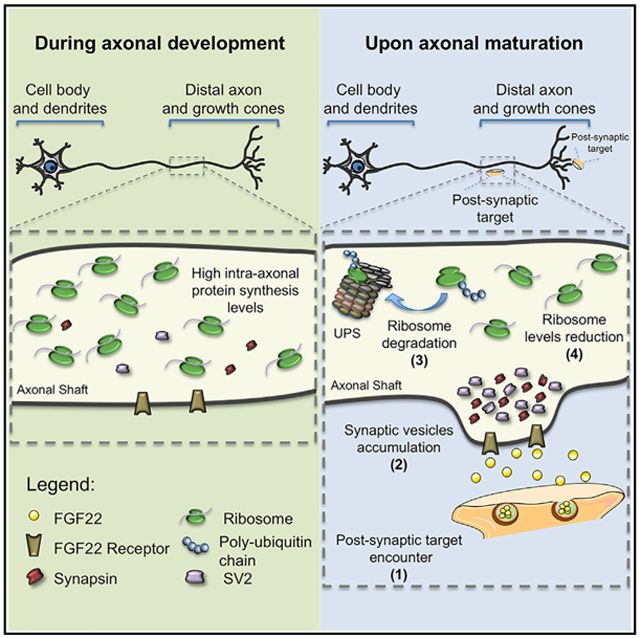
(H and I) Proposed model for ribosomal regulation in distal axons during axonal development (H) and upon axonal maturation (I). Upon axonal contact with a postsynaptic target or with synaptogenic molecules such as FGF22 (1), SVs accumulate within the nascent presynaptic button (2). After the immature axon establishes functional synapses, ribosome levels decrease significantly (3). A mechanism that is dependent on the ubiquitin proteasome system (UPS) is shown (4).
Because the UPS is the major degradation pathway used by cells to eliminate specific target proteins (Haas and Broadie, 2008), we also investigated whether the UPS was used by axons for ribosome clearance. To test this possibility, we used MG132 and epoxomicin, two well-known inhibitors of the UPS. MG132 blocks the proteolytic activity of the 26S proteasome complex by inhibiting the degradation of ubiquitin-conjugated proteins (Baptista et al., 2010), and epoxomicin binds the catalytic subunits of the proteasome potently inhibiting its chymotrypsin-like activity (Meng et al., 1999).
In these experiments, rat spinal motor neurons were cultured in microfluidic chambers, and the axonal compartment was treated with FGF22 at DIV 4. RibP P0 levels were assessed by immunocytochemistry as before. As seen previously, FGF22 significantly decreased the number of RibP P0 puncta per axon length (Figure 5E, 68.2%, p < 0.05; Figure 5G, 75.9%, p < 0.05). However, this effect was prevented by co-application of MG132 or epoxomicin (105%, p < 0.01 and 96.29%, respectively; p < 0.05 when compared to FGF22-treated condition) (Figures 5B, 5C, 5E, and 5G). MG132 or epoxomicin alone did not alter the ribosomal levels (Figures 5B, 5C, 5E, and 5G), and no neuronal degeneration due to MG132 or epoxomicin treatment was observed. Together these data indicate that UPS-mediated degradation is required for the decrease in ribosomal components induced by synapse formation.
DISCUSSION
In this study, we demonstrate that axonal ribosome levels decrease during the earliest stages of neuronal development in vivo and in vitro. We find that this drop in ribosome levels is not strictly determined by a temporally activated program but is instead actively triggered by pathways that lead synapse formation. This loss of ribosomes is selective to axons, as ribosome levels and nascent ribosome assembly in the cell body are unaffected by synaptogenic signals in axons. Last, we show that this process is dependent on the UPS but not on autophagy. This work identifies synaptogenesis as the long elusive trigger by which neurons change their axonal ribosomal composition during maturation (Figures 5H and 5I).
Although synaptogenic stimuli are thought to induce numerous local alterations that lead to the assembly of the presynaptic terminal (Phillips et al., 2001), our results suggest that these stimuli also elicit signals that are needed for an axonal “maturation” process. This involves transitioning axons from an elongation and guidance phase characterized by relatively high ribosome and mRNA levels to a phase that lacks a requirement for high levels of local translation. Local translation is particularly important since it is required for axonal elongation and the rapid morphological responses of the growth cone environmental guidance cues (Hengst et al., 2006; Yoon et al., 2009). However, once the axon has encountered its targets, these processes are no longer needed and may be detrimental to the axon terminal, which needs to form a physically stable connection. Thus, synaptogenesis may be the ideal developmental step for ribosome elimination since it marks the step at which the need for local translation is markedly reduced.
Our in vivo studies demonstrate that axonal ribosomes disappear in CPNs after PND7-PND12. This time period matches the time at which CPN axons reach the contralateral hemisphere and ramify their axons to establish synaptic contacts. Thus, our data demonstrate that axonal ribosome loss begins at the developmental stage coincident with the documented synaptogenesis peak for these neurons (Fame et al., 2011; Semple et al., 2013). Synaptic pruning is described to occur in a later stage (starting after PND 20–21) (Semple et al., 2013). Therefore, this later synapse-removal pathway is unlikely to be the trigger for ribosome removal. Consistent with ribosome removal during the earliest steps after axonal contact with target cells in vivo, we also found that the decrease in ribosome levels starts soon after presynaptic terminal formation in cultured neurons (Figure S3K). Thus, ribosome removal is an early step in synaptogenesis signaling.
Our in vitro experiments allowed us to precisely control the induction of synaptogesis, either by selective axonal application of a physiologically relevant synaptogenic molecule (FGF22) or cellular synaptic targets cells. We observed a reduction in the axonal levels of all the tested RibPs (RibP P0, RibP S6, and RibP L10a) and rRNA 5.8S. We also observed an overall partial decline in intra-axonal translation, demonstrated by the decrease in p-4E-BP1 levels and by the reduction in protein synthesis levels used a puromycin-based assay. Interestingly, we did not observe a complete loss of ribosomes from axons. This is in accordance with recent screens showing that mature axons still contain mRNAs and retain the ability to translate proteins (Gumy et al., 2011; Zivraj et al., 2010). In addition, Shigeoka et al. (2016) using translating ribosome affinity purification (TRAP) showed that the pool of mRNAs changes between developing and adult mice (Shigeoka et al., 2016). The authors observed a reduction in the axonal translatome during development, which is in line with our observations. This study describes that during developmental stages the axonal proteome is related with axon growth, axonal branching, and synaptogenesis, while in the adult stage the predominant transcripts are related with neurotransmission and axon survival (Shigeoka et al., 2016).
Notably, in untreated cultures maintained up to DIV12/15, no change in ribosome levels was observed. This indicates that ribosome loss does not occur spontaneously and does not occur as a consequence of a predetermined developmental program. Instead, an exogenous signal, such as synaptogenic signals, is needed to initiate a program of ribosome depletion from axons.
