

Abstract
There has been a steadily increasing appreciation of the fact that the relationship between protein sequence and structure is often sufficiently ambiguous to allow a single sequence to adopt alternate, stable folds. Living organisms have been able to utilize such metamorphic proteins in remarkable and unanticipated ways. HIV-1 reverse transcriptase is among the earliest such proteins identified and remains a unique example in which a functional heterodimer contains two, alternately-folded polymerase domains. Structural characterization of the p66 precursor protein combined with NMR spectroscopic and molecular modeling studies have provided insights into the factors underlying the metamorphic transition and the subunit-specific programmed unfolding step required to expose the protease cleavage site within the Ribonuclease H domain, supporting the conversion of the p66/p66' precursor into the mature p66/p51 heterodimer.
Graphical Abstract
eTOC blurb
Economy of the RT coding sequence is achieved at the expense of a complex maturation pathway that includes multiple structural rearrangements. These not only produce the active RT heterodimer, but also regulate the point at which the buried cleavage site in the RNase H domain becomes exposed for proteolytic processing.
I: Background
Metamorphic proteins - characterized by sufficient sequence-structure ambiguity to support at least two distinct, stable folds, are increasingly being identified and new functional roles discovered (Goodchild et al., 2011; Murzin, 2008). Examples include the cytokine lymphotactin (Volkman et al., 2009), redox-sensitive transcription factors such as OxyR (Choi et al., 2001), the DNA single-strand break repair scaffold XRCC1 (Cuneo and London, 2010), the chloride ion channel CLIC1 (Littler et al., 2004), the RNA polymerase (Tahirov et al., 2002), the spindle checkpoint protein Mad2 (Skinner et al., 2008), the HIV-1 capsid maturation switches (Wagner et al., 2016), and the cyanobacterial circadian clock protein KaiB (Tseng et al., 2017). Recent analyses indicate that this class of proteins may be more common than generally appreciated (Porter and Looger, 2018).
All of the above examples represent functional expansion of a single gene sequence at the level of folding, much as post-translational modifications can introduce new or modified protein functional characteristics (Jeffery, 2016). The driving force for the evolution of such structures is particularly strong for organisms that are subject to genome size constraints for which maximizing the information density of the coding sequence becomes critical. This constraint is most important for RNA viruses that are subject to both chemical and biochemical instability that limit the mean genome size to approximately 9,000 bases (Belshaw et al., 2007).
Among the earliest metamorphic proteins characterized, HIV-1 reverse transcriptase (RT) remains unique in that two alternately folded forms of the polymerase domain are both present in the p66/p51 RT heterodimer (Kohlstaedt et al., 1992; Wang et al., 1994). Maturation from a p66 monomer to a p66/p51 heterodimer is a multi-step process, the characterization of which has required additional structural and spectroscopic investigations (London, 2016) and references therein. The steps involved include an initial metamorphic transition followed by formation of an asymmetric p66/p66' homodimer, additional conformational rearrangements, and a subsequent, subunit-specific programmed unfolding of the RH domain in one subunit that renders it proteolytically labile. These studies have revealed the remarkable behavior of an HIV pharmaceutical target of central importance, and elucidated maturation intermediates that may serve as alternative targets for drug interference while providing broader insights in the area of protein folding/unfolding.
The three stable states
Each subunit of the mature p66/p51 RT heterodimer contains a polymerase domain. In p66, the domain adopts an active structure with a solvent-exposed catalytic site that is accessible to substrates, while in p51, the polymerase domain is more compact and the important catalytic residues are buried in the interior. These structural differences are most easily observed by color coding the polymerase subdomains: fingers (green), palm (blue), thumb (orange), and connection (magenta) (Figure 1). In order to simplify the presentation, we have not adhered to a rigorous distinction between domains and polymerase subdomains. In the p51 polymerase domain, the connection forms a large, non-covalent interface with the fingers/palm (the terminology containing the slash refers to the fact that the fingers and palm sequences are discontinuous and cannot be separated). Formation of this interface results in substantially greater buried hydrophobicity. This characteristic led Steitz and coworkers to conclude that this form of the polymerase is more stable than the extended form observed in the p66 subunit, resulting in the predictions that the monomeric forms of both p51 and p66 would adopt the more compact, p51-type structure observed in the p51 subunit of RT and that both of the homodimers formed would be asymmetric (Wang et al., 1994).
