

Abstract
Objectives:
Pneumocystis jirovecii pneumonia (PJP) is an important cause of morbidity and mortality in HIV-positive patients. Polymorphisms in immune genes are increasingly reported to influence susceptibility to fungal infections. We analysed the role of 21 single nucleotide polymorphisms from 19 candidate genes on PJP development in patients from the Swiss HIV Cohort Study.
Design and methods:
The analysis included patients with a nadir CD4+ T-cell count less than 200 cells/μl, divided into a discovery (N = 1645) and a replication (N = 1861) cohort. The associations were analysed by using cumulative incidence curves as well as competing risk regression over 18 years, starting from the estimated date of HIV infection, considering death a competing risk, with censoring at lost follow-up, and assuming the dominant mode of inheritance.
Results:
The minor allele of rs2243250 in IL-4 was associated with the risk of PJP in the discovery cohort (cumulative incidence 0.18 versus 0.12, P = 0.002). This association was replicated in the validation cohort (0.16 versus 0.12, P = 0.02). It was still significant in multivariate models, adjusted for HIV transmission mode, viral load, CD4+ T cells slope, age, antiretroviral therapy, tobacco smoking, hepatitis C virus coinfection, year of cohort entry and PJP prophylaxis (global subhazard ratio 1.42, 95% confidence interval 1.17–1.73, P = 0.0004).
Conclusion:
Our data suggest rs2243250, a single nucleotide polymorphism known to influence IL-4 production, is associated with susceptibility to PJP in HIV-positive patients.
Keywords: genetic susceptibility, HIV infection, immunity, IL-4, Pneumocystis jirovecii pneumonia
Introduction
Pneumocystis jirovecii is an opportunistic fungus causing severe pneumonia in HIV/AIDS and other populations of immunosuppressed patients [1]. It is one of the most common AIDS-defining conditions and an important cause of AIDS-related deaths [2–4]. P. jirovecii pneumonia (PJP) typically manifests among individuals with a CD4+ T-cell count of less than 200/μl, in particular when the HIV viral load is elevated [5]. Other factors such as ethnicity [6] and HIV transmission mode [7] have been reported to alter susceptibility to PJP in some studies, but this was not universally confirmed [8].
Increasing evidence suggests that polymorphisms in host immune genes influence the course of infections due to fungal pathogens. Single nucleodide polymorphisms (SNPs) in genes encoding pattern recognition receptors (PRRs) such as pentraxin 3 [9–11] and Dectin-1 [12–14] are emerging as reliable predictors of the future occurrence of invasive aspergillosis among onco-hematological patients as well as hematopoietic stem cell and solid organ transplant recipients [15,16]. Similarly, polymorphisms in genes encoding cytokines were associated with both invasive aspergillosis (IL1B [17,18]) and candidiasis (TNFα [19], IL-4 [20]). Fewer studies examined the role of immune gene polymorphisms in susceptilbly to PJP or AIDS progression. One study associated low producing mannose binding lectin 2 (MBL2) haplotype [21] with PJP infection in a small cohort of HIV-infected individuals. Other polymorphisms apparently associated with PJP in HIV-positive patients are in fact markers of rapid progression to AIDS [22–24].
In this study, we analysed the role of polymorphisms from 21 candidate genes encoding relevant fungal PRRs and cytokines/chemokines with regards to the predisposition to PJP in the patients from the Swiss HIV Cohort Study (SHCS).
Methods
Study cohort and design
The SHCS (www.shcs.ch) is a prospective observational multicenter cohort of seven Swiss hospitals (Basel, Bern, Geneva, Lausanne, Zurich, Lugano and St. Gallen [25]). More than 20 000 HIV-infected patients have been enrolled in Switzerland since 1988 [25,26]. The clinical stage of the patients was defined according to the 1993 classification system for HIV infection of the Centers for Disease Control and Prevention [27]. Demographic characteristics including age, duration of HIV infection, CD4+ T-cell count nadir, opportunistic infections, HIV maximal viral load and antiretroviral therapy used were extracted from the SHCS clinical database [28]. Written informed consent was obtained from all patients, including consent for the genetic studies. All patients whose CD4+ T-cell count was of less than 200 cells/μl for at least 3 months were selected. Patients were randomly stratified into a discovery group and a validation group at a 1 : 1 ratio. Additional patients who were entered into the cohort after the randomization process were added to the validation group.