Our study also demonstrates that the UPS mediates ribosomal clearance. Interestingly, the UPS has been implicated in the clearance of excess ribosome protein subunits and to maintain protein homeostasis in yeast and in human-derived cell lines (Hinnebusch, 2009; Sung et al., 2016; Stavreva et al., 2006). Thus, a similar mechanism may occur in axons to reduce the excess translational capacity of axons after synapse formation, which marks the stage in neuronal development when the high protein synthesis capacity is no longer required. Notably, autophagy does not appear to be required for ribosome removal despite the use of this pathway for ribosome removal in nutrient-depleted conditions in yeast (Kraft et al., 2008). Thus, the UPS system appears to be the primary pathway for ribosome removal in axons; however, since we also observed a reduction in rRNA, we can hypothesize that other mechanism (e.g., axonal transport) might also be involved in this process. Interestingly, we observed a time-dependent increase in ribosomes in the cell body (Figure 1F). This might be due to an increased need in protein synthesis in the cell body level, in contrast to what occurs in distal axons, and this could possibly be achieved also by altered ribosomal transport.
Understanding the pathways that determine ribosome levels in axons may be particularly important in order to improve the regenerative capacity of injured axons. Injured axons appear to acquire features of developing axons, including accumulating mRNA and ribosomes (Court et al., 2008; Gumy et al., 2010; Twiss and Fainzilber, 2009; Michaelevski et al., 2010), suggesting the local translation may facilitate axonal growth and reinnervation. Indeed, Segal and collaborators observed that neurotrophins induce the transport of bclw mRNA to axons, where is locally translated, preventing axonal degeneration (Cosker et al., 2013), and adult bclw−/− mice have increased paclitaxel-induced neuropathy (Pease-Raissi et al., 2017). These and other studies suggest that adult neurons rely on axonal mRNA translation for maintaining axonal function and for axonal regeneration after injury. Although our studies focused on the regulation of ribosomes in a context of synaptogenesis during development, it will be important to establish whether the translational capacity of axons in adult neurons can be enhanced by targeting the UPS-regulated pathways described here and whether this approach could improve axon regeneration in vivo.
One open question is how the transmembrane (synaptogenic) receptors covey the message for ribosome degradation to the axoplasm. It will be important to identify the specific E3 ubiquitin ligases that are required for ribosome degradation. Additionally, synaptogenesis typically occurs only in specific regions of the axon, especially axonal termini. However, ribosome removal is seen along the entire length of the axon. It will be important to determine how the local synaptogenic signals elicit an axon-wide ribosome removal pathway and whether this requires a retrograde signal to deplete ribosomes throughout the length of the axon.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ramiro D. Almeida (ramirodalmeida@gmail.com).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
For in utero electroporation: Timed-pregnant Sprague Dawley rats (Harlan Italy SRL, Correzzana, Italy) were used and the experiments performed in accordance with the guidelines established by the European Community Council Directive 2010/63/EU of 22 September 2010, were approved by IIT licensing and Italian Ministry of Health. After electroporation, embryos developed normally and both delivered males and females were used for the experiments. 30 animals were used in the study.
For primary cultures: Wistar-Han rat females (Charles River, Barcelona, Spain) were housed (two per cage) and maintained in a controlled environment at 22–24°C with 55% humidity, on a 12 h light/dark cycle and fed with regular rodent’s chow and tap water ad libitum. E13.5 rat embryos (males and females) were used for spinal cord dissection. All manipulations were done after approval from the CNC Animal Welfare Committee (ORBEA_34_2013/28022013) and from the Portuguese national authority for animal experimentation, Direção Geral de Veterinária (DGAV: 0421/000/000/2013), and in accordance with the approved guidelines and regulations on animal care and experimentation stated in the European Union Directive 2010/63/EU. For this study 81 animals were used.
Cell lines
Mouse myoblast (C2C12; ATCC® CRL1772TM) and HEK293 (ATCC® CRL-1573) cell lines were grown in 75 cm2 tissue flasks in DMEM (Sigma) containing 10% heat inactivated fetal bovine serum (FBS) and pen/strep (100 U/ml). C2C12 myotube formation was enhanced by replacing the growth medium with DMEM containing 2% horse serum (HS). Both cell lines (C2C12 and HEK293) were maintained at 37°C in a humidified incubator under an atmosphere of 95% air and 5% CO2.
Cell lines used for viral production
HEK293T (ATCC® CRL-3216) and BHK-21 (ATCC® CCL-10) cell lines were grown in 75 cm2 tissue flasks in DMEM (Sigma) containing 10% heat inactivated fetal bovine serum (FBS) and pen/strep (100 U/ml). Both cell lines (HEK293T and BHK-21) were maintained at 37°C in a humidified incubator under an atmosphere of 95% air and 5% CO2.
METHOD DETAILS
Constructs and viral production
Ribosomal protein (RibP) L10a was cloned as previously described (Heiman et al., 2008). Briefly, L10a was PCR-amplified from a mouse brain cDNA library and cloned into the EcoRI and BamHI sites of C1-EGFP vector. The selected EGFP-L10a clone was sequenced and verified to conformity against the reference mRNA GenBank: BC083346. The resulting pEGFP-C1-L10a sequence then was used to generate a plasmid for lentiviral-mediated expression of EGFP-L10a fusion protein. The EGFP-L10a coding sequence was amplified by PCR and cloned into pRRLSIN.cPPt.PGK-GFP.WPRE vector, using the BamHI and Sall sites, replacing the existent GFP, by the In-Fusion® HD Cloning Kit (Clontech, Mountain View, USA). To generate a plasmid for sindbis-mediated viral expression of EGFP-L10a fusion protein, EGFP-L10a coding sequence was amplified by PCR and cloned into pSinRep5 vector (Invitrogen), using the MluI and SphI sites, by the In-Fusion® HD Cloning Kit (Clontech, Mountain View, USA). For in utero electroporation experiments, EGFP-L10a was amplified by PCR and cloned into pCAGGS-IRES-EGFP expression vector, using the NotI and BamHI sites, replacing the existent IRES-EGFP. tdTomato was amplified by PCR and cloned into pCAGGS-IRES-EGFP, using the NotI and BamHI sites, replacing the existent EGFP. pCAGGS expression vector contains a modified chicken β-actin promoter with a cytomegalovirus immediate early enhancer, conferring high and long-lasting expression in vivo (Cancedda et al., 2007).