Figure 1. Alternate structures of the RT polymerase domain.

Ribbon diagrams of the HIV-1 RT p66 subunit (A), the p51 subunit (B), and the p66 monomer (p66M) (C). The polymerase subdomains are color coded: fingers (green), palm (blue), thumb (orange), connection (magenta), and the RH domain is brown. The polymerase domain in the p66 subunit contains a short β-sheet formed from the palm domain C-terminus and the connection domain N-terminus that is absent in the monomer and the p51 subunit. The structure of the polymerase domain in p66M is fairly similar to that of p51C, with the major differences being the unfolding of helix αM and the disorder of segments connecting the thumb to the palm and connection subdomains, so that its orientation is variable. The F440∣Y441 cleavage site (yellow) in the RH domain in both the extended structure (A) and the p66 monomer (C) is inaccessible to the solvent and to the protease.
This prediction of Wang et al. (Wang et al., 1994) was largely borne out in studies following a mutational strategy in which residues Lys219-Met230 were deleted (Zheng et al., 2014). This segment forms a disordered loop in p51 but is structurally and functionally important in the extended polymerase domain present in the p66 subunit (Ding et al., 1998). The p51 construct lacking these palm loop residues, p51ΔPL, does not dimerize allowing a crystal structure of the monomer to be determined (Zheng et al., 2014; Zheng et al., 2015). This result contrasts with many point mutations which in our experience reduce, but do not block dimer formation. Although the structure of p51ΔPL is generally similar to that of the p51 subunit of RT, there are two major differences: 1) the residues linking the thumb to the palm and connection domains are disordered, so that the orientation of the thumb subdomain is unconstrained, and 2) the C-terminal helix of the connection domain, αM, that occupies the space between the connection and thumb, is unraveled. Additional NMR studies with p66ΔPL and with other p66 constructs under conditions favoring the monomer indicated that there are no interface contacts between the polymerase and RH domains in the monomer, leading to the p66M structure shown in Figure 1C.
As summarized below, each of the three different structures shown in Figure 1 plays a specific role in the RT maturation process, and in particular, the structural difference between the p51 subunit and the p66 monomer plays a critical role in determining at what point in the maturation process the RH domain becomes susceptible to proteolytic cleavage.
Overview of RT maturation
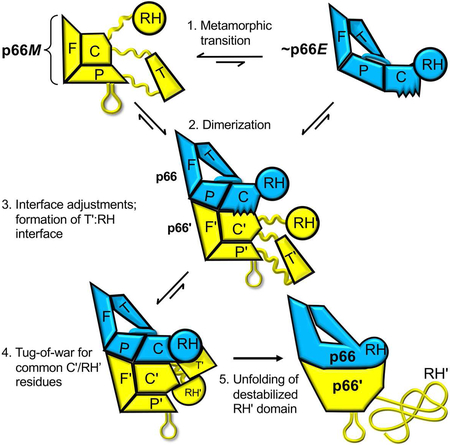
Figure 2 summarizes the basic steps involved in formation of the p66/p51 heterodimer from the p66 monomer. The process is initiated by dissociation of the connection domain from the fingers/palm in p66M (step 1). This dissociation releases multiple interdomain constraints that allow the remaining intra-domain constraints to become more dominant, supporting subsequent maturation steps while reducing the rate of fingers/palm - connection re-association. These domain re-arrangements and interactions create a large, solvent-exposed hydrophobic surface that supports initial homodimer formation with a second monomer, (step 2), so that the initially formed p66/p66' homodimer exists as a structural heterodimer, sometimes referred to in the literature as an asymmetric homodimer. Note that subsequent to dimerization, it becomes possible to distinguish the two subunits of the homodimer, and we introduce primes to denote the subunit formed directly from the monomer without initial domain rearrangements that is destined for proteolytic processing. The homodimer may dissociate, reversing the process, or undergo further adjustments that include more significant conformational changes in the palm and connection domains, and subsequent formation of the additional subunit interface between the p66 RH domain and the p66' thumb' domain (step 3) that is not present in the initially-formed homodimer. The unraveled C-terminal segment of the connection domain in p66' that connects the polymerase' and RH' domains then occupies the available channel that has been created in between these two domains in p66' (step 4) and waits patiently for the rate-limiting unraveling of N-terminal residues in the supernumerary RH' domain. Upon release of Tyr427' and a few adjacent residues, helix αM' in the p66' subunit extends by incorporating these residues, and the stability of the resulting structure forces the truncated and destabilized RH' domain to persist for a longer period of time with increasing likelihood of more extensive unfolding – a process which is likely supported by additional factors in the virion (Ilina et al., 2018; London, 2016). These transitions are discussed in greater detail below.
