

Abstract
Synthesis of a variety of sugar lactols (hemiacetals) has been accomplished in moderate to excellent yields by using bromine-mediated oxidation of thioglycosides. It was found that acetonitrile is the optimal solvent for this oxidation reaction. This approach involving bromine as oxidant is superior to that using N-bromosuccimide (NBS) which produces byproduct succinimide often difficult to separate from the lactol products.
Keywords: sugar lactols, hemiacetals, bromine, oxidation, thioglycosides
Introduction
Glycosyl hydroxides, i.e. sugar lactols or hemiacetals, serve as important glycosyl donors (1-hydroxy glycosyl donors) and synthetic intermediates for chemical synthesis of complex biologically significant carbohydrate molecules.[1–5] Back in 1890s, sugars lactols were first used as glycosyl donors in the well-known Fischer glycosylation.[6–7] Recently, Gin[8–14] and Bennett[15–16] reported dehydrative glycosylations using 1-hydroxy glycosyl donors. In addition, in 1970s Schmidt pioneered the studies of stereoselective synthesis of oligosaccharides and glycoconjugates via anomeric O-alkylation of sugar lactols.[17–22] Based on Schmidt’s work, recently our group[23–27] and others[28–33] resurrected the use of anomeric O-alkylation for stereoselective construction of challenging glycosidic linkages. Specifically, our group disclosed stereoselective synthesis of several classes of challenging glycosides via anomeric O-alkylation of the corresponding sugar hemiacetals, e.g. 2-deoxy-β-glycosdies,[23–24] 2-deoxy-α-glycosdies,[25] as well as β-mannosides.[26–27] Furthermore, sugar hemiacetals are used as precursors for the preparation of commonly used glycosyl donors, e.g. trichloroacetimidates,[34] N-(phenyl)trifluoroacetimidates,[35] phosphites and phosphates,[36–37] or ortho-alkynylbenzoates,[38] carbonates,[39–42] sulfonates,[43–44] and glycosyl esters.[45–46]
Typically, lactols (2) are obtained by methanolysis of glycosyl esters (1, X = OCOR),[47–48] hydrolysis of glycosyl halides (1, X = halides)[49–50] or methyl glycosides (1, X = OMe),[51] or oxidation of thioglycosides (3)[52–53] (a and b, Scheme 1). Among those methods, oxidation of readily available and stable thioglycosides (3) to lactols is one of the most popular strategies for preparation of sugar hemiacetals and N-bromosuccimide (NBS) is usually employed as the oxidant (b, Scheme 1). Despite its mild nature, the reaction employing NBS generates the byproduct succinimide which oftentimes is difficult to separate from the desired lactol products, especially when the lactols become quite polar. Therefore, our group has been searching for alternative oxidant to facilitate the purification process.
Scheme 1.

Representative strategies for preparation of sugar lactols.
Bromine is often used to oxidize thioglycosides to the corresponding glycosyl bromide donors for the synthesis of various oligosaccharides and glycoconjugates.[54–57] In addition, there was limited report that sugar hemiacetals can be prepared by bromine-mediated oxidation of thioglycosides via a two-step sequence: 1) bromine-mediated oxidation of thioglycosides in dry dichloromethane to the corresponding glycosyl bromide; 2) hydrolysis of isolated glycosyl bromide.[58] Use of bromine as oxidant is advantageous as it can avoid the production of difficultly separable byproducts. However, this two-step procedure may not be suitable for carbohydrate substrates containing acid-sensitive protecting groups or free hydroxyl groups. We wondered if it is possible to develop a one-pot oxidation/hydrolysis under suitable conditions for the preparation of sugar hemiacetals from thioglycosides using bromine as the oxidant. In this Communication, we wish to report the facile synthesis of sugar lactols via bromine-mediated oxidation of thioglycosides in a mixture of acetonitrile and water (c, Scheme 1).
Results and Discussion
Phenyl 3,4,6-tri-O-benzyl-1-thio-α-D-mannopyranoside (4, Table 1) was chosen as the substrate for preparing 3,4,6-tri-O-benzyl-D-mannose 5 under bromine-mediated oxidation. It is worth noting that a stock solution of bromine in dichloromethane (5.0 M) was prepared for use instead of pure bromine due to easy handling and safety concerns.[59] Initially, we used a mixture of acetone and water (10/1, v/v), the solvent system for NBS-mediated oxidation of thioglycosides, for bromine-mediated oxidation. This reaction was sluggish at 0 °C and took 24 hours to complete and 3,4,6-tri-O-benzyl-D-mannose 5 was isolated in 83% yield (α/β=3/1, entry 1, Table 1). The slow reaction rate may be due to that Br2 is also able to α–brominate acetone, which competes with the oxidation of thioglycosides. Addition of bromine solution at lower temperature followed by warming up to 0 °C did not help (entry 2). Switching to dichloromethane, the solvent used for preparation of glycosyl bromide by bromine oxidation of thioglycosides, did not work well and complex mixtures were obtained (entries 3 and 4). Use of THF as solvent gave similar results as acetone, albeit the reaction was found to be a little faster (entry 5). Finally, we found acetonitrile was the optimal solvent for this reaction which was complete in two hours and gave almost quantitative yield of the corresponding lactol 5 (entry 6). The method was also applied to 0.2 mmol and 1 gram scale of thioglycoside 4 which furnished the desired lactol 5 in 98% and 91% yield, respectively (entry 6). Application of previously reported two-step procedure for this oxidation–hydrolysis only gave 34% yield of the lactol 5 (entry 7). In comparison, use of NBS as the oxidant instead of bromine afforded lactol 5 in 82% yield (entry 8).
Table 1.
Synthesis of 3,4,6-tri-O-benzyl-D-mannose by bromine-mediated oxidation of corresponding thioglycoside.a

| ||
|---|---|---|
| Entry | Solvent/Temp/Timeb | Yield,c α/β ratiod |
| 1 | acetone/H2O (10/1, v/v), 0 °C, 24 h | 83%, 3/1 |
| 2 | acetone/H2O (10/1, v/v), −30 to 0 °C, 26 h | 79%, 4/1 |
| 3 | CH2Cl2/H2O (10/1, v/v), −30 to 0 °C, 3.5 h | trace |
| 4 | CH2Cl2/H2O (10/1, v/v), 0 °C, 3 h | trace |
| 5 | THF/H2O (10/1, v/v), 0 °C, 12 h | 81%, 4/1 |
| 6 | CH3CN/H2O (10/1, v/v), 0 °C, 2 h | 97% (98%,e 91%f), 4/1 |
| 7 | Br2, CH2Cl2, 0 °C, 0.5 h; then acetone/H2O (4/1, v/v), RT, 2 hg | 34% |
| 8 | NBS (3.0 eq.), acetone/H2O (10/1, v/v), 0 °C, 2 h | 82% |
All reactions were performed using 0.1 mmol of phenyl 3,4,6-tri-O-benzyl-1-thio-D-mannopyranoside 4 (1.0 eq.) and 60 μL of 5.0 M Br2 in CH2Cl2 (3.0 eq.) in 0.5 mL organic solvent and 50 μL H2O unless otherwise noted;
When TLC showed the substrate was completely consumed;
Isolated yield;
Determined by 1H NMR;
Isolated yield obtained on 0.2 mmol scale.