Definite and presumptive PJP infections were defined according to standard definitions [29]. Briefly, a definitive diagnosis required the identification of the pathogen from respiratory samples by cytology/microscopy or histology. The presumptive diagnosis was made on a combination of clinical signs/symptoms and radiological findings (http://www.shcs.ch/122–4-cdc-category-c-diagnoses#4.2.1). The CD4+ T-cell loss rate was calculated for each individuals using a linear regression of time on the square root of CD4+ T-cell counts as described elsewhere [30]. Unknown HIV-infection dates were estimated by using a joint back calculation model as described elsewhere [31].
Genotyping
A total of 21 SNPs from 19 genes were selected based on a systematic literature review, including SNPs previously associated with fungal infections. Genomic DNA was extracted from cell pellets or whole blood with use of a MagNA Pure LC DNA Isolation Kit (Roche Applied Science, Munich, Germany) according to the manufacturer's protocols. The SNPs were part of a customized Golden Gate Genotyping Assay (Veracode technology, Illumina) or were genotyped using a Competitive Allele Specific PCR system (KBioscience/LGC Genomics; http://www.lgcgenomics.com). Genotype data were analyzed on a BeadXpress Reader or a KlusterKaller software (KBioscience/LGC Genomics) according to the standard protocols and quality controls [32].
Statistical analysis
Statistical analyses were performed in Stata 15.1 (StataCorp LLC, College Station, Texas, USA). Cumulative incidence of PJP was assessed over a 18 years period starting at the estimated date of the HIV infection with censoring at last follow-up and considering death as a competing event, by using stcrreg implemented in Stata. For simplicity a dominant mode of inheritance was assumed for each SNP and the first episode of PJP was considered. Multivariate analyses were performed by using stcrreg, with adjustment for co-variables possibly associated with PJP, considering a cut-off P value of 0.1 in the univariate analyses. CD4+ T-cell counts were accounted for either by using the CD4+ slope before antiretroviral therapy (as described above) or as a time-varying covariable. Other variables such as hepatitis C virus (HCV) or hepatitis B virus infection, as well as antiretroviral and anti-Pneumocystis carinii pneumonia (PCP) drugs were accounted for either as present/absent at any time during follow-up (e.g demographic tables) or as time-varying covariables (time-dependent analyses). Associations were first analysed among patients from the discovery cohort and, when significant, replicated in the validation cohort. The linkage disequilibrium and Hardy–Weinberg equilibrium (HWE) tests were assessed by using the pwld and hwe softwares implemented in Stata. Bonferroni's correction was used to adjust data for the number of tests included in the models. MBL2 haplotypes were phased using PHASE software version 2.1 (University of Washington, Seattle, Washington, USA).
Results
A total of 3506 Caucasian individuals were included (1645 in the discovery and 1861 in the replication study, Table 1), among whom 470 developed PJP (413 definite and 57 presumptive). Patient characteristics were equally distributed in the discovery and the replication studies, with a mean age of 33 years (range 10–74) at time of cohort entry, a male predominance (77%), a mean CD4+ T-cell nadir count of 90.5 cells/μl (range 0–199) and a mean maximal log viral load of 5.20 copies/ml (range 1–8). HIV infection was acquired by male-male sexual contact in 40%, by heterosexual contact in 31% and by intravenous drug use in 26%.
Table 1.
Demographic characteristic of the patients.