Figure 2. Schematic illustration of the main steps involved in RT maturation.

Step 1: Dissociation of the connection domain from the fingers/palm is followed by a series of conformational changes that constitute the metamorphic transition of the polymerase domain, altering the structure to a family of p66E-like forms, that can dimerize with the predominant p66M species. 2. an initial dimer is formed, followed by conformational adjustments at the interface that involve primarily the connection and palm subdomains. The ribbon diagram to the right illustrates some of the necessary conformational adjustments of the connection subdomain. 3. The subunit interface between the thumb' and the RH domain is formed, and the unfolded residues from the C-terminus of the p66' connection' domain occupy the channel between the connection' and thumb' domains. 4. Inherent instability of RH' domain releases Tyr427' and adjacent residues that are then captured by an extending helix αM'. 5. The destabilized RH' domain unfolds, exposing the cleavage site. Straightening of α-helix E in the palm subdomain (cyan) is one of the drivers for the metamorphic transition.
The metamorphic transition, p66M → p66E; thermodynamics vs. kinetics
As illustrated in Figure 1, the p66 monomer (p66M) contains a globular complex of the fingers/palm:connection subdomains, with the thumb (T) and RH domains connected by flexible segments. In the above, the colon represents the non-covalent interface between the fingers/palm and the connection subdomains. This structure is well supported by both crystallographic and NMR studies of Ile-labeled p66M (Zheng et al., 2014). In contrast with the methyl labeled NMR studies, the size of RT makes 1H-15N TROSY spectra of U-[2H,15N]p66 considerably more difficult to interpret, and previous studies have led to divergent interpretations (Sharaf et al., 2014; Zheng et al., 2017). However, Zheng et al. (Zheng et al., 2017) have concluded that this approach also yields results consistent with the monomer structure shown in Figure 1C. Despite its thermodynamic stability, the fingers/palm:connection interface does not persist indefinitely. We have previously described this structure as "spring-loaded", because upon dissociation, removal of the interface constraints allows the component subdomains to rapidly express conformational preferences that differ from those observed in the complex. The difference is most immediately apparent for the fingers/palm, for which the two subdomains rotate away from each other, adopting a more expanded conformation similar to that in the p66 subunit of RT (Figure 3) (Unge et al., 1994; Zheng et al., 2015). This expansion appears to result in part from the preference of α-helix E to eliminate the kink near Phe160, adopting a more standard helical geometry after the interdomain constraints have been removed (Zheng et al., 2017). Straightening of the helix may also contribute to dimerization, since αE is located at the subunit interface.
Figure 3. Drivers for the metamorphic transition.

After dissociation of the connection subdomain, the fingers/palm subdomains rotate away from each other, and the palm C-terminus forms a short, amphiphilic β-sheet that lies against the palm domain β-sheet (β6-β9-β10). The tips of the fingers and thumb form a weak interface that helps to position the system for dimer formation. A ribbon diagram of the fingers – thumb interface is shown at the upper right. The initial p66/p66' homodimer is a structural heterodimer that is formed from a p66E-like structure and the p66M monomer structure. The ribbon diagram to the lower left corresponds to the initial homodimer with color-coded p66' subdomains; in the ribbon diagram to the lower right, the p66 subdomains are color coded, and the overlaid initial connection domain conformation is indicated in light pink.