Isolated yield on a 1 gram scale.
see reference 58.
With the optimal condition established, the bromine-mediated oxidation of thioglycosides was applied to the synthesis of a variety of sugar hemiacetals (Table 2). In general, sugar hemiacetals were obtained in good to excellent yields. It was also found that both aryl (e.g. phenyl) and alkyl (e.g. ethyl) thioglycosides can be oxidized smoothly to afford the corresponding lactols in good yields (entry 1). Various functional groups, such as carbamate, amide, imide, azide, and carboxylic ester, are tolerated. For thioglycosides containing acid-sensitive protecting groups, such as silyl ethers (entry 2), benzylidene acetals (entries 10–12), and 1,2-diketals (entry 13), solid sodium bicarbonate needs to be added to buffer the acidity of the reaction in order to achieve good yields. Interestingly, methoxyethoxymethyl (MEM) ether was found to be stable in this type of reaction and no sodium bicarbonate was added necessarily (entry 9). Obviously, deoxy sugars, such as 6-deoxy and 2,6-dideoxy sugars, are very reactive substrates towards bromine oxidation (entries 2–4). Sugar substrates with acyl protecting groups, i.e. “disarmed donors”, including thioglycoside of peracetylated N-acetyl neuraminic acid (18), D-mannose-derived thioglycoside (30) and phenyl 2,3,4,6-tetra-O-acetyl-1-thio-β-glucopyranoside (32), were found to be relatively unreactive and produced corresponding hemiacetals in moderate yields, probably due to the inductive effect. Allowing the reaction to proceed for an additional period of time or adding more Br2 did not help, probably due to the decomposition of the starting materials or lactols over time (entries 7, 13 and 14).
Table 2.
Synthesis of various sugar lactols by bromine-mediated oxidation of thioglycosides.a

| |||
|---|---|---|---|
| Entry | Thioglycosides | Lactols | Yield,b
α/β ratio,c Reaction Condition, Time |
| 1 |  |
 |
84%, 2/1 from 6a, A, 2 h; 86%, 2/1 from 6b, A, 2 h |
| 2 |  |
 |
95%, 1.4/1, B, 1 h |
| 3 |  |
 |
90%, 1/1, A, 2 h |
| 4 |  |
 |
93%, 2/1, A, 2 h |
| 5 |  |
 |
94%, α only, A, 2 h |
| 6 |  |
 |
84%, 2.5/1, A, 2 h |
| 7 | 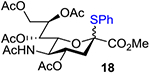 |
 |
66%,d 1/10, A, 12 h; 63%,e 1/10, A, 12 h |
| 8 |  |
 |
75%, 10/1, A, 4 h |
| 9 |  |
 |
81%, 2.5/1, A, 3 h |
| 10 |  |
 |
15%, 2/1, A, 2 h; 72%, 2/1, B, 3 h |
| 11 |  |
 |
64%, β only, B, 5 h |
| 12 |  |
 |
61%, 1.4/1, B, 5 h |
| 13 |  |
 |
60%,f >20/1, B, 18 h; 55%,g, >20/1, B, 36 h; 59%h |
| 14 |  |
 |
20%,i 4/1, 2 h; 38%,j 4/1, 12 h |
| 15 |  |
 |
86%, 2/1, 3 h |
All reactions were performed using 0.2 mmol of thioglycosides (1.0 eq.) and 120 μL of 5.0 M Br2 in CH2Cl2 (3.0 eq.) in 1 mL acetonitrile and 0.1 mL H2O at 0 °C;
Isolated yield;
Determined by 1H NMR;
29% of starting material was recovered;
4.5 eq. of bromine was used. Trace amount of starting material was recovered;
31% of starting material was recovered;
The reaction was stirred for 36 hours and 24% of starting material was recovered;
NBS (3.0 eq.) was used and the reaction was stirred for 18 hours and 29% of starting material was recovered;
64% of starting material was recovered;
24% of starting material was recovered.
In conclusion, an approach for the synthesis of sugar hemiacetals has been developed via bromine-mediated oxidation of thioglycosides in a mixture of acetonitrile/water. Various functional groups and protecting groups are tolerated and a wide range of sugar lactols were obtained in moderate to excellent yields. This method is advantageous as it avoids the production of byproducts difficult to separate.
Experimental
Materials and Methods
Proton and carbon nuclear magnetic resonance spectra (1H NMR and 13C NMR) were recorded on either Bruker 600 (1H NMR-600 MHz; 13C NMR 150) or INOVA 600 (1H NMR-600 MHz; 13C NMR-150 MHz) at ambient temperature with CDCl3 as the solvent unless otherwise stated. Chemical shifts are reported in parts per million relative to residual protic solvent internal standard CDCl3: 1H NMR at δ 7.26, 13C NMR at δ 77.36. Data for 1H NMR are reported as follows: chemical shift, integration, multiplicity (app = apparent, par obsc = partially obscure, ovrlp = overlapping, s = singlet, d = doublet, dd = doublet of doublet, t = triplet, q = quartet, m = multiplet) and coupling constants in Hertz. All 13C NMR spectra were recorded with complete proton decoupling. High resolution mass spectra (HRMS) were acquired on a Waters Acuity Premiere XE TOF LC-MS by electrospray ionization. Optical rotations were measured with Autopol-IV digital polarimeter; concentrations are expressed as g/100 mL.
All reagents and chemicals were purchased from Acros Organics, Sigma Aldrich, Fisher Scientific, Alfa Aesar, and Strem Chemicals and used without further purification. THF, methylene chloride, toluene, and diethyl ether were purified by passing through two packed columns of neutral alumina (Innovative Technology). Anhydrous DMF and benzene were purchased from Acros Organics and Sigma-Aldrich and used without further drying. All reactions were carried out in oven-dried glassware under an argon atmosphere unless otherwise noted. Analytical thin layer chromatography was performed using 0.25 mm silica gel 60-F plates. Flash column chromatography was performed using 200–400 mesh silica gel (Scientific Absorbents, Inc.). Yields refer to chromatographically and spectroscopically pure materials, unless otherwise stated.
General procedure for synthesis of sugar lactols by bromine-mediated oxidation of thioglycosides.
General procedure A:
Thioglycosides (0.2 mmol) were dissolved by 1.0 mL acetonitrile in a 10 mL flask, followed by the addition of 0.1 mL water. The mixture was stirred at 0 °C for 30 min. 120 μL of Br2 (3.0 eq.) solution in CH2Cl2 (5.0 M) was added dropwise and then the yellow mixture was stirred at this temperature until the TLC showed that the substrate was completely consumed. Then the mixture was diluted by 5 mL saturated NaHCO3 aqueous solution and evaporated under reduced pressure to remove acetonitrile. The residue was extracted by CH2Cl2 (10 mL × 3, for lactol 11 10% n-butanol was used together). The organic layers were combined and dried over anhydrous Na2SO4. The filtrate was concentrated and purified by flash column chromatography to give the desired lactols.