For generation of lentivirus, HEK293T cells were transfected, using the calcium phosphate transfection protocol (Almeida et al., 2005), with the lentiviral expression vector (pRRLSIN.cPPt.PGK-EGFP-L10a.WPRE) and three lentiviral packaging vectors pLP1, pLP2 and pLP-VSVG, for the expression of gag/pol genes, rev gene and vesicular stomatitis virus G (VSVG) envelope glycoprotein gene, respectively. The supernatant containing virus particles was collected at 24 h, 48 h and 60 h after transfection and concentrated at 22,000 RPM, using an SW40Ti rotor (Beckman Coulter ultracentrifuge), for 2 h at 22°C. The viral pellet was then re-suspended in PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4, pH 7.4) with 1% BSA and stored at −80°C. After virus transduction, protein expression was detected 48-60 hours later. For live-imaging experiments, cells were incubated with the virus for 48-72 h before imaging.
Generation of sindbis virus was performed accordingly to Pinto et al. (2016a). Briefly, the pSinRep construct expressing EGFP-L10a and the helper plasmid DH26S were linearized with NotI and properly treated for the removal of RNase contamination. Synthesis of RNA from linearized DNAs was performed by in vitro transcription using the mMESSAGE mMACHINE SP6 kit (Invitrogen). BHK-21 cells were electroporated with 12 μg DH26S RNA and 12 μg of the desired pSinRep RNA, and production of virus was allowed to occur for 24–36 h. Supernatant was then collected, and virus particles were purified by centrifugation at 22,000 RPM, using an SW40Ti rotor (Beckman Coulter ultracentrifuge), for 2 h and 20 min at 15°C. The viral pellet was then resuspended in PBS with 1% BSA and stored at −80°C. The virus titer was determined in BHK-21 cells, and the volume of virus for infection was adjusted so that > 85% of neurons were transduced. For expression of EGFP-L10a in microfluidic devices, expression was allowed to occur for 20-24 h.
In utero electroporation
Standard bipolar in utero electroporation of the somatosensory cortex was performed as previously described (Bony et al., 2013; Szczurkowska et al., 2016). Timed-pregnant Sprague Dawley rats (Harlan Italy SRL, Correzzana, Italy) were anaesthetized at E17.5 with isoflurane (induction, 3.5%; surgery, 2.5%), and uterine horns were exposed by laparotomy. Expression vectors (1–2 μg μl−1/Vector in water) and dye Fast Green (0.3 mg ml−1; Sigma, St. Louis, MO) were injected (5–6 μl) through the uterine wall into one of the embryo’s lateral ventricle by a 30-G needle (Pic indolor, Grandate, Italy). Each embryo’s head was held between tweezer-type electrodes (10 mm diameter; Nepa Gene, Chiba, Japan) across the uterus and five electrical pulses (amplitude, 50 V; duration, 50 ms; intervals, 100 ms) were delivered with a square-wave electroporation generator (CUY21EDIT; Nepa Gene). Uterine horns were returned into the abdominal cavity, and embryos continued their normal development.
Slice histology
Postnatal brains (PND 7, PND 12, PND 21) were fixed by transcardial perfusion of 4% PFA. Brains were sectioned coronally 80 μm thick with a vibratome (Leica VT1000S). Free-floating slices were permeabilized and blocked with PBS containing 0.3% Triton X-100, 10% NGS and 0.2% BSA. When used, primary antibodies were incubated in PBS containing 5% NGS and 0.1% BSA (chicken anti-GFP 1:500 (AbCam)). Immunostaining was then detected using Alexa fluorescent secondary antibody 1:600 (Invitrogen) in PBS containing 5% NGS. Samples were mounted in Vectashield (Vector Laboratories, Inc., Burlingame, CA) and examined using widefield (Zeiss Axiovert 200) and confocal microscopy (Zeiss LSM 710).
Western Blot
Protein levels analysis was performed by western blot as previously described (Costa et al., 2012; Shin et al., 2010), with minor modifications. Corpus callosum (C.C.) midline area region or CPN cell body (C.B.) region was dissected from electroporated brains with different postnatal days (PND 7, PND 12, PND 21) (Figures 1D and 1E). Protein was then extracted from tissue (tissue cube with approximately 27 mm3), containing CPN axons or cell bodies (filled with tdTomato; red) and EGFP tagged ribosomes (green), in extraction solution (SDS 2% w/v in PBS, 6% β-mercaptoethanol) supplemented with cOmplete-Protease Inhibitor Cocktail (Roche). Then, samples were incubated on ice for 5 min, mixing by vortex every minute, and then boiled a 100°C for 20 min. Afterward, samples were incubated at 80°C for additional 2 h with agitation at 750 rpm using a thermomixer (Eppendorf, Hamburg, Germany). Lysates were then sonicated and centrifuged at 14,000 × g for 15 min at 4°C and the supernatant collected. Protein quantification was performed by the BCA assay, and samples (60 μg) were denatured with 6× denaturating buffer (500 mM Tris, 600 mM DTT, 10% (w/v) SDS, 30% (v/v) glycerol and 0.012% (w/v) bromophenol blue) and boiled at 95°C for additional 3 min. Equal amounts of each protein sample were separated by electrophoresis in a 4%–20% gradient polyacrylamide gel 1.5 mm thick in a tris-glycine-SDS buffer (25 mM Tris, 192 mM glycine, 0.1% w/v SDS, pH 8.3). Proteins were then electroblotted (at 100 V for 2 h, at 4°C) onto PVDF membranes (Amersham Pharmacia Biotech, Buckinghamshire, UK). Membranes were then washed once with TBS-T (TBS (150 mM NaCl, 25 mM Tris–HCl; pH 7.6) with 0.1% (v/v) Tween 20), and then blocked for 1 h at room temperature in TBS-T containing 5% (w/v) nonfat dry milk. Membranes were subsequently incubated overnight at 4°C with the primary antibodies (chicken anti-GFP 1:500 (AbCam) or rabbit anti-RFP/mCherry 1:5,000 (Rockland)) diluted in TBS-T containing 1% w/v non-fat dry milk. Membranes were again washed 3 × with TBS-T and then incubated in TBS-T with 1% (w/v) nonfat dry milk for 2 h at room temperature with HR-peroxidase-conjugated anti-chicken or anti-rabbit secondary antibody at a dilution of 1:20,000 (Abcam). Membranes were then incubated with ECL Plus reagent (Pierce) for 5–10 min and immunoreactive bands were detected on an imageQuant LAS 4000 mini Imaging System, under linear exposure conditions. Quantification was performed using ImageJ software. Whenever necessary, membranes were stripped with Restore Plus buffer (Thermofisher scientific) and reprobed.
Preparation of microfluidic devices for neuronal culture
Microfluidic devices consist of a molded poly-dimethylsiloxane (PDMS) chamber (20 mm × 25 mm), assembled in a glass coverslip (Taylor et al., 2005). This system is composed by a somal and an axonal compartment, each 1.5 mm wide and 7 mm long. Both compartments are separated by a set of microgrooves (450 μm long, 10 μm wide). The height difference between microgrooves (3 μm) and compartments (100 μm), combined with a minimal volume difference between the two sides leads to a fluidic isolation between the two compartments.