In addition, a structural comparison of p66M and p66E indicates that the C-terminal segment of the palm subdomain undergoes a significant structural change, from an extended structure that runs along the surface of p66M and includes the disordered loop mentioned above, to a more compact, 3-stranded amphiphilic β-sheet structure in p66E. CD analysis of the isolated segment (residues 226-241) indicates that it adopts a conformation more similar to that in the p66 compared with the p51 subunit, and the β-sheet structure is likely stabilized by hydrophobic interactions of the sidechains (Zheng et al., 2017). This amphiphilic β-sheet which corresponds to β12-β13-β14 in p66E forms a β-sandwich with the hydrophobic surface of the β6-β9-β10 sheet in the palm. Occasional separation of these two β-sheets, perhaps linked to motion of the thumb subdomain (Ivetac and McCammon, 2009) creates the non-nucleoside RT inhibitor (NNRTI) binding pocket that surprisingly is not present in the unliganded enzyme (Hsiou et al., 1996).
Formation of this β-sheet structure also creates a subdomain interface that facilitates positioning of the adjacent thumb so that the tips of the fingers and thumb make a small, hydrophobic contact that competes with re-association of the connection subdomain and helps to position the domains optimally for dimer formation (Figure 3). The non-conservative L289K mutation in the thumbnail has been shown to inhibit dimerization (Goel et al., 1993; Zheng et al., 2010), probably by interfering with the formation of the hydrophobic fingers:thumb interface.
Hence, although the globular fingers/palm:connection structure shown in Figure 1C is more stable, the changes summarized in Figure 3 compete with re-association of the fingers/palm and connection subdomains, retarding the re-association rate. The effect is to extend the time period during which the structurally transformed monomer may associate with a second monomer that is in the ground state.
The mechanism shown in figure 2 hinges on the ability of the metamorphic structure to adopt a group of relatively unstable structures for a sufficient time period to allow a stable dimer to form. Or, form a thermodynamic perspective, on an ideal balance between the thermodynamic forces favoring the monomer and the kinetic factors protracting the lifetime of the p66E structure. Stabilization of the transiently re-organized p66M structure is contingent on formation of an initial dimer that follows the creation of a large interface that can interact with p66M. The requirement for structural rearrangement prior to dimer formation is consistent with the inability of p66M lacking the palm loop noted above to dimerize (Zheng et al., 2015), and corresponds to a conformational selection process in which the predominant p66M selects a p66E-like structure for dimerization (Figure 2). This conclusion is also supported by kinetic studies of Barkley and coworkers (Braz et al., 2010; Venezia et al., 2009).
Dimerization and interface maturation
The metamorphic transition discussed above must create a sufficient interface to support initial dimerization with p66M. It is, however, likely that additional conformational changes, particularly involving segments near the domain and subunit interfaces, undergo subsequent adjustments from an initial p66E-like structure to p66E. The exposed hydrophobic surfaces created by the fingers/palm:connection dissociation provide initial high affinity, low specificity surfaces that allow these rearrangements to occur, further enhancing dimer stability. A comparison of the domain/subdomain structures indicates that the fingers, thumb, and RH domains exhibit fairly minimal changes, while the palm and connection undergo more significant conformational alterations. Changes in the palm subdomain include formation of a short β-sheet by the C-terminal segment discussed above, as well as adjustments at the interface involving residues 91-103, that are disordered in the RT216 structure (Unge et al., 1994). This segment interacts with residues in the p66' β7-β8 loop upon formation of the dimer (Pandey et al., 2001).
The ribbon diagram in Figure 2 (middle, right) illustrates how the p66 connection domain must adjust from its monomer conformation (pink) to the conformation observed in the mature heterodimer (magenta). The transition involves elimination of the bend in helix αL and large movements of the subsequent loop containing Trp406. Straightening of αL creates a binding site for Lys331' in the p66' subunit. The β18-αK loop at the RH domain interface also shows significant variation. It is unclear to what extent these changes may precede, accompany or follow dimerization.
Formation of the RH:thumb' interface present in mature RT occurs on a somewhat slower time scale. However, NMR observations, particularly for Ile434 located at this interface, indicate that it is formed within the first hour after dimerization occurs, and likely on a significantly shorter time scale (Zheng et al., 2015). Formation of this interface is likely a cooperative process in which the connection:RH, connection':thumb', the connection:connection' and the RH:thumb' interface are mutually supportive. The cooperative nature of the connection'-thumb' interactions is suggested by the monomer structure in which both the αM' helix and thumb' orientation are disordered (compare Figures 1B and C).