General procedure B:
Thioglycosides (0.2 mmol) were dissolved by 1.0 mL acetonitrile in a 10 mL flask, followed by the addition of 0.1 mL water and 168 mg of NaHCO3 (10.0 eq.). The mixture was stirred at 0 °C for 30 min. 120 μL of Br2 (3.0 eq.) solution in CH2Cl2 (5.0 M) was added dropwise and then the yellow mixture was stirred at this temperature until the TLC showed that the substrate was completely consumed. Then the mixture was diluted by 5 mL saturated NaHCO3 aqueous solution and evaporated under reduced pressure to remove acetonitrile. The residue was extracted by CH2Cl2 (10 mL × 3). The organic layers were combined and dried over anhydrous Na2SO4. The filtrate was concentrated and purified by flash column chromatography to give the desired lactol.
3,4,6-Tri-O-benzyl-α/β-D-mannopyranose (5).
Lactol 5 was prepared from thioglycoside 4[60] (109 mg, 0.201 mmol) following the general procedure A. The crude reaction mixture was purified by flash column chromatography (Hexanes/EtOAc = 2/1 to CH2Cl2/MeOH = 15/1) to give 88.8 mg (0.197 mmol, 98%) of lactol 5 (α/β = 4/1) as colorless syrup. 1H and 13C NMR data of 5 were in agreement with reported ones.[61]
2,3,4,6-Tetra-O-benzyl-α/β-D-glucopyranose (7).
Lactol 7 was prepared from thioglycoside 6[62] (127 mg, 0.201 mmol) following the general procedure A. The crude reaction mixture was purified by flash column chromatography (Hexanes/EtOAc = 5/1 to CH2Cl2/MeOH = 20/1) to give 90.8 mg (0.168 mmol, 84%) of lactol 7 (α/β = 2/1) as white solids. 1H and 13C NMR data of 7 were in agreement with reported ones.[63]
3,4-Di-O-tert-butyldimethylsilyl-2,6-dideoxy-α/β-D-glucopyranose (9).
Lactol 9 was prepared from thioglycoside 8[64] (93.7 mg, 0.2 mmol) following the general procedure B. The crude reaction mixture was purified by flash column chromatography (Hexanes/EtOAc = 35/1) to give 71.5 mg (0.19 mmol, 95%) of lactol 9 (α/β = 1.4/1) as colorless syrup. = +61.5 (c 0.1, CHCl3). 1H NMR (600 MHz, CDCl3) δ 5.28 (d, J = 2.9 Hz, 1H), 4.85 (ddd, J = 8.8, 6.1, 2.2 Hz, 0.6H), 4.01 (ddd, J = 10.5, 7.6, 4.4 Hz, 1H), 3.89 (dq, J = 8.5, 6.5 Hz, 1H), 3.69 (dddd, J = 10.8, 7.8, 4.6, 1.0 Hz, 0.6H), 3.41 (d, J = 6.7 Hz, 0.6H), 3.37 – 3.28 (m, 0.6H), 3.26 – 3.11 (m, 1.6H), 2.81 – 2.60 (m, 1H), 2.24 (ddd, J = 12.8, 4.7, 2.2 Hz, 0.6H), 2.16 – 2.01 (m, 1H), 1.68 (ddd, J = 13.6, 10.2, 3.6 Hz, 1H), 1.62 – 1.52 (m, 0.6H), 1.31 (d, J = 6.4 Hz, 1.8H), 1.26 (d, J = 6.5 Hz, 3H), 0.97 – 0.86 (m, 29H), 0.17 – 0.02 (m, 19H). 13C NMR (150 MHz, CDCl3) δ 93.66, 91.60, 77.90, 77.44, 73.06, 72.86, 70.44, 69.90, 41.82, 39.27, 26.40, 26.37, 26.22, 19.10, 18.78, 18.44, 18.42, 18.22, 18.19, −2.69, −2.79, −2.97, −3.15, −3.81, −3.93, −4.12, −4.26. ESIHRMS calculated for C18H40O4Si2Na [M+Na]+ 399.2363, found 399.2363.
2-O-Methyl-4-O-benzyl-6-deoxy-α/β-D-allopyranose (11).
Lactol 11 was prepared from thioglycoside 10[64] (69.7 mg, 0.194 mmol) following the general procedure A. The crude reaction mixture was purified by flash column chromatography (CH2Cl2/MeOH = 10/1) to give 46.9 mg (0.175 mmol, 90%) of lactol 11 (α/β = 1/1) as colorless syrup. = +128.5 (c 0.1, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.38 – 7.27 (m, 10H), 5.18 (s, 2H), 4.96 (dd, J = 7.9, 3.6 Hz, 1H), 4.68 – 4.64 (m, 2H), 4.55 – 4.51 (m, 2H), 4.45 (m, 1H), 4.39 (t, J = 2.9 Hz, 1H), 4.19 (s, 1H), 4.08 (m, 1H), 3.89 (m, 1H), 3.51 (s, 3H), 3.47 (s, 3H), 3.27 (s, 1H), 3.18 (m, 1H), 3.10 – 3.06 (m, 2H), 2.95 (dd, J = 7.8, 2.9 Hz, 1H), 2.58 (s, 1H), 1.28 (d, J = 6.2 Hz, 3H), 1.26 (d, J = 6.2 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ 137.48, 137.23, 128.70, 128.60, 128.33, 128.14, 128.08, 128.02, 93.68, 91.61, 81.15, 79.87, 79.43, 76.27, 71.52, 71.46, 68.27, 67.53, 65.44, 61.28, 58.16, 56.72, 17.91, 17.76. ESIHRMS calculated for C14H20O5Na [M+Na]+ 291.1208, found 291.1213.
2,3,4-Tri-O-benzyl-6-deoxy-α/β-L-galactopyranose (13).
Lactol 13 was prepared from thioglycoside 12[65] (108.9 mg, 0.201 mmol) following the general procedure A. The crude reaction mixture was purified by flash column chromatography (Hexanes/EtOAc = 2/1) to give 78.7 mg (0.181 mmol, 90%) of lactol 13 (α/β = 2/1) as colorless syrup. 1H and 13C NMR data of 13 were in agreement with reported ones.[66]
2-N-(Benzyloxy)carbonyl-3,4,6-tri-O-benzyl-2-deoxy-2-amino-α-D-mannopyranose (15).