Microfluidic devices were prepared and assembled as previously described (Pinto et al., 2016b; Neto et al., 2014; Martins et al., 2017). Coverslips were double-coated with poly-D-lysine (Milipore) 0.1 mg/ml overnight at 37°C and laminin I (Cultrex) 10 μg/ml for 2 h at 37°C. Cell were plated in the somal compartment in motor neuron growth media (see below). During treatments, a minimal volume difference between the somal compartment and the axonal compartment (~25 μl) was maintained to prevent the diffusion of the applied factors from the axonal to the somal compartment.
Primary cultures
Primary cultures of rat embryonic spinal motor neurons (MN) were prepared as previously described (Henderson et al., 1995), with minor modifications. Briefly, after the dissection, embryonic spinal cords from Wistar rats embryos (E13.5), were cut in small fragments and dissociated in 0.025% trypsin w/v in Hank’s balanced salt solution (HBSS) (5.36 mM KCl, 0.44 mM KH2PO4, 137 mM NaCl, 4.16 mM NaHCO3, 0.34 mM Na2HPO4.2H2O, 5 mM glucose, 1 mM sodium pyruvate, 10 mM HEPES and 0,001% phenol red) for 8 min at 37°C. Immediately after trypsin incubation, fragments were transferred to the dissociation solution [Leibovitz’s L-15 Medium (GIBCO) plus 0.4% dialyzed BSA (w/v) and 0.1 mg/ml deoxyribonuclease] and mechanically dissociated with a fire-polished Pasteur pipette. Afterward, cell suspension was placed over a 4% BSA (w/v) cushion and centrifuged for 5 min, 470 × g at room temperature. Pelleted cells were then re-suspended in incomplete culture medium [Neurobasal medium (GIBCO) supplemented with 2% B27 (GIBCO), 2% Horse Serum (GIBCO), 0.5 mM glutamine (GIBCO), 25 μM glutamate (GIBCO), 0.025 mM 2-mercaptoethanol (Sigma) and 1× penicillin-streptomycin solution (GIBCO)]. Re-suspended cells were then placed over a 6.5% OptiPrep (Sigma-Aldrich) solution (v/v in Leibovitz’s L-15 medium; GIBCO) and centrifuged for 15 min, 830 × g at room temperature. After OptiPrep density gradient centrifugation, the motor neuron turbid band, at the medium-Optiprep interface, was collected, diluted up to 10 mL in incomplete culture medium and again centrifuged for 5 min, 470 × g at room temperature over a 4% BSA (w/v) cushion. All centrifugations were carried with no acceleration and no break to reduce the vibration. Pelleted cells were re-suspended in complete culture medium [incomplete culture medium supplemented with 0.1 ng/mL glial derived neurotrophic factor (GDNF) (PeproTech), 0.5 ng/ml mL ciliary neurotrophic factor (CNTF) (PeproTech), 1 ng/mL brain derived neurotrophic factor (BDNF) (PeproTech)]. Purified motor neurons were plated in the somal compartment of microfluidic chambers and allowed to attach for 2 h and then complete culture medium was added. Cells were maintained at 37°C in a humidified incubator under an atmosphere of 95% air and 5% CO2. At DIV 2, the mitotic inhibitor 5-fluoro-2′-deoxiuridina (5-FDU) (10 μM) was added to reduce contamination with glia cells. Cells were allowed to grow and, unless otherwise indicated, experiments were performed at DIV 4/5.
Cell Lines and Co-cultures
For co-cultures with spinal motor neurons (MN), at MN DIV 1/2, C2C12 muscle fibers or HEK293 cells were gently detached from the flask, using 0.025% Trypsin (sigma) in PBS solution, washed with growth medium and then added to the chamber axonal compartment. MN axons were allowed to contact with C2C12 fibers or HEK293 cells for at least 3 days before processing the co-culture for immunocytochemistry. To evaluate axonal ribosomal proteins, at DIV 4/5 MN only (somal compartment of the microfluidic chamber) were infected with virus containing EGFP-L10a 20-24h prior to the experiment terminus. Cells were then fixed and immunocytochemistry was performed.
Treatments
Induction of presynaptic differentiation. At DIV 3/4 recombinant human FGF22 (R&D) (Umemori et al., 2004) was added to rat motor neuron axons. FGF22 was used at a 2 nM concentration and applied in conditioned medium for 14 h, unless otherwise indicated.
Drug treatments
Cell treatment with ubiquitin proteasome system (UPS) and autophagy inhibitor was performed in conditioned medium for a period of 6 h at DIV 3/4. When cells were co-treated with FGF22, inhibitors were applied in the last 6 h of the FGF22 14 h stimulus. Equal amounts of drug solvent were added to control conditions (Ctr/Veh). The proteasome inhibitors MG132 (Baptista et al., 2010) or epoxomicin (Meng et al., 1999), were applied at 1 μM. The autophagy inhibitors 3-MA (Wu et al., 2010) or leupeptin (Kovács et al., 1982; Goo et al., 2017), were applied at 10 mM and 200 μM, respectively.
Viral infection. For lentivirus, at DIV 1/2 motor neuron cell bodies were infected with pRRLSIN.cPPt.PGK-EGFP-L10a.WPRE lentivirus for 48-72 hours. Then, FGF22 was added to axonal compartment for 14 h to induce presynaptic differentiation. For sindbis-mediated viral expression of EGFP-L10a, at DIV 4/5 the virus was applied for 20-24h to MNs only (somal compartment of the microfluidic chamber). Cells were then fixed and immunocytochemistry was performed.
Immunocytochemistry
Cells were then fixed for 10 minutes with 4% paraformaldehyde (in PBS with 4% sucrose) at room temperature, rinsed 3 times for 5 minutes each with ice-cold PBS and permeabilized in PBS with 0.25% Triton X-100 for 5 minutes at room temperature. Cells were then washed once with PBS and blocked with 3% BSA (in PBS) for 30 minutes at room temperature (Martins et al., 2017). Then, cells were incubated, either overnight at 4°C or for 2 h at 37°C, with the mix of primary antibodies diluted in 3% BSA (in PBS), primary antibodies were then washed 3 times with PBS, and cells incubated for 1 h with Alexa fluorescent secondary antibodies diluted (1:1,000; Invitrogen) in 3% BSA, at room temperature. Finally, cells were washed twice in PBS with 0.1% Triton X-100 and once with PBS, 5 minutes each. Coverslips were then mounted in prolong mounting media with or without DAPI (Molecular probes). Preparations were cured overnight at 4°C, protected from light, sealed with nailpolish and kept at 4°C until microscopy analysis. Depending on the experiment the following antibodies and dilutions were used: chicken anti-neurofilament (1:1,000; Millipore), chicken anti-tau (1:1,000; Abcam), rabbit anti-Synapsin I (1:4,000; Millipore), mouse Anti-SV2 (1:1,000; Developmental Studies Hybridoma Bank), rabbit anti-ribosomal protein S6 (1:50; Cell Signaling Technology), human anti-ribosomal protein P0 (1:1,000; Immunovision), mouse anti-ribosomal RNA 5.8S (Y10B; 1:250; Thermo Scientific), mouse anti-GFP (1:250; Roche Diagnostics), chicken anti-MAP-2 (1:10,000; Abcam), rabbit anti-p-4E-BP1 (Ser65/Thr70; 1:1,000; Santa Cruz Biotechnology), anti-α-Bungarotoxin(Alexa Fluor 647 conjugate; 1:1,000, Invitrogen). For anti-ribosomal protein S6, Pierce Immunostain Enhancer (Thermo Fisher Scientific) was also used to improve signal.