Programmed unfolding of the RH domain and proteolysis
Biochemical and structural studies of the isolated RT RH domain have demonstrated that the F440∣Y441 cleavage site that serves as the HIV-1 protease substrate is located on the central β-strand near the center of the domain (Davies et al., 1991; Himmel et al., 2009; Himmel et al., 2014; Hostomska et al., 1991; Kirschberg et al., 2009) (Figure 1). Formation of a stable RH domain requires an additional 14 residues beginning at Tyr427 rather than at Tyr441 (Becerra et al., 1991; Davies et al., 1991; Hostomska et al., 1991). Multiple crystal structures of the isolated RH domain all include Tyr427 (Davies et al., 1991; Himmel et al., 2009; Himmel et al., 2014; Kirschberg et al., 2009). The stable, isolated RH domain that results is resistant to cleavage by HIV-1 protease (Hostomska et al., 1991; Tomasselli et al., 1993). In contrast, cleavage of the p66/p66' at F440∣Y441 does not produce a stable RH domain, but rather a group of peptides that subsequently undergoes further proteolysis (Tomasselli et al., 1993). Conversion of p66 to p51 is controlled by the accessibility of the cleavage site, and must occur either prior to domain folding or subsequent to a specific unfolding step. Excessive processing of p66 can result from destabilizing mutations within the RNase H domain, including those located in the segment between Tyr427 and Tyr441 (Abram and Parniak, 2005; Hostomska et al., 1991; Navarro et al., 2001; Slack et al., 2015), and seriously impairs viral viability by resulting in excessive formation of p51 relative to p66. Not only is excessive processing of p66 a potential problem for the virus, but processing must halt when 50% of p66 has been cleaved - a difficult constraint for an enzyme to achieve.
The solution to the problem of p66 processing is intrinsic to the structures shown in Figure 1. The cleavage site within the RH domain lies buried in the folded RH domain in the p66 monomer (Figure 1C) preventing premature processing. The step 1 metamorphic transition does not alter this situation, so the cleavage site remains protected. Subsequent to dimerization, and maturation of the p66/p66' interface, a channel between the connection' and thumb' domain in p66' is created and the subunit moves toward completing the structural transition by forming helix αM' within this channel (Figure 4). At this point, an apparent conflict arises since the helix seeks to incorporate several residues that also form the N-terminus of the stable RH domain. No such conflict exists in the p66 subunit, in which the corresponding residues extend from the connection to the RH domain without conflict (Figure 4). This leads to a functional tug-of-war between the polymerase' and the RH' domains of p66' for common residues, a conflict that is completely dependent on the alternative structures of the polymerase domain. In the monomer, the RH domain wins the competition, retaining residues beginning with Tyr427, while in the final stage of dimer maturation, the polymerase' wins the tug-of-war, resulting in RH' destabilization.
Figure 4. Subunit-specific "tug-of-war" for N-terminal RH domain residues.

In the p66 subunit (A), the C-terminal segment of the connection domain (magenta) adopts an extended conformation that links the polymerase (gray) and RH (brown) domains. The N-terminal segment of the RH domain from Tyr427-Tyr440 is colored yellow. In the p66' subunit (B), The polymerase domain C-terminal residues form a helix (magenta) that extends to include several residues derived from the RH' domain (yellow) when this domain is transiently unfolded. In the structure shown (pdb: 1RTJ, (Esnouf et al., 1995)), 8 hydrogen bonds are shown between residues derived from the RH' domain and the polymerase' domain, and additional interactions include the hydrophobic interface between Leu429 and Trp426. In both panels, the polymerase domain is gray, except for the C-terminal residues (magenta); the N-terminal RH domain residues Tyir427-Phe440 are yellow, and the remaining RH domain residues are brown. In panel B, the sidechain conformations of Q242 and Q428 were adjusted relative to structure 1RTJ to optimize the H-bond interactions. The schematics on the left illustrate the inclusion of RH' domain N-terminal residues (yellow) into the polymerase' domain.