| Discovery, N = 1645 | Replication, N = 1861 | All patients, N = 3506 | |
| Variable | N (%) | N (%) | N (%) |
| Age at cohort entry (mean years; range) | 32.5 (10–73) | 33 (13–74) | 32.8 (10–74) |
| Male sex | 1273 (77) | 1425 (77) | 2698 (77) |
| ART/HAART therapy at any time | 1641 (99) | 1856 (99) | 3495 (99) |
| HIV maximal viral load (mean RNA log10 copies/ml; range)a | 5.20 (2–8) | 5.21 (1–8) | 5.20 (1–8) |
| Nadir CD4+ T-cell count (mean cells/μl; range)b | 89.9 (0–199) | 91.0 (0–199) | 90.5 (0–199) |
| CD4+ slope before ART/HAART initiation (mean; range)c | −2.12 (−7–1) | −2.16 (−6–2) | −2.14 (−7–2) |
| PJPd | 240 (15) | 260 (14) | 500 (14) |
| At presentation | 135 | 162 | 297 |
| During follow-up | 105 | 98 | 203 |
| Type of HIV transmission | |||
| Male–male sexual contact | 681 (41) | 725 (39) | 1406 (40) |
| Heterosexual contact | 462 (28) | 611 (33) | 1071 (31) |
| Intravenous drug user | 446 (27) | 462 (25) | 908 (26) |
| Other/unknown | 58 (4) | 63 (3) | 121 (3) |
| HCV coinfectione | 551 (33) | 616 (33) | 1165 (33) |
| Active HBV infectionf | 64 (4) | 82 (4) | 146 (4) |
| Tobacco smokersg | 985 (60) | 1118 (60) | 2103 (60) |
ART, antiretroviral therapy; HBV, hepatitis b virus; HCV, hepatitis C virus; PJP, Pneumocystis jirovecii pneumonia.
aMean maximal HIV RNA load, was missing in two and seven patients in the discovery and replication cohort, respectively.
bLowest level of a CD4+ T-cell count.
cRate of CD4+ depletion in the absence of HAART, was missing in 24 and 25 patients in the discovery and replication cohort, respectively.
dAmong PJP cases, 202 (84%) were definitive and 38 (16%) presumptive in the discovery cohort and 237 (91%) definitive and 23 (9%) presumptive in the replication cohort.
eReflected by HCV serology.
fHBV serostatus, defined by the presence of HBsAg in the blood.
gAt cohort entry: more than 10 packet unit year.
All the SNPs were at the HWE equilibrium and had minor allele frequencies (MAF) comparable to the ones known for the white population (Supplementary Table S1). In the discovery cohort, associations (P < 0.05) were observed for four polymorphisms in four genes, including rs2243250 in IL-4 [cumulative incidence (CI) 0.18 versus 0.12, P = 0.002, Fig. 1a], rs4252125 in plasminogen (CI 0.11 versus 0.16, P = 0.005), rs16910526 in Dectin-1 (CLEC7A; CI 0.08 versus 0.14, P = 0.01) and rs17886395 in surfactant protein A (CI 0.10 versus 0.15, P = 0.03, Table 2).
Fig. 1.
Cumulative incidence of Pneumocystis jirovecii pneumonia according to IL-4 rs2243250 in the discovery [(a) n = 1426 patients with available genotypes] and replication [(b) n = 1832] studies.