Bio-orthogonal labeling assay
Intra-axonal translation was assessed using the O-propargyl-puromycin (OPP) labeling method, followed by Click-It chemistry detection of nascent proteins (Slomnicki et al., 2016; Liu et al., 2012). Labeling was performed according to the manufacturer’s instructions (Click-It™ Plus OPP Alexa Fluor™ 488 Protein Synthesis Assay Kit, Molecular Probes, C10269) and axons analyzed by widefield fluorescence microscopy.
Fluorescence microscopy and quantification
Fluorescent images were taken with a Plan-Neofluar 63 × oil objective (1.4 NA) in inverted microscope Zeiss Axiovert 200 equipped with an AxioCam HRm camera and ZEN Blue 2011 software. Images were acquired under identical settings (exposure time and fluorescence light intensity were kept constant in each experiment). In microfluidic chambers, images of random fields of view (FOVs) were acquired from distal axons in the axonal compartment (without including the microgrooves) and clusters/puncta quantified with ImageJ software. Briefly, all images were converted to 8-bit for quantification purposes. Then, the axonal marker image was used to randomly select populations of axons to quantify. Axonal length was determined by performing analysis (ImageJ plugin “Analyze skeleton”) of a “skeletonized” version of the axonal marker. The sum of the length of all the axonal branches identified in an image was used as the axonal length. Correspondent images of synaptic or ribosomal markers were thresholded and a particle analysis was performed to calculate puncta number, area, integrated density. These values were divided by axonal length, unless otherwise indicated in figure legends. Quantification process was performed in a condition-blind manner. All images were processed and prepared for presentation using ImageJ (NIH), Photoshop and Illustrator (Adobe) software programs.
QUANTIFICATION AND STATISTICAL ANALYSIS
For each independent experiment, data was normalized to the mean of control group (and expressed as % of control). Results are presented as averaged values ± SEM of at least 3 independent experiments, unless otherwise indicated in figure caption. In microscopy experiments, each independent experiment consisted in the analysis of at least 12 fields of view (FOV) (n = 12), acquired in condition-blind manner, containing several isolated axons.
For Figure 1F at least 2 animals were used per condition (PND 7, n = 2; PND 12, n = 3; PND 21, n = 10), while for Figure 1G at least 9 animals were used per condition (PND 7, n = 9; PND 12, n = 8; PND 21, n = 9). For fig, 2O, in each independent experiment, at least 12 fields of view (FOV) were analyzed per condition (DIV 1, n = 24; DIV 4, n = 36; DIV 7, n = 36; DIV 12, n = 36; DIV 15, n = 12).
Graphs and statistical analysis were performed in GraphPad Prism 6 software. Statistical significance was assessed by parametric tests. Unpaired two-tailed Student’s t test was performed for the comparison of two groups, while analysis of variance (ANOVA), followed by Dunnett’s or Tukey’s post hoc, used for comparisons between multiple groups. A p < 0.05 value was considered statistically significant.
DATA AND CODE AVAILABILITY
This study did not generate/analyze datasets or codes.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chicken anti-GFP | Abcam | Cat# ab13970; RRID: AB_300798 |
| Rabbit anti-RFP/mCherry | Rockland | Cat# 600-401-379; RRID: AB_2209751 |
| Chicken anti-neurofilament H (NF-H) | Abcam | Cat# ab4680; RRID: AB_304560 |
| Chicken anti-neurofilament M (NF-M) | Merk/Milipore | Cat# AB5735; RRID: AB_240806 |
| Chicken anti-Tau | Abcam | Cat# ab75714; RRID: AB_1310734 |
| Rabbit anti-Synapsin I | Millipore | Cat# AB1543; RRID: AB_2200400 |
| Mouse Anti-SV2 (MIgG1) | DSHB | Cat# AB2315387; RRID: AB_2315387 |
| Rabbit anti-ribosomal protein S6 (5G10) | Cell Signaling | Cat# 2217; RRID: AB_331355 |
| Human anti-ribosomal protein P0 | Immunovision | Cat# HPO-0100 |
| Mouse anti-ribosomal RNA 5.8 s (Y10B) | Thermo scientific | Cat# MA1-13017; RRID: AB_10979967 |
| Mouse anti-GFP | Roche Diagnostics | Cat# 11814460001; RRID: AB_390913 |
| Chicken anti-MAP2 | Abcam | Cat# ab5392; RRID: AB_2138153 |
| Rabbit anti-p-4E-BP1 (Ser65/Thr70) | Santa Cruz Biotech. | Cat# sc-12884; RRID: AB_667679 |
| α-Bungarotoxin, Alexa Fluor 647 conjugate | Invitrogen | Cat# B35450 |
| Alexa fluor secondary Ab goat anti-mouse 488 | Invitrogen | Cat# A32723; RRID: AB_2633275 |
| Alexa fluor secondary Ab goat anti-mouse 568 | Invitrogen | Cat# A11031; RRID: AB_144696 |
| Alexa fluor secondary Ab goat anti-rabbit 488 | Invitrogen | Cat# A11034; RRID: AB_2576217 |
| Alexa fluor secondary Ab goat anti-rabbit 568 | Invitrogen | Cat# A11036; RRID: AB_10563566 |
| Alexa fluor secondary Ab goat anti-chicken 405 | Abcam | Cat# ab175674 |
| Alexa fluor secondary Ab goat anti-chicken 488 | Invitrogen | Cat# A11039; RRID: AB_2534096 |
| Alexa fluor secondary Ab goat anti-chicken 568 | Invitrogen | Cat# A11041; RRID: AB_2534098 |
| Alexa fluor secondary Ab goat anti-chicken 647 | Invitrogen | Cat# A21449; RRID: AB_2535866 |
| Alexa fluor secondary Ab goat anti-human 488 | Invitrogen | Cat# A11013; RRID: AB_2534080 |
| HR-peroxidase-conjugated anti-chicken secondary Ab | Abcam | Cat# ab97135; RRID: AB_10680105 |
| HR-peroxidase-conjugated anti-rabbit secondary Ab | Abcam | Cat# ab6721; RRID: AB_955447 |
| Bacterial and Virus Strains | ||
| Sindbis Virus (EGFP-L10a) | This paper | n/a |
| Lenti-Virus (EGFP-L10a) | This paper | n/a |
| Biological Samples | ||
| n/a | n/a | n/a |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Pierce Immunostain Enhancer | Thermo Scientific | Cat# 46644 |
| cOmplete-Protease Inhibitor Cocktail | Roche | Cat# 11697498001 |
| MG132 | Merk | Cat# 474790 |
| Epoxomicin | Enzo | Cat# BML-PI127-0100 |
| 3-MA | Merk | Cat# M9281 |
| Leupeptin | Merk | Cat# L2884 |
| ECL Plus reagent | Pierce | Cat# 32132 |
| Poly-D-Lysine | Milipore | Cat# A-003-E |
| Mouse Laminin I | Cultrex, Trevigen | Cat# 3400-010-02 |
| Optiprep | Sigma aldrich | Cat# 1556 |
| Leibovitz’s L15 medium | GIBCO | Cat# 11415064 |
| GDNF | PeproTech | Cat# 167450-10-A |
| CTNF | PeproTech | Cat# 167450-13-A |