Lactol 15 was prepared from thioglycoside 14 (134.4 mg, 0.199 mmol) following the general procedure A. The crude reaction mixture was purified by flash column chromatography (Hexanes/EtOAc = 3/1 to 1/1) to give 109.4 mg (0.187 mmol, 94%) of lactol 15 (α only) as white solids. = +89.0 (c 0.1, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.40 – 7.28 (m, 18H), 7.16 (dd, J = 7.0, 2.3 Hz, 2H), 5.34 – 5.28 (m, 1H), 5.26 (d, J = 8.8 Hz, 1H), 5.15 (s, 2H), 4.87 (d, J = 10.8 Hz, 1H), 4.79 (d, J = 11.0 Hz, 1H), 4.59 (d, J = 12.2 Hz, 1H), 4.52 (d, J = 11.1 Hz, 1H), 4.47 (d, J = 12.2 Hz, 1H), 4.45 – 4.38 (m, 2H), 4.18 (d, J = 3.9 Hz, 1H), 4.14 (dd, J = 9.4, 4.5 Hz, 1H), 4.06 (ddd, J = 9.9, 4.8, 2.1 Hz, 1H), 3.66 (dd, J = 10.5, 4.8 Hz, 1H), 3.62 (dd, J = 10.4, 2.3 Hz, 1H), 3.56 (t, J = 9.6 Hz, 1H). 13C NMR (150 MHz, CDCl3) δ 156.55, 138.24, 138.04, 137.63, 136.29, 128.63, 128.50, 128.44, 128.40, 128.29, 128.26, 128.24, 128.19, 128.02, 127.90, 127.76, 93.83, 77.47, 75.15, 74.29, 73.44, 71.20, 70.31, 68.85, 67.12, 51.54. ESIHRMS calculated for C35H37NO7Na [M+Na]+ 606.2468, found 606.2469.
2-N-Acetyl-3,4,6-tri-O-benzyl-2-deoxy-2-amino-α/β-D-mannopyranose (17).
Lactol 17 was prepared from thioglycoside 16[67] (116.6 mg, 0.2 mmol) following the general procedure A. The crude reaction mixture was purified by flash column chromatography (CH2Cl2/MeOH=100/1) to give 84.4 mg (0.167 mmol, 84%) of lactol 17 (α/β = 2.5/1) as white solid. 1H and 13C NMR data of 17 were in agreement with reported ones.[68]
Methyl 4,7,8,9-tetra-O-acetyl-3,5-dideoxy-5-acetamido-α/β-D-glycero-D-galacto-2-nonulopyranosonate (19).
Lactol 19 was prepared from thioglycoside 18[69] (118.0 mg, 0.202 mmol) following the general procedure A. The crude reaction mixture was purified by flash column chromatography (Hexanes/Acetone = 3/2) to give 65.0 mg (0.132 mmol, 65%) of lactol 19 (α/β = 1/10) as white solids with 34.2 mg (29%) of thioglycoside 18 recovered. 1H and 13C NMR data of 19 were in agreement with reported ones.[70]
Benzyl (3,4-di-O-benzyl-α/β-D-mannopyranose) uronate (21).
Lactol 21 was prepared from thioglycoside 20 (106.3 mg, 0.191 mmol) following the general procedure A. The crude reaction mixture was purified by flash column chromatography (Hexanes/EtOAc = 2/1 to CH2Cl2/MeOH = 15/1) to give 66.5 mg (0.143 mmol, 75%) of lactol 21 (α/β = 10/1) as white solids. = +35.3 (c 0.1, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.34 – 7.25 (m, 15H), 7.23 – 7.20 (m, 2H), 5.38 (t, J = 4.1 Hz, 1H), 5.17 (d, J = 12.1 Hz, 0.1H), 5.12 (d, J = 12.1 Hz, 1H), 5.04 (d, J = 12.2 Hz, 1H), 4.83 (d, J = 9.8 Hz, 0.1H), 4.71 (d, J = 11.0 Hz, 0.1H), 4.68 (d, J = 11.2 Hz, 1H), 4.66 – 4.61 (m, 0.2H), 4.58 – 4.55 (m, 3H), 4.52 (d, J = 6.8 Hz, 1H), 4.44 (t, J = 10.5 Hz, 0.1H), 4.15 (t, J = 7.0 Hz, 1H), 4.10 (t, J = 8.1 Hz, 0.1H), 4.05 (d, J = 8.0 Hz, 0.1H), 4.00 (s, 0.1H), 3.95 (t, J = 3.7 Hz, 1H), 3.92 (dd, J = 7.1, 3.2 Hz, 1H), 3.65 (d, J = 4.7 Hz, 1H), 2.63 (d, J = 5.4 Hz, 1H). 13C NMR (150 MHz, CDCl3) δ 169.60, 169.43, 137.82, 137.74, 137.62, 137.39, 135.19, 135.03, 128.74, 128.72, 128.70, 128.67, 128.64, 128.54, 128.49, 128.25, 128.11, 127.99, 127.97, 127.91, 127.89, 94.43, 93.82, 79.86, 78.21, 75.42, 75.30, 74.57, 74.33, 74.03, 72.50, 72.42, 72.11, 68.94, 67.93, 67.64, 67.37. ESIHRMS calculated for C27H28O7Na [M+Na]+ 487.1733, found 487.1732.
3,4-Di-O-benzyl-6-O-(methoxyethoxy)methyl-α/β-D-mannopyranose (23).
Lactol 23 was prepared from thioglycoside 22 (109.8 mg, 0.203 mmol) following the general procedure A. The crude reaction mixture was purified by flash column chromatography (Hexanes/EtOAc = 1/1 to CH2Cl2/MeOH = 15/1) to give 74.1 mg (0.165 mmol, 81%) of lactol 23 (α/β = 2.5/1) as colorless syrup. = +91.0 (c 0.1, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.39 – 7.27 (m, 14H), 5.27 (d, J = 1.7 Hz, 1H), 4.92 (d, J = 10.9 Hz, 0.4H), 4.89 (d, J = 11.0 Hz, 1H), 4.77 – 4.66 (m, 6.4H), 4.63 (d, J = 10.9 Hz, 0.4H), 4.59 (d, J = 11.0 Hz, 1H), 4.11 (ddd, J = 9.3, 6.7, 2.1 Hz, 1H), 4.06 (dd, J = 3.3, 1.8 Hz, 2H), 4.01 (dd, J = 3.3, 1.2 Hz, 0.4H), 3.97 (dd, J = 9.1, 3.2 Hz, 1H), 3.87 – 3.59 (m, 8H), 3.56 – 3.49 (m, 3H), 3.43 (ddd, J = 9.8, 4.7, 2.3 Hz, 0.4H), 3.35 (d, J = 5.7 Hz, 4.2H). 13C NMR (150 MHz, CDCl3) δ 138.34, 138.24, 137.94, 137.72, 128.66, 128.63, 128.53, 128.49, 128.12, 128.03, 127.98, 127.96, 127.92, 127.91, 127.82, 96.32, 96.17, 94.21, 93.88, 81.73, 79.84, 75.19, 75.13, 74.92, 74.46, 73.99, 72.04, 71.93, 71.80, 70.59, 68.97, 68.64, 68.33, 67.16, 67.10, 67.08, 59.07, 59.04. ESIHRMS calculated for C27H32O8Na [M+Na]+ 471.1995, found 471.1978.