The structural conflict rests on the strong dependence of RH domain stability on a few N-terminal residues – particularly Tyr427. Support for this conclusion comes from observations on destabilization of an N-terminally truncated RH domain, RHΔNT, in which the first two residues: Tyr427 and Gln428, were deleted, and Leu429 mutated to methionine (L429M) (Zheng et al., 2014), and from studies of a domain-swapped RH dimer in which the monomerdimer interconversion rate is greatly accelerated in constructs lacking Tyr427 (Zheng et al., 2016). The interconversion rate is negligible for the RH domain at 25 °C, but sufficiently rapid for RHΔNT to render isolation of either form in a pure state challenging.
Maturation of HIV-2 reverse transcriptase
Studies of HIV-2 reverse transcriptase, including data related to its maturation, are limited, however the structure of the HIV-2 RT heterodimer has been reported (pdb: 1MU2, (Ren et al., 2002). A structural comparison indicates that all of the critical elements involved in maturation of the HIV-1 enzyme are present in the HIV-2 enzyme as well (London, 2016). One interesting difference arises because the analogous F440∣Y441 cleavage site in the HIV-2 enzyme is resistant to HIV-2 protease, so that the protein is cleaved at a different site, M484∣A485 (Fan et al., 1995). This result is consistent with the maturation steps outlined above in which the polymerase'-RH' tug-of-war leads to RH' domain unfolding in the HIV-2 RT homodimer, but the actual site of cleavage is determined by the selectivity of the corresponding protease once the domain unfolds. As is observed for HIV-1 RT (Tomasselli et al., 1993), maturation does not release an intact RH domain, but fragments that are further proteolyzed. Remarkably, the HIV-2 RT p68/p68' homodimer precursor is cleaved at F440∣Y441 by the HIV-1 protease (Fan et al., 1995). The ability of the HIV-1 and HIV-2 proteases to cleave two sites separated by 44 residues, each of which is buried in the folded RH domain, provides strong support for an HIV-2 RT maturation pathway that also entails complete, subunit-selective unfolding of one RH domain.
Conclusions
As summarized above, RT maturation is a story of ambiguity and conflict. There is conflict between the interdomain structural restraints and the conformational preferences of the isolated subdomains that drive many of the rearrangements characterizing the initial metamorphic transition. The conflict between thermodynamic and kinetic factors supports the extended lifetime of the unstable, conformationally-altered polymerase domain that allows dimer formation and subsequent conformational adjustments before the thermodynamic stability of the monomer is re-asserted. There is intrinsic instability inherent in the RH domain resulting from linker strain and perhaps other factors that, when coupled with the additional competition between the polymerase and RH domain for common residues, is sufficient to result in RH domain unfolding, exposing the initially buried cleavage site. Nevertheless, all of these highly unusual behavior patterns are merely atypical combinations of conventional protein folding tendencies.
Highlights.
RT structural maturation proceeds through an asymmetric p66/p66' homodimer
An initial metamorphic transition forms the active p66 polymerase domain
Subsequently, a second metamorphic transition destabilizes the p66' RNase H domain
Programmed RNase H domain unfolding prevents premature over-processing of p66
ACKNOWLEDGEMENTS
The author is grateful to Dr. Alexander Foo, Dr. Geoff Mueller, Dr. Jason Williams, and Dr. Lars Pedersen for a critical reading of this manuscript, and for the many research contributions of Dr. Xunhai Zheng. This work was supported by Research Project Number ZIA-ES050147 in the Intramural Research Program of the National Institute of Environmental Health Sciences, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests. The author declares no competing interests.