| BDNF | PeproTech | Cat# 167450-02-B |
| FGF22 | R&D | Cat# 3867-FG-025 |
| Critical Commercial Assays | ||
| Click-It Plus OPP Alexa Fluor 488 Protein Synthesis Assay Kit | Invitrogen / Molecular probes | Cat# C10269 |
| mMESSAGE mMACHINE SP6 Transcription Kit | Invitrogen | Cat# AM1340 |
| In-Fusion® HD Cloning Kit | Clontech | Cat# 638909 |
| Deposited Data | ||
| n/a | n/a | n/a |
| Experimental Models: Cell Lines | ||
| HEK293 | ATCC | Cat# 293 [HEK293] (ATCC® CRL-1573) |
| HEK293T | ATCC | Cat# 293T (ATCC® CRL-3216) |
| Mouse myoblast (C2C12) | ATCC | Cat# C2C12 (ATCC® CRL1772TM) |
| BHK-21 | ATCC | BHK-21 [C-13] (ATCC® CCL-10) |
| Experimental Models: Organisms/Strains | ||
| Sprague Dawley rats |
Szczurkowska et al., 2016; Harlan Italy SRL, Correzzana, Italy |
n/a |
| Wistar-Han rats | Charles River, Barcelona, Spain | Strain Code 273 |
| Recombinant DNA | ||
| pRRLSIN.cPPT.PGK-GFP.WPRE (lentiviral vector) | Addgene | Cat# 12252 |
| pRRLSIN.cPPT.PGK-EGFP-L10a.WPRE | This paper | n/a |
| pSinRep5 | Invitrogen | Cat# C-180402 |
| pSinRep5-EGFP-L10a | This paper | n/a |
| pCAGGS-IRES-EGFP | Cancedda et al., 2007 | n/a |
| pCAGGS-IRES-Tomato | Cancedda et al., 2007 | n/a |
| pCAGGS-EGFP-L10a | This paper | n/a |
| Software and Algorithms | ||
| ImageJ / Fiji (V1.47q) | NIH | https://imagej.nih.gov/ij/download.html |
| GraphPad Prism 6 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Photoshop CC | Adobe | https://www.adobe.com |
| Illustrator CC | Adobe | https://www.adobe.com |
| Endnote | Clarivate analytics | https://www.endnote.com |
Highlights.
Synapse formation induces intra-axonal ribosomal decrease
Ribosomes are degraded by ubiquitin proteasome system upon axonal maturation
ACKNOWLEDGMENTS
This work was funded by FEDER - Fundo Europeu de Desenvolvimento Regional, through COMPETE 2020 - Operational Programme for Competitiveness and Internationalisation and Portuguese national funds via FCT - Fundação para a Ciência e a Tecnologia, under the projects PTDC/SAU-NEU/104100/2008, EXPL/NEU-NMC/0541/2012, UID/BIM/04501/2013, UID/NEU/04539/2019, POCI-01-0145-FEDER-007628, and CENTRO-01-0145-FEDER-000008:BrainHealth 2020, individual grants SFRH/BPD/84593/2012 (R.O.C.), PD/BD/114170/2016 (L.F.M.), SFRH/BPD/115546/2016 (M.M.), SFRH/BD/77789/2011 (J.R.P.), and SFRH/BD/139368/2018 (D.T.), and by Marie Curie Actions - IRG - 7th Research Framework Programme, and NIH grants R35NS111631 and NS056306 (S.R.J.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.06.080.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPORTING CITATIONS
The following references appear in the Supplemental Information: Abou Elela and Nazar (1997); Hutchinson et al., (2011); Uchiumi and Kominami (1997).
REFERENCES
- Abou Elela S, and Nazar RN (1997). Role of the 5.8S rRNA in ribosome translocation. Nucleic Acids Res. 25, 1788–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida RD, Manadas BJ, Melo CV, Gomes JR, Mendes CS, Grãos MM, Carvalho RF, Carvalho AP, and Duarte CB (2005). Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ. 12, 1329–1343. [DOI] [PubMed] [Google Scholar]
- Baptista MS, Melo CV, Armelão M, Herrmann D, Pimentel DO, Leal G, Caldeira MV, Bahr BA, Bengtson M, Almeida RD, and Duarte CB (2010). Role of the proteasome in excitotoxicity-induced cleavage of glutamic acid decarboxylase in cultured hippocampal neurons. PLoS ONE 5, e10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassell GJ, Singer RH, and Kosik KS (1994). Association of poly(A) mRNA with microtubules in cultured neurons. Neuron 12, 571–582. [DOI] [PubMed] [Google Scholar]
- Bassell GJ, Zhang H, Byrd AL, Femino AM, Singer RH, Taneja KL, Lifshitz LM, Herman IM, and Kosik KS (1998). Sorting of beta-actin mRNA and protein to neurites and growth cones in culture. J. Neurosci. 18, 251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederer T, Sara Y, Mozhayeva M, Atasoy D, Liu X, Kavalali ET, and Südhof TC (2002). SynCAM, a synaptic adhesion molecule that drives synapse assembly. Science 297, 1525–1531. [DOI] [PubMed] [Google Scholar]
- Bony G, Szczurkowska J, Tamagno I, Shelly M, Contestabile A, and Cancedda L (2013). Non-hyperpolarizing GABAB receptor activation regulates neuronal migration and neurite growth and specification by cAMP/ LKB1. Nat. Commun. 4, 1800. [DOI] [PubMed] [Google Scholar]
- Campbell DS, and Holt CE (2001). Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron 32, 1013–1026. [DOI] [PubMed] [Google Scholar]
- Campbell DS, and Holt CE (2003). Apoptotic pathway and MAPKs differentially regulate chemotropic responses of retinal growth cones. Neuron 37, 939–952. [DOI] [PubMed] [Google Scholar]
- Cancedda L, Fiumelli H, Chen K, and Poo MM (2007). Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J. Neurosci. 27, 5224–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioni JM, Koppers M, and Holt CE (2018). Molecular control of local translation in axon development and maintenance. Curr. Opin. Neurobiol. 51, 86–94. [DOI] [PubMed] [Google Scholar]
- Cosker KE, Pazyra-Murphy MF, Fenstermacher SJ, and Segal RA (2013). Target-derived neurotrophins coordinate transcription and transport of bclw to prevent axonal degeneration. J. Neurosci. 33, 5195–5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RO, Lacor PN, Ferreira IL, Resende R, Auberson YP, Klein WL, Oliveira CR, Rego AC, and Pereira CM (2012). Endoplasmic reticulum stress occurs downstream of GluN2B subunit of N-methyl-d-aspartate receptor in mature hippocampal cultures treated with amyloid-β oligomers. Aging Cell 11, 823–833. [DOI] [PubMed] [Google Scholar]