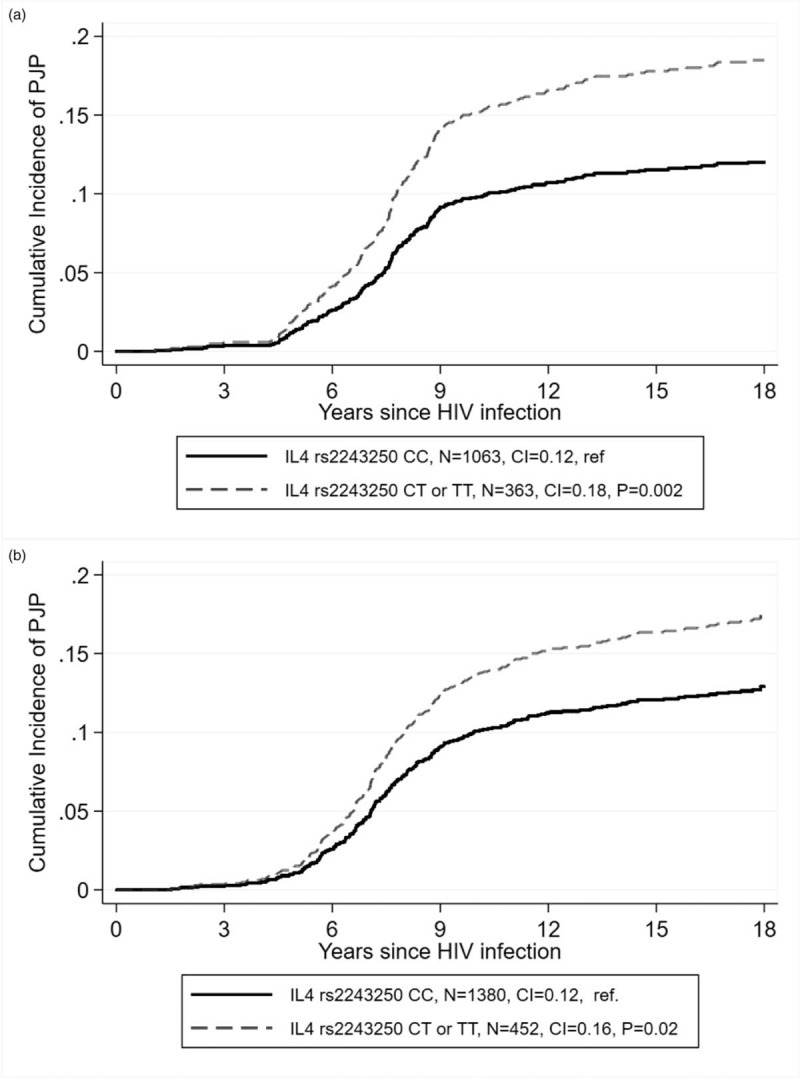
Graphs were performed using the cumulative incidence function in stcurve after competing risk regression with stcrred, considering death as competing risk (Stata).
Table 2.
Cumulative incidence of Pneumocystis jirovecii pneumonia according to candidate gene polymorphisms in HIV-positive patients from Swiss HIV Cohort Study cohort.
| Discovery study, N = 1645 | Replication study, N = 1861 | ||||||||||
| Cum. Incid. | Cum. Incid. | ||||||||||
| Gene | rs number | nt|aa change | MAF | Na | WT | MUT | Pb | Na | WT | MUT | Pb |
| Pattern recognition receptors | |||||||||||
| CLEC7A | rs16910526 | Y238X | 0.08 | 1639 | 0.08 | 0.14 | 0.01 | ||||
| TLR3 | rs3775291 | L412F | 0.29 | 1632 | 0.15 | 0.12 | 0.1 | ||||
| TLR1 | rs5743611 | R80T | 0.08 | 1631 | 0.15 | 0.13 | 0.3 | ||||
| MBL2 | Haplotype | Low MBL | 0.27 | 1548 | 0.11 | 0.14 | 0.3 | ||||
| PTX3 | rs3816527 | A48D | 0.40 | 1531 | 0.13 | 0.14 | 0.5 | ||||
| TLR2 | rs5743708 | R753Q | 0.02 | 918 | 0.12 | 0.14 | 0.7 | ||||
| TLR6 | rs5743810 | S249P | 0.36 | 1623 | 0.14 | 0.13 | 0.8 | ||||
| TLR1 | rs5743604 | S602I | 0.33 | 1622 | 0.13 | 0.13 | 0.8 | ||||
| TLR4 | rs4986790 | D299G | 0.05 | 1623 | 0.13 | 0.13 | 0.9 | ||||
| Cytokines/Chemokines and other genes | |||||||||||
| IL4 | rs224333250c | −590 C/T | 0.14 | 1426 | 0.18 | 0.12 | 0.002d | 1832 | 0.16 | 0.12 | 0.02 |
| PLG | rs4252125 | D472N | 0.31 | 1632 | 0.11 | 0.16 | 0.005e | 1839 | 0.14 | 0.12 | 0.2 |
| SPA2 | rs17886395 | A91P | 0.14 | 1590 | 0.10 | 0.15 | 0.03 | ||||
| IL1A | rs1800587 | −889 C/T | 0.28 | 1598 | 0.15 | 0.12 | 0.05 | ||||
| TNFα | rs1800629 | −308 G/A | 0.13 | 1465 | 0.11 | 0.14 | 0.08 | ||||
| IL1B | rs1143627 | −31 T/C | 0.34 | 1633 | 0.12 | 0.15 | 0.1 | ||||
| IL4RA | rs1805015 | S503P | 0.15 | 1624 | 0.15 | 0.12 | 0.1 | ||||
| IL19 | rs1800896 | −1082 A/G | 0.44 | 1634 | 0.13 | 0.14 | 0.5 | ||||
| CXCL10 | rs3921 | 1642 G/C | 0.43 | 1634 | 0.13 | 0.14 | 0.6 | ||||
| DEFB1 | rs1800972 | −44 C/G | 0.19 | 1612 | 0.14 | 0.13 | 0.7 | ||||
| FCGR2A | rs1801274 | R131H | 0.48 | 1614 | 0.14 | 0.13 | 0.9 | ||||
CI, confidence interval; CLEC7A, C-type lectin domain 7, also known as Dectin-1; CXCL10, CXC-chemokine ligand-10; DEFB1, human beta-defensin 1; FCGR2A, Fc Fragment of IgG receptor IIa; HR, hazard ratio; IL, interleukin; IL4RA, IL4 receptor subunit alpha; LD, linkage disequilibrium; MAF, minor allele frequency; MBL2, mannose binding lectin 2; PJP, Pneumocystis jirovecii pneumonia; PLG, plasminogen; PTX3, pentraxin 3; SHCS, Swiss HIV cohort study; SPA2, surfactant protein A2; TLR, Toll-like receptor; WT, wild type.