REFERENCES
- Abram ME, and Parniak MA (2005). Virion instability of human immunodeficiency virus type 1 reverse transcriptase (RT) mutated in the protease cleavage site between RT p51 and the RT RNase H domain. J Virol 79, 11952–11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra SP, Kumar A, Lewis MS, et al. (1991). Protein-protein interactions of HIV-1 reverse transcriptase: implication of central and C-terminal regions in subunit binding. Biochemistry-Us 30, 11707–11719. [DOI] [PubMed] [Google Scholar]
- Belshaw R, Pybus OG, and Rambaut A (2007). The evolution of genome compression and genomic novelty in RNA viruses. Genome research 17, 1496–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braz VA, Holladay LA, and Barkley MD (2010). Efavirenz binding to HIV-1 reverse transcriptase monomers and dimers. Biochemistry-Us 49, 601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Kim S, Mukhopadhyay P, et al. (2001). Structural basis of the redox switch in the OxyR transcription factor. Cell 105, 103–113. [DOI] [PubMed] [Google Scholar]
- Cuneo MJ, and London RE (2010). Oxidation state of the XRCC1 N-terminal domain regulates DNA polymerase beta binding affinity. Proc Natl Acad Sci U S A 107, 6805–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JF 2nd, Hostomska Z, Hostomsky Z, et al. (1991). Crystal structure of the ribonuclease H domain of HIV-1 reverse transcriptase. Science 252, 88–95. [DOI] [PubMed] [Google Scholar]
- Ding J, Das K, Hsiou Y, et al. (1998). Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 A resolution. J Mol Biol 284, 1095–1111. [DOI] [PubMed] [Google Scholar]
- Esnouf R, Ren J, Ross C, et al. (1995). Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat Struct Biol 2, 303–308. [DOI] [PubMed] [Google Scholar]
- Fan N, Rank KB, Leone JW, et al. (1995). The differential processing of homodimers of reverse transcriptases from human immunodeficiency viruses type 1 and 2 is a consequence of the distinct specificities of the viral proteases. J Biol Chem 270, 13573–13579. [PubMed] [Google Scholar]
- Goel R, Beard WA, Kumar A, et al. (1993). Structure/function studies of HIV-1(1) reverse transcriptase: dimerization-defective mutant L289K. Biochemistry-Us 32, 13012–13018. [DOI] [PubMed] [Google Scholar]
- Goodchild SC, Curmi PMG, and Brown LJ (2011). Structural gymnastics of multifunctional metamorphic proteins. Biophys Rev 3, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmel DM, Maegley KA, Pauly TA, et al. (2009). Structure of HIV-1 reverse transcriptase with the inhibitor beta-Thujaplicinol bound at the RNase H active site. Structure 17, 1625–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmel DM, Myshakina NS, Ilina T, et al. (2014). Structure of a dihydroxycoumarin active-site inhibitor in complex with the RNase H domain of HIV-1 reverse transcriptase and structure-activity analysis of inhibitor analogs. J Mol Biol 426, 2617–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostomska Z, Matthews DA, Davies JF, 2nd, et al. (1991). Proteolytic release and crystallization of the RNase H domain of human immunodeficiency virus type 1 reverse transcriptase. J Biol Chem 266, 14697–14702. [PubMed] [Google Scholar]
- Hsiou Y, Ding J, Das K, et al. (1996). Structure of unliganded HIV-1 reverse transcriptase at 2.7 angstrom resolution: Implications of conformational changes for polymerization and inhibition mechanisms. Structure 4, 853–860. [DOI] [PubMed] [Google Scholar]
- Ilina TV, Slack RL, Elder JH, et al. (2018). Effect of tRNA on the Maturation of HIV-1 Reverse Transcriptase. J Mol Biol 430, 1891–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivetac A, and McCammon JA (2009). Elucidating the inhibition mechanism of HIV-1 non-nucleoside reverse transcriptase inhibitors through multicopy molecular dynamics simulations. J Mol Biol 388, 644–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery CJ (2016). Protein species and moonlighting proteins: Very small changes in a protein's covalent structure can change its biochemical function. J Proteomics 134, 19–24. [DOI] [PubMed] [Google Scholar]
- Kirschberg TA, Balakrishnan M, Squires NH, et al. (2009). RNase H active site inhibitors of human immunodeficiency virus type 1 reverse transcriptase: design, biochemical activity, and structural information. J Med Chem 52, 5781–5784. [DOI] [PubMed] [Google Scholar]
- Kohlstaedt LA, Wang J, Friedman JM, et al. (1992). Crystal-Structure at 3.5 Angstrom Resolution of Hiv-1 Reverse-Transcriptase Complexed with an Inhibitor. Science 256, 1783–1790. [DOI] [PubMed] [Google Scholar]