3-O-Benzyl-4,6-O-[(R)-phenylmethylene]-α/β-D-mannopyranose (25).
Lactol 25 was prepared from thioglycoside 24[71] (91.4 mg, 0.203 mmol) following the general procedure B. The crude reaction mixture was purified by flash column chromatography (Hexanes/Acetone = 2/1 to CH2Cl2/MeOH = 20/1) to give 52.3 mg (0.146 mmol, 72%) of lactol 25 (α/β = 2/1) as colorless syrup. 1H and 13C NMR data of 25 were in agreement with reported ones.[26]
3-O-Benzyl-4,6-O-[(R)-phenylmethylene]-2-deoxy-2-phthalimido-β-D-glucopyranose (27).
Lactol 27 was prepared from thioglycoside 26[72] (113.6 mg, 0.196 mmol) following the general procedure B. The crude reaction mixture was purified by flash column chromatography (Hexanes/Acetone = 4/1 to 1/1) to give 61.2 mg (0.126 mmol, 64%) of lactol 27 (β only) as white solids. 1H and 13C NMR data of 27 were in agreement with reported ones.[73]
3-O-Benzyl-4,6-O-[(R)-phenylmethylene]-2-deoxy-2-azido-α/β-D-mannopyranose (29).
Lactol 29 was prepared from thioglycoside 28[74] (95 mg, 0.2 mmol) following the general procedure B. The crude reaction mixture was purified by flash column chromatography (Hexanes/EtOAc = 7/1) to give 47 mg (0.122 mmol, 61%) of lactol 29 (α/β = 1.4/1) as colorless syrup. = +34.0 (c 0.1, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.49 (ddd, J = 7.3, 3.8, 1.8 Hz, 3H), 7.43 – 7.27 (m, 14H), 5.63 (s, 1H), 5.61 (s, 0.7H), 5.17 (dd, J = 3.4, 1.5 Hz, 1H), 4.95 – 4.90 (m, 1.7H), 4.80 – 4.71 (m, 2.4H), 4.32 (dd, J = 10.5, 4.9 Hz, 0.7H), 4.23 (dd, J = 10.3, 4.9 Hz, 1H), 4.19 (dd, J = 9.7, 3.7 Hz, 1H), 4.13 (t, J = 9.5 Hz, 1H), 4.08 – 4.00 (m, 3.5H), 3.87 (dd, J = 9.6, 3.7 Hz, 0.7H), 3.82 (td, J = 10.3, 2.2 Hz, 1.7H), 3.60 (d, J = 11.7 Hz, 0.7H), 3.36 (td, J = 9.7, 4.9 Hz, 0.7H), 2.78 (d, J = 3.5 Hz, 1H). 13C NMR (150 MHz, CDCl3) δ 138.11, 137.76, 137.47, 137.29, 129.19, 129.11, 128.67, 128.56, 128.41, 128.37, 128.13, 127.90, 127.78, 127.66, 126.17, 126.12, 101.75, 101.67, 94.15, 93.28, 79.28, 78.61, 77.57, 75.29, 73.50, 73.45, 68.83, 68.50, 67.13, 65.09, 64.07, 62.99. ESIHRMS calculated for C20H21N3O5Na [M+Na]+ 406.1379, found 406.1379.
2,6-Di-O-benzoyl-3,4-O-[(1S,2S)-1,2-dimethoxy-1,2-dimethyl-1,2-ethanediyl]-α/β-D-mannopyranose (31).
Lactol 31 was prepared from thioglycoside 30 (119.3 mg, 0.201 mmol) following the general procedure B. The crude reaction mixture was purified by flash column chromatography (Hexanes/EtOAc = 4/1 to 1/1) to give 60.9 mg (0.121 mmol, 60%) of lactol 31 (α/β > 20/1) as colorless syrup with 37.1 mg (31%) of thioglycoside 30 recovered. = +123.5 (c 0.1, CHCl3). 1H NMR (600 MHz, CDCl3) δ 8.08 – 8.02 (m, 4H), 7.60 – 7.52 (m, 2H), 7.38 (q, J = 7.7 Hz, 4H), 5.39 (dd, J = 4.0, 1.6 Hz, 1H), 5.34 (dd, J = 3.0, 1.7 Hz, 1H), 4.62 (dd, J = 12.0, 2.2 Hz, 1H), 4.56 (dd, J = 12.0, 3.5 Hz, 1H), 4.48 (t, J = 10.2 Hz, 1H), 4.38 (dd, J = 10.3, 3.0 Hz, 1H), 4.34 (dt, J = 10.0, 2.8 Hz, 1H), 3.69 (dd, J = 4.0, 1.6 Hz, 1H), 3.29 (s, 3H), 3.19 (s, 3H), 1.29 (s, 3H), 1.24 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 166.59, 166.10, 133.23, 133.21, 130.27, 130.06, 129.97, 129.75, 128.51, 128.49, 100.39, 100.10, 92.98, 71.56, 68.99, 65.86, 63.48, 62.67, 48.28, 48.05, 17.86, 17.76. ESIHRMS calculated for C26H20O10Na [M+Na]+ 525.1737, found 525.1743.
2,3,4,6-Tetra-O-acetyl-α/β-D-glucopyranose (33).
Lactol 33 was prepared from thioglycoside 32[75] (88.6 mg, 0.201 mmol) following the general procedure A. The crude reaction mixture was purified by flash column chromatography (Hexanes/Acetone = 2/1) to give 26.9 mg (0.077 mmol, 38%) of lactol 33 (α/β = 4/1) as colorless syrup with 21.3 mg (24%) of thioglycoside 32 recovered. 1H and 13C NMR data of 33 were in agreement with reported ones.[76]
2,3,6-Tri-O-benzyl-4-O-(2,3,4,6-tetra-O-beznyl-β-D-galactopyranosyl)-α/β-D-glucopyranose (35).
Lactol 35 was prepared from thioglycoside 34[77] (207.0 mg, 0.194 mmol) following the general procedure A. The crude reaction mixture was purified by flash column chromatography (Hexanes/EtOAc = 3/1) to give 163.0 mg (0.167 mmol, 86%) of lactol 35 (α/β = 2/1) as white solids. 1H and 13C NMR data of 35 were in agreement with reported ones.[78]
Acknowledgements
We are grateful to National Science Foundation (CHE-1464787), National Institutes of Health Common Fund Glycosciences Program (U01GM125289), and The University of Toledo for supporting this research.
Funding
National Science Foundation [CHE-1464787]; National Institutes of Health Common Fund Glycosciences Program [U01GM125289].