- Court FA, Hendriks WT, MacGillavry HD, Alvarez J, and van Minnen J (2008). Schwann cell to axon transfer of ribosomes: toward a novel understanding of the role of glia in the nervous system. J. Neurosci. 28, 11024–11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox LJ, Hengst U, Gurskaya NG, Lukyanov KA, and Jaffrey SR (2008). Intra-axonal translation and retrograde trafficking of CREB promotes neuronal survival. Nat. Cell Biol. 10, 149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristovão G, Pinto MJ, Cunha RA, Almeida RD, and Gomes CA (2014). Activation of microglia bolsters synapse formation. Front. Cell. Neurosci. 8, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, et al. (2008). Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 135, 749–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fame RM, MacDonald JL, and Macklis JD (2011). Development, specification, and diversity of callosal projection neurons. Trends Neurosci. 34, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Kus L, and Heintz N (2010). Rapid bacterial artificial chromosome modification for large-scale mouse transgenesis. Nat. Protoc. 5, 1678–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goo MS, Sancho L, Slepak N, Boassa D, Deerinck TJ, Ellisman MH, Bloodgood BL, and Patrick GN (2017). Activity-dependent trafficking of lysosomes in dendrites and dendritic spines. J. Cell Biol. 216, 2499–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumy LF, Tan CL, and Fawcett JW (2010). The role of local protein synthesis and degradation in axon regeneration. Exp. Neurol. 223, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumy LF, Yeo GS, Tung YC, Zivraj KH, Willis D, Coppola G, Lam BY, Twiss JL, Holt CE, and Fawcett JW (2011). Transcriptome analysis of embryonic and adult sensory axons reveals changes in mRNA repertoire localization. RNA 17, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas KF, and Broadie K (2008). Roles of ubiquitination at the synapse. Biochim. Biophys. Acta 1779, 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suárez-Fariñas M, Schwarz C, Stephan DA, Surmeier DJ, et al. (2008). A translational profiling approach for the molecular characterization of CNS cell types. Cell 135, 738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Kulicke R, Fenster RJ, Greengard P, and Heintz N (2014). Cell type-specific mRNA purification by translating ribosome affinity purification (TRAP). Nat. Protoc. 9, 1282–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson CE, Bloch-Gallego E, and Camu W (1995). Purified embryonic motoneurons In Nerve Cell Culture: A Practical Approach, Cohen JWG, ed. (Oxford: ), pp. 69–81. [Google Scholar]
- Hengst U, Cox LJ, Macosko EZ, and Jaffrey SR (2006). Functional and selective RNA interference in developing axons and growth cones. J. Neurosci. 26, 5727–5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG (2009). Active destruction of defective ribosomes by a ubiquitin ligase involved in DNA repair. Genes Dev. 23, 891–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson JA, Shanware NP, Chang H, and Tibbetts RS (2011). Regulation of ribosomal protein S6 phosphorylation by casein kinase 1 and protein phosphatase 1. J. Biol. Chem. 286, 8688–8696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman R, Banker G, and Steward O (1994). Development of subcellular mRNA compartmentation in hippocampal neurons in culture. J. Neurosci. 14, 1130–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovács AL, Reith A, and Seglen PO (1982). Accumulation of autophagosomes after inhibition of hepatocytic protein degradation by vinblastine, leupeptin or a lysosomotropic amine. Exp. Cell Res. 137, 191–201. [DOI] [PubMed] [Google Scholar]
- Kraft C, Deplazes A, Sohrmann M, and Peter M (2008). Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat. Cell Biol. 10, 602–610. [DOI] [PubMed] [Google Scholar]
- Kumar SS, and Huguenard JR (2001). Properties of excitatory synaptic connections mediated by the corpus callosum in the developing rat neocortex. J. Neurophysiol. 86, 2973–2985. [DOI] [PubMed] [Google Scholar]
- Kun A, Otero L, Sotelo-Silveira JR, and Sotelo JR (2007). Ribosomal distributions in axons of mammalian myelinated fibers. J. Neurosci. Res. 85, 2087–2098. [DOI] [PubMed] [Google Scholar]
- Liu J, Xu Y, Stoleru D, and Salic A (2012). Imaging protein synthesis in cells and tissues with an alkyne analog of puromycin. Proc. Natl. Acad. Sci. USA 109, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyles V, Zhao Y, and Martin KC (2006). Synapse formation and mRNA localization in cultured Aplysia neurons. Neuron 49, 349–356. [DOI] [PubMed] [Google Scholar]
- Madison RD, McGee C, Rawson R, and Robinson GA (2014). Extracellular vesicles from a muscle cell line (C2C12) enhance cell survival and neurite outgrowth of a motor neuron cell line (NSC-34) J. Extracell. Vesicles, Published online February 19, 2014 10.3402/jev.v3.22865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin NR, Passey SL, Player DJ, Mudera V, Baar K, Greensmith L, and Lewis MP (2015). Neuromuscular Junction Formation in Tissue-Engineered Skeletal Muscle Augments Contractile Function and Improves Cytoskeletal Organization. Tissue Eng. Part A 21, 2595–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins LF, Costa RO, Pedro JR, Aguiar P, Serra SC, Teixeira FG, Sousa N, Salgado AJ, and Almeida RD (2017). Mesenchymal stem cells secretome-induced axonal outgrowth is mediated by BDNF. Sci. Rep. 7, 4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, and Crews CM (1999). Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Natl. Acad. Sci. USA 96, 10403–10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merianda TT, Coleman J, Kim HH, Kumar Sahoo P, Gomes C, Brito-Vargas P, Rauvala H, Blesch A, Yoo S, and Twiss JL (2015). Axonal amphoterin mRNA is regulated by translational control and enhances axon outgrowth. J. Neurosci. 35, 5693–5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelevski I, Medzihradszky KF, Lynn A, Burlingame AL, and Fainzilber M (2010). Axonal transport proteomics reveals mobilization of translation machinery to the lesion site in injured sciatic nerve. Mol. Cell. Proteomics 9, 976–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neto E, Alves CJ, Sousa DM, Alencastre IS, Lourenço AH, Leitão L, Ryu HR, Jeon NL, Fernandes R, Aguiar P, et al. (2014). Sensory neurons and osteoblasts: close partners in a microfluidic platform. Integr. Biol. 6, 586–595. [DOI] [PubMed] [Google Scholar]