- Littler DR, Harrop SJ, Fairlie WD, et al. (2004). The intracellular chloride ion channel protein CLIC1 undergoes a redox-controlled structural transition. J Biol Chem 279, 9298–9305. [DOI] [PubMed] [Google Scholar]
- London RE (2016). Structural Maturation of HIV-1 Reverse Transcriptase-A Metamorphic Solution to Genomic Instability. Viruses 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin AG (2008). Biochemistry. Metamorphic proteins. Science 320, 1725–1726. [DOI] [PubMed] [Google Scholar]
- Navarro JM, Damier L, Boretto J, et al. (2001). Glutamic residue 438 within the protease-sensitive subdomain of HIV-1 reverse transcriptase is critical for heterodimer processing in viral particles. Virology 290, 300–308. [DOI] [PubMed] [Google Scholar]
- Pandey PK, Kaushik N, Talele TT, et al. (2001). The beta7-beta8 loop of the p51 subunit in the heterodimeric (p66/p51) human immunodeficiency virus type 1 reverse transcriptase is essential for the catalytic function of the p66 subunit. Biochemistry-Us 40, 9505–9512. [DOI] [PubMed] [Google Scholar]
- Porter LL, and Looger LL (2018). Extant fold-switching proteins are widespread. Proc Natl Acad Sci U S A 115, 5968–5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Bird LE, Chamberlain PP, et al. (2002). Structure of HIV-2 reverse transcriptase at 2.35-A resolution and the mechanism of resistance to non-nucleoside inhibitors. Proc Natl Acad Sci U S A 99, 14410–14415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharaf NG, Poliner E, Slack RL, et al. (2014). The p66 immature precursor of HIV-1 reverse transcriptase. Proteins 82, 2343–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner JJ, Wood S, Shorter J, et al. (2008). The Mad2 partial unfolding model: regulating mitosis through Mad2 conformational switching. J Cell Biol 183, 761–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack RL, Spiriti J, Ahn J, et al. (2015). Structural integrity of the ribonuclease H domain in HIV-1 reverse transcriptase. Proteins 83, 1526–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahirov TH, Temiakov D, Anikin M, et al. (2002). Structure of a T7 RNA polymerase elongation complex at 2.9 A resolution. Nature 420, 43–50. [DOI] [PubMed] [Google Scholar]
- Tomasselli AG, Sarcich JL, Barrett LJ, et al. (1993). Human immunodeficiency virus type-1 reverse transcriptase and ribonuclease H as substrates of the viral protease. Protein Sci 2, 2167–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng R, Goularte NF, Chavan A, et al. (2017). Structural basis of the day-night transition in a bacterial circadian clock. Science 355, 1174–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unge T, Knight S, Bhikhabhai, R, et al. (1994). 2.2 A resolution structure of the amino-terminal half of HIV-1 reverse transcriptase (fingers and palm subdomains). Structure 2, 953–961. [DOI] [PubMed] [Google Scholar]
- Venezia CF, Meany BJ, Braz VA, et al. (2009). Kinetics of Association and Dissociation of HIV-1 Reverse Transcriptase Subunits. Biochemistry-Us 48, 9084–9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman BF, Liu TY, and Peterson FC (2009). Chapter 3. Lymphotactin structural dynamics. Methods Enzymol 461, 51–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JM, Zadrozny KK, Chrustowicz, J, et al. (2016). Crystal structure of an HIV assembly and maturation switch. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Smerdon SJ, Jager J, et al. (1994). Structural Basis of Asymmetry in the Human-Immunodeficiency-Virus Type-1 Reverse-Transcriptase Heterodimer. P Natl Acad Sci USA 91, 7242–7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Mueller GA, Cuneo MJ, et al. (2010). Homodimerization of the p51 subunit of HIV-1 reverse transcriptase. Biochemistry-Us 49, 2821–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Mueller GA, Kim K, et al. (2017). Identification of drivers for the metamorphic transition of HIV-1 reverse transcriptase. Biochem J 474, 3321–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Pedersen LC, Gabel SA, et al. (2014). Selective unfolding of one Ribonuclease H domain of HIV reverse transcriptase is linked to homodimer formation. Nucleic acids research 42, 5361–5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Pedersen LC, Gabel SA, et al. (2016). Unfolding the HIV-1 reverse transcriptase RNase H domain - how to lose a molecular tug-of-war. Nucleic acids research 44, 1776–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Perera L, Mueller GA, et al. (2015). Asymmetric conformational maturation of HIV-1 reverse transcriptase. Elife 4, e06359. [DOI] [PMC free article] [PubMed] [Google Scholar]