References
- [1].Koto S; Sato T; Morishima N; Zen S The glucosylation of several alcohols with tetra-O-benzyl-α-D-glucopyranose and a mixture of p-nitrobenzenesulfonyl chloride, silver trifluoromethanesulfonate, and triethylamine. Bull. Chem. Soc. Jpn 1980, 53, 1761–1762. [Google Scholar]
- [2].Susaki H Glycosidation with 1-hydroxy sugars as glycosyl donors promoted by trimethylsilyl chloride and zinc triflate. Chem. Pharm. Bull 1994, 42, 2. [Google Scholar]
- [3].Suda S; Mukaiyama T Stereoselective synthesis of 1,2-trans-ribofuranosides from 1-hydroxy sugars by the use of [1,2-benzenediolato(2-)-O,O’]oxotitanium and trifluoromethanesulfonic anhydride. Chem. Lett 1991, 431–434. [Google Scholar]
- [4].Uchiro H; Mukaiyama T Trityl salt catalyzed stereoselective glycosylation of alcohols with 1-hydroxyribofuranose. Chem. Lett 1996, 79–80. [Google Scholar]
- [5].Inanaga J; Yokoyama Y; Hanamoto T Catalytic O-glycosylation and S-glycosylation of 1-hydroxy sugars. J. Chem. Soc., Chem. Commun 1993, 1090–1091. [Google Scholar]
- [6].Izumi M; Fukase K; Kusumoto S TMSCl as a mild and effective source of acidic catalysis in Fischer glycosidation and use of propargyl glycoside for anomeric protection. Biosci. Biotechnol. Biochem 2002, 66, 211–214. [DOI] [PubMed] [Google Scholar]
- [7].Fischer E Ueber die glucoside der alkohole. Chem. Ber 1893, 26, 2400–2412. [Google Scholar]
- [8].Garcia BA; Poole JL; Gin DY Direct glycosylations with 1-hydroxy glycosyl donors using trifluoromethanesulfonic anhydride and diphenyl sulfoxide. J. Am. Chem. Soc 1997, 119, 7597–7598. [Google Scholar]
- [9].Garcia BA; Gin DY Dehydrative glycosylation with activated diphenyl sulfonium reagents. Scope, mode of C(1)-hemiacetal activation, and detection of reactive glycosyl intermediates. J. Am. Chem. Soc 2000, 122, 4269–4279. [Google Scholar]
- [10].Nguyen HM; Poole JL; Gin DY Chemoselective iterative dehydrative glycosylation. Angew. Chem., Int. Ed 2001, 40, 414–417. [DOI] [PubMed] [Google Scholar]
- [11].Kim YJ; Gin DY Synthesis of the trisaccharide portion of the immunologic adjuvant QS-21A via sulfonium-mediated oxidative and dehydrative glycosylation. Org. Lett 2001, 3, 1801–1804. [DOI] [PubMed] [Google Scholar]
- [12].Nguyen HM; Chen Y; Duron SG; Gin DY Sulfide-mediated dehydrative glycosylation. J. Am. Chem. Soc 2001, 123, 8766–8772. [DOI] [PubMed] [Google Scholar]
- [13].Gin D Dehydrative glycosylation with 1-hydroxy donors. J. Carbohydr. Chem 2002, 21, 645–665. [Google Scholar]
- [14].Boebel TA; Gin DY Probing the mechanism of sulfoxide-catalyzed hemiacetal activation in dehydrative glycosylation. J. Org. Chem 2005, 70, 5818–5826. [DOI] [PubMed] [Google Scholar]
- [15].Nogueira JM; Nguyen SH; Bennett CS Cyclopropenium cation promoted dehydrative glycosylations using 2-deoxy- and 2,6-dideoxy-sugar donors. Org. Lett 2011, 13, 2814–2817. [DOI] [PubMed] [Google Scholar]
- [16].Nogueira JM; Bylsma M; Bright DK; Bennett CS Reagent-controlled α-selective dehydrative glycosylation of 2,6-dideoxy- and 2,3,6-trideoxy sugars. Angew. Chem., Int. Ed 2016, 55, 10088–10092. [DOI] [PubMed] [Google Scholar]
- [17].Schmidt RR; Michel J Direct O-glycosyl trichloroacetimidate formation - nucleophilicity of the anomeric oxygen atom. Tetrahedron Lett 1984, 25, 821–824. [Google Scholar]
- [18].Schmidt RR New methods for the synthesis of glycosides and oligosaccharides—are there alternatives to the Koenigs-Knorr method? Angew. Chem., Int. Ed 1986, 25, 212–235. [Google Scholar]
- [19].Schmidt RR Recent developments in the synthesis of glycoconjugates. Pure Appl. Chem 1989, 61, 1257–1270. [Google Scholar]
- [20].Schmidt RR; Klotz W Glycoside bond formation via anomeric O-alkylation: how many protective groups are required? Synlett 1991, 168–170. [Google Scholar]
- [21].Tsvetkov YE; Klotz W; Schmidt RR Disaccharide synthesis via anomeric O-alkylation. Liebigs Ann. Chem 1992, 371–375. [Google Scholar]
- [22].Schmidt RR The anomeric O-alkylation and the trichloroacetimidate method - versatile strategies for glycoside bond formation. Frontiers in Natural Product Research 1996, 1 (Modern Methods in Carbohydrate Synthesis), 20–54. [Google Scholar]
- [23].Zhu D; Baryal KN; Adhikari S; Zhu J Direct synthesis of 2-deoxy-β-glycosides via anomeric O-alkylation with secondary electrophiles. J. Am. Chem. Soc 2014, 136, 3172–3175. [DOI] [PubMed] [Google Scholar]
- [24].Li X; Woodward J; Hourani A; Zhu D; Ayoub S; Zhu J Synthesis of the 2-deoxy trisaccharide glycal of antitumor antibiotics landomycins A and E. Carbohydr. Res 2016, 430, 54–58. [DOI] [PubMed] [Google Scholar]
- [25].Zhu D; Adhikari S; Baryal KN; Abdullah BN; Zhu J Stereoselective synthesis of α-Digitoxosides and α-Boivinosides via chelation-controlled anomeric O-alkylation. J. Carbohydr. Chem 2014, 33, 438–451. [Google Scholar]
- [26].Nguyen H; Zhu D; Li X; Zhu J Stereoselective construction of β-mannopyranosides by anomeric O-Alkylation: Synthesis of the trisaccharide core of N-linked glycans. Angew. Chem., Int. Ed 2016, 55, 4767–4771. [DOI] [PubMed] [Google Scholar]
- [27].Bhetuwal BR; Woodward J; Li X; Zhu J Stereoselective β-mannosylation via anomeric O-alkylation: Concise synthesis of β-D-Xyl-(l→2)-β-D-Man-(1→4)-α-D-Glc-OMe, a trisaccharide oligomer of the hyriopsis schlegelii glycosphingolipid. J. Carbohydr. Chem 2017, 36, 162–172. [Google Scholar]
- [28].Lubineau A; Escher S; Alais J; Bonnaffé D Stereoselectivity control in anomeric O-alkylation. Application to the synthesis of C2 symmetric glycoconjugates. Tetrahedron Lett. 1997, 38, 4087–4090. [Google Scholar]
- [29].Trewartha G; Burrows JN; Barrett AGM Glycosidation via conjugate addition of anomeric alkoxides to nitroalkenes and nitrosoalkenes. Tetrahedron Lett. 2005, 46, 3553–3556. [Google Scholar]