- Pease-Raissi SE, Pazyra-Murphy MF, Li Y, Wachter F, Fukuda Y, Fenstermacher SJ, Barclay LA, Bird GH, Walensky LD, and Segal RA (2017). Paclitaxel reduces axonal Bclw to initiate IP3R1-dependent axon degeneration. Neuron 96, 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BE, and Huber KM (2006). Current advances in local protein synthesis and synaptic plasticity. J. Neurosci. 26, 7147–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips GR, Huang JK, Wang Y, Tanaka H, Shapiro L, Zhang W, Shan WS, Arndt K, Frank M, Gordon RE, et al. (2001). The presynaptic particle web: ultrastructure, composition, dissolution, and reconstitution. Neuron 32, 63–77. [DOI] [PubMed] [Google Scholar]
- Pinto MJ, and Almeida RD (2016). Puzzling out presynaptic differentiation. J. Neurochem. 139, 921–942. [DOI] [PubMed] [Google Scholar]
- Pinto MJ, Alves PL, Martins L, Pedro JR, Ryu HR, Jeon NL, Taylor AM, and Almeida RD (2016a). The proteasome controls presynaptic differentiation through modulation of an on-site pool of polyubiquitinated conjugates. J. Cell Biol. 212, 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto MJ, Pedro JR, Costa RO, and Almeida RD (2016b). Visualizing K48 Ubiquitination during Presynaptic Formation By Ubiquitination-Induced Fluorescence Complementation (UiFC). Front. Mol. Neurosci. 9, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper M, and Holt C (2004). RNA translation in axons. Annu. Rev. Cell Dev. Biol. 20, 505–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, and Serafini T (2000). Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 101, 657–669. [DOI] [PubMed] [Google Scholar]
- Semple BD, Blomgren K, Gimlin K, Ferriero DM, and Noble-Haeusslein LJ (2013). Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 106–107, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeoka T, Jung H, Jung J, Turner-Bridger B, Ohk J, Lin JQ, Amieux PS, and Holt CE (2016). Dynamic Axonal Translation in Developing and Mature Visual Circuits. Cell 166, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SB, Almeida RD, Gerona-Navarro G, Bracken C, and Jaffrey SR (2010). Assembling ligands in situ using bioorthogonal boronate ester synthesis. Chem. Biol. 17, 1171–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slomnicki LP, Pietrzak M, Vashishta A, Jones J, Lynch N, Elliot S, Poulos E, Malicote D, Morris BE, Hallgren J, and Hetman M (2016). Requirement of Neuronal Ribosome Synthesis for Growth and Maintenance of the Dendritic Tree. J. Biol. Chem. 291, 5721–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavreva DA, Kawasaki M, Dundr M, Koberna K, Müller WG, Tsujimura-Takahashi T, Komatsu W, Hayano T, Isobe T, Raska I, et al. (2006). Potential roles for ubiquitin and the proteasome during ribosome biogenesis. Mol. Cell. Biol. 26, 5131–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC (2000). The synaptic vesicle cycle revisited. Neuron 28, 317–320. [DOI] [PubMed] [Google Scholar]
- Sung MK, Reitsma JM, Sweredoski MJ, Hess S, and Deshaies RJ (2016). Ribosomal proteins produced in excess are degraded by the ubiquitin-proteasome system. Mol. Biol. Cell 27, 2642–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczurkowska J, Cwetsch AW, dal Maschio M, Ghezzi D, Ratto GM, and Cancedda L (2016). Targeted in vivo genetic manipulation of the mouse or rat brain by in utero electroporation with a triple-electrode probe. Nat. Protoc. 11, 399–12. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, and Jeon NL (2005). A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat. Methods 2, 599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Berchtold NC, Perreau VM, Tu CH, Li Jeon N, and Cotman CW (2009). Axonal mRNA in uninjured and regenerating cortical mammalian axons. J. Neurosci. 29, 4697–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terauchi A, Johnson-Venkatesh EM, Toth AB, Javed D, Sutton MA, and Umemori H (2010). Distinct FGFs promote differentiation of excitatory and inhibitory synapses. Nature 465, 783–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twiss JL, and Fainzilber M (2009). Ribosomes in axons–scrounging from the neighbors? Trends Cell Biol. 19, 236–243. [DOI] [PubMed] [Google Scholar]
- Uchiumi T, and Kominami R (1997). Binding of mammalian ribosomal protein complex P0.P1.P2 and protein L12 to the GTPase-associated domain of 28 S ribosomal RNA and effect on the accessibility to anti-28 S RNA autoantibody. J. Biol. Chem. 272, 3302–3308. [DOI] [PubMed] [Google Scholar]
- Umemori H, Linhoff MW, Ornitz DM, and Sanes JR (2004). FGF22 and its close relatives are presynaptic organizing molecules in the mammalian brain. Cell 118, 257–270. [DOI] [PubMed] [Google Scholar]
- Verma P, Chierzi S, Codd AM, Campbell DS, Meyer RL, Holt CE, and Fawcett JW (2005). Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J. Neurosci. 25, 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BA, Hengst U, Kim HJ, Jeon NL, Schmidt EF, Heintz N, Milner TA, and Jaffrey SR (2012). Reprogramming axonal behavior by axon-specific viral transduction. Gene Ther. 19, 947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welshhans K, and Bassell GJ (2011). Netrin-1-induced local β-actin synthesis and growth cone guidance requires zipcode binding protein 1. J. Neurosci. 31, 9800–9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KY, Hengst U, Cox LJ, Macosko EZ, Jeromin A, Urquhart ER, and Jaffrey SR (2005). Local translation of RhoA regulates growth cone collapse. Nature 436, 1020–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, and Shen HM (2010). Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 285, 10850–10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon BC, Zivraj KH, and Holt CE (2009). Local translation and mRNA trafficking in axon pathfinding. Results Probl. Cell Differ. 48, 269–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen M, Huang X, Bamber B, and Jin Y (2000). Regulation of presynaptic terminal organization by C. elegans RPM-1, a putative guanine nucleotide exchanger with a RING-H2 finger domain. Neuron 26, 331–343. [DOI] [PubMed] [Google Scholar]
- Zivraj KH, Tung YC, Piper M, Gumy L, Fawcett JW, Yeo GS, and Holt CE (2010). Subcellular profiling reveals distinct and developmentally regulated repertoire of growth cone mRNAs. J. Neurosci. 30, 15464–15478. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets or codes.