- [30].Ryan DA; Gin DY Ring-opening of aziridine-2-carboxamides with carbohydrate C1-O-nucleophiles. Stereoselective preparation of α- and β-O-glycosyl serine conjugates. J. Am. Chem. Soc 2008, 130, 15228–15229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Morris WJ; Shair MD Stereoselective synthesis of 2-deoxy-β-glycosides using anomeric O-alkylation/arylation. Org. Lett 2009, 11, 9–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pertel SS; Gorkunenko OA; Kakayan ES; Chirva VJ Synthesis of 1,2-cis- and 1,2-trans-glycosides of 2-acetamido-4,6-O-benzylidene-2-deoxy-D-glucopyranose by anomeric O-alkylation. Carbohydr. Res 2011, 346, 685–688. [DOI] [PubMed] [Google Scholar]
- [33].Li X; Berry N; Saybolt K; Ahmed U; Yuan Y Stereoselective β-mannosylation via anomeric O-alkylation: Formal synthesis of potent calcium signal modulator acremomannolipin A. Tetrahedron Lett. 2017, 58, 2069–2072. [Google Scholar]
- [34].Schmidt RR; Michel J Facile synthesis of α-O-glycosyl and β-O-glycosyl imidates - preparation of glycosides and disaccharides. Angew. Chem., Int. Ed 1980, 19, 731–732. [Google Scholar]
- [35].Yu B; Tao HC Glycosyl trifluoroacetimidates. Part 1: Preparation and application as new glycosyl donors. Tetrahedron Lett. 2001, 42, 2405–2407. [Google Scholar]
- [36].Sim MM; Kondo H; Wong CH Synthesis of dibenzyl glycosyl phosphites using dibenzyl N,N-diethylphosphoramidite as phosphitylating reagent: an effective route to glycosyl phosphates, nucleotides, and glycosides. J. Am. Chem. Soc 1993, 115, 2260–2267. [Google Scholar]
- [37].Garcia BA; Gin DY Synthesis of glycosyl-1-phosphates via dehydrative glycosylation. Org. Lett 2000, 2, 2135–2138. [DOI] [PubMed] [Google Scholar]
- [38].Li Y; Yang Y; Yu B An efficient glycosylation protocol with glycosyl ortho-alkynylbenzoates as donors under the catalysis of Ph3PAuOTf. Tetrahedron Lett. 2008, 49, 3604–3608. [Google Scholar]
- [39].Babu RS; O’Doherty GA A palladium-catalyzed glycosylation reaction: the de novo synthesis of natural and unnatural glycosides. J. Am. Chem. Soc 2003, 125, 12406–12407. [DOI] [PubMed] [Google Scholar]
- [40].Babu RS; Zhou M; O’Doherty GA De novo synthesis of oligosaccharides using a palladium-catalyzed glycosylation reaction. J. Am. Chem. Soc 2004, 126, 3428–3429. [DOI] [PubMed] [Google Scholar]
- [41].Mishra B; Neralkar M; Hotha S Stable alkynyl glycosyl carbonates: Catalytic anomeric activation and synthesis of a tridecasaccharide reminiscent of mycobacterium tuberculosis cell wall lipoarabinomannan. Angew. Chem., Int. Ed 2016, 55, 7786–7791. [DOI] [PubMed] [Google Scholar]
- [42].Yu X; O’Doherty GA De novo synthesis in carbohydrate chemistry: From furans to monosaccharides and oligosaccharides. ACS Symposium Series 2008, 990 (Chemical Glycobiology), 3–28. [Google Scholar]
- [43].Issa JP; Lloyd D; Steliotes E; Bennett CS Reagent controlled β-specific dehydrative glycosylation reactions with 2-deoxy-sugars. Org. Lett 2013, 15, 4170–4173. [DOI] [PubMed] [Google Scholar]
- [44].Issa JP; Bennett CS A reagent-controlled SN2-glycosylation for the direct synthesis of β-linked 2-deoxy-sugars. J. Am. Chem. Soc 2014, 136, 5740–5744. [DOI] [PubMed] [Google Scholar]
- [45].Barrett AGM; Bezuidenhoudt BCB A convenient stereoselective Synthesis of α-glycosyl esters. Heterocycles 1989, 28, 209–212. [Google Scholar]
- [46].Lim D; Fairbanks AJ Selective anomeric acetylation of unprotected sugars in water. Chem. Sci 2017, 8, 1896–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Deng LH; Tsybina P; Gregg KJ; Mosi R; Zandberg WF; Boraston AB; Vocadlo DJ Synthesis of 4-methylumbelliferyl α-D-mannopyranosyl-(1→6)-β-D-mannopyranoside and development of a coupled fluorescent assay for GH125 exo-α−1,6-mannosidases. Biorg. Med. Chem 2013, 21, 4839–4845. [DOI] [PubMed] [Google Scholar]
- [48].El-Badri MH; Willenbring D; Tantillo DJ; Gervay-Hague J Mechanistic studies on the stereoselective formation of β-mannosides from mannosyl iodides using α-deuterium kinetic isotope effects. J. Org. Chem 2007, 72, 4663–4672. [DOI] [PubMed] [Google Scholar]
- [49].Singhamahapatra A; Sahoo L; Paul KJV; Varghese B; Loganathan D Improved synthesis of per-O-acetylated Cl hydroxyglycopyranose and structural study as non-covalent organic framework. Tetrahedron Lett. 2013, 54, 6121–6124. [Google Scholar]
- [50].Pilgrim W; Murphy PV SnCl(4)- and TiCl(4)-catalyzed anomerization of acylated O- and S-glycosides: analysis of factors that lead to higher alpha:beta anomer ratios and reaction rates. J. Org. Chem 2010, 75, 6747–6755. [DOI] [PubMed] [Google Scholar]
- [51].Peng RG; VanNieuwenhze MS Construction of the DEF-ring system of nogalamycin and menogaril via an efficient Suzuki-Miyaura reaction. Tetrahedron Lett. 2017, 58, 2236–2239. [Google Scholar]
- [52].Gomez AM; Company MD; Agocs A; Uriel C; Valverde S; Lopez JC Synthesis of 2,3 : 4,6-di-O-isopropylidene-D-allopyranose from D-glucose. Carbohydr. Res 2005, 340, 1872–1875. [DOI] [PubMed] [Google Scholar]
- [53].Yamamoto K; Yakushiji F; Matsumaru T; Ichikawa S Total Synthesis of Tunicamycin V. Org Lett 2018, 20, 256–259. [DOI] [PubMed] [Google Scholar]
- [54].Singh Y; Wang T; Geringer SA; Stine KJ; Demchenko AV Regenerative glycosylation. J. Org. Chem 2018, 83, 374–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Emmadi M; Khan N; Lykke L; Reppe K; S GP; Lisboa MP; Wienhold SM; Witzenrath M; Pereira CL; Seeberger PHA Streptococcus pneumoniae type 2 oligosaccharide glycoconjugate elicits opsonic antibodies and is protective in an animal model of invasive pneumococcal disease. J. Am. Chem. Soc 2017, 139, 14783–14791. [DOI] [PubMed] [Google Scholar]
- [56].Lu X; Kovac P Chemical synthesis of the galacturonic acid containing pentasaccharide antigen of the O-specific polysaccharide of vibrio cholerae O139 and its five fragments. J. Org. Chem 2016, 81, 6374–6394. [DOI] [PubMed] [Google Scholar]
- [57].Hederos M; Konradsson P Synthesis of the trypanosoma cruzi LPPG heptasaccharyl myo-inositol. J. Am. Chem. Soc 2006, 128, 3414–3419. [DOI] [PubMed] [Google Scholar]
- [58].Paulsen H; Heume M; Nurnberger H Synthesis of branched nonasaccharide sequence of bisected structure of N-glycoproteins. Carbohydr. Res 1990, 200, 127–166. [DOI] [PubMed] [Google Scholar]
- [59].At large scale, bromine may be used directly for this reaction via slow addition.
- [60].Sanapala SR; Kulkarni SS Chemical synthesis of asparagine-linked archaeal N-glycan from methanothermus fervidus. Chem. Eur. J 2014, 20, 3578–3583. [DOI] [PubMed] [Google Scholar]
- [61].Jonke S; Liu K-G; Schmidt, R. R. Solid-phase oligosaccharide synthesis of a small library of N-glycans. Chem. Eur. J 2006, 12, 1274–1290. [DOI] [PubMed] [Google Scholar]
- [62].France RR; Rees NV; Wadhawan JD; Fairbanks AJ; Compton RG Selective activation of glycosyl donors utilising electrochemical techniques: a study of the thermodynamic oxidation potentials of a range of chalcoglycosides. Org. Biomol. Chem 2004, 2, 2188–2194. [DOI] [PubMed] [Google Scholar]
- [63].Zeng J; Vedachalam S; Xiang S; Liu X-W Direct C-glycosylation of organotrifluoroborates with glycosyl fluorides and its application to the total synthesis of (+)-Varitriol. Org. Lett 2011, 13, 42–45. [DOI] [PubMed] [Google Scholar]
- [64].Acharya PP; Baryal KN; Reno CE; Zhu J Synthesis of S-linked trisaccharide glycal of derhodinosylurdamycin A: Discovery of alkyl thiocyanate as an efficient electrophile for stereoselective sulfenylation of 2-deoxy glycosyl lithium. Carbohydr. Res 2017, 448, 103–109. [DOI] [PubMed] [Google Scholar]
- [65].Li C-Y; Liu G-J; Dua W; Zhang Y; Xing G-W A novel O-fucosylation strategy preactivated by (p-Tol)2SO/Tf2O and its application for the synthesis of Lewis blood group antigen Lewisa. Tetrahedron Lett. 2017, 58, 2109–2112. [Google Scholar]
- [66].Matwiejuk M; Thiem J New method for regioselective glycosylation employing saccharide oxyanions. Eur. J. Org. Chem 2011, 5860–5878. [Google Scholar]
- [67].Arihara R; Kakita K; Suzuki N; Nakamura S; Hashimoto S Glycosylation with 2-acetamido-2-deoxyglycosyl donors at a low temperature: Scope of the non-oxazoline method. J. Org. Chem 2015, 80. [DOI] [PubMed] [Google Scholar]
- [68].Ayadi E; Czernecki S; Xie J Convenient preparation of perbenzylated 2-azido and 2-N-acetylamino-2-deoxy-D-hexono-1,5-lactones by oxidation of the corresponding lactols. J. Carbohydr. Chem 1996, 15, 191–199. [Google Scholar]
- [69].Liao GC; Zhou ZF; Guo ZW Synthesis and immunological study of α−2,9-oligosialic acid conjugates as anti-group C meningitis vaccines. Chem. Commun 2015, 51, 9647–9650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Nagorny P; Fasching B; Li X; Chen G; Aussedat B; Danishefsky SJ Toward fully synthetic homogeneous β-human follicle-stimulating hormone (β-hFSH) with a biantennary N-linked dodecasaccharide. synthesis of β-hFSH with chitobiose units at the natural linkage sites. J. Am. Chem. Soc 2009, 131, 5792–5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Ohira S; Yamaguchi Y; Takahashi T; Tanaka H The chemoselective O-glycosylation of alcohols in the presence of a phosphate diester and its application to the synthesis of oligomannosylated phosphatidyl inositols. Tetrahedron 2015, 71, 6602–6611. [Google Scholar]
- [72].Gampe CM; Tsukamoto H; Wang TS; Walker S; Kahne D Modular synthesis of diphospholipid oligosaccharide fragments of the bacterial cell wall and their use to study the mechanism of moenomycin and other antibiotics. Tetrahedron 2011, 67, 9771–9778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Chiara JL; Garcia A; Cristobal-Lumbroso G Ketone-imide versus ketone-oxime reductive cross-coupling promoted by samarium diiodide: new mechanistic insight gained from a failed aminocyclopentitol synthesis. J. Org. Chem 2005, 70, 4142–4151. [DOI] [PubMed] [Google Scholar]
- [74].Litjens REJN; Leeuwenburgh MA; van der Marel GA; van Boom JH A novel approach towards the stereoselective synthesis of 2-azido-2-deoxy-β-D-mannosides. Tetrahedron Lett. 2001, 42, 8693–8696. [Google Scholar]
- [75].D’Angelo KA; Taylor MS Borinic acid catalyzed stereo- and regioselective couplings of glycosyl methanesulfonates. J. Am. Chem. Soc 2016, 138, 11058–11066. [DOI] [PubMed] [Google Scholar]
- [76].Baddeley TC; Harrison WTA; Howie RA; Skakle JMS; Wardell JL Co-crystallised alpha- and beta-anomers of 2,3,4,6-tetra-acetyl-D-glucopyranose, in which the alpha- and beta-Anomers are randomly distributed in the hydrogen-bonded chains of molecules. J. Chem. Crystallogr 2007, 37, 853–858. [Google Scholar]
- [77].McGarvey GJ; LeClair CA; Schmidtmann BA Studies on the stereoselective synthesis of C-allyl glycosides. Org Lett 2008, 10, 4727–4730. [DOI] [PubMed] [Google Scholar]
- [78].Yamanoi T; Misawa N; Matsuda S; Watanabe M Preparation of partially benzylated mono-, di-, and trisaccharides by selective cleavage of the beta-fructofuranosidic linkage in fully benzylated sucrose and sucrose-related oligosaccharides under acidic conditions. Carbohydr. Res 2008, 343, 1366–1372. [DOI] [PubMed] [Google Scholar]