aN stands for the number of available genotypes for each SNP (after quality testing).
bAssocaitions were analysed by using stcrreg, considering dominant mode of inheritance (patients homo- and heterozygous for the rare allele are compared to the others).
cBecause some genotypes were missing, the association was also run for rs2070874, which is in strong LD with rs2243250 (R2 = 0.96). The P value for rs2243250 was 0.0008.
dP = 0.047 and P = 0.016 for rs2243250 and rs2070874, respectively, after Bonferroni correction (21 tests).
eP = 0.099 after Bonferroni correction (21 tests).
Among those, only one association was significant after Bonferroni correction for multiple testing (21 tests, rs2243250 in IL-4). This association was also significant in the replication cohort (CI 0.16 versus 0.12, P = 0.02; Fig. 1b). Furthermore, the association was still significant in a multivariable regression model in both the discovery (subhazard ratio, SHR = 1.43, 95% confidence interval 1.07–1.92, P = 0.02) and replication (SHR = 1.42, 95% confidence interval 1.08–1.85, P = 0.01, Table 3) studies. In the combined cohorts after adjustment for the maximal HIV viral load, antiretroviral therapy, CD4+ slope, age at estimated time of HIV infection, PJP prophylaxis, tobacco use, HCV coinfection, period of cohort entry as well as the mode of HIV transmission, the association was more significant (SHR = 1.42, 95% confidence interval 1.17–1.73, P = 0.0004). The association between PJP and rs2243250 were significant when the presumptive PJP cases were removed from the model (SHR = 1.36, 95% confidence interval 1.10–1.68, P = 0.004), and when CD4+ T cells were accounted for as a time-dependent covariates instead of a slope (SHR = 1.41, 95% confidence interval 1.14–1.75, P = 0.00016, Supplementary Table 3).
Table 3.
Multivariate analysis of factors associated with Pneumocystis jirovecii pneumonia.
| Discovery studya, N = 1424 | Replication studya, N = 1825 | All patientsa, N = 3210 | |||||||
| SHR | 95% CI | Pa | SHR | 95% CI | Pa | SHR | 95% CI | Pa | |
| Ageb | 1.00 | 0.99–1.02 | 0.8 | 1.01 | 1.00–1.02 | 0.06 | 1.01 | 1.00–1.02 | 0.1 |
| Male sex | 0.78 | 0.53–1.15 | 0.2 | 0.95 | 0.69–1.30 | 0.7 | 0.87 | 0.68–1.11 | 0.3 |
| CD4+ slopec | 0.17 | 0.11–0.26 | <0.0001 | 0.16 | 0.11–0.24 | <0.0001 | 0.17 | 0.13–0.22 | <0.0001 |
| Maximal HIV RNA (log copies/ml) | 1.37 | 1.11–1.69 | 0.003 | 1.67 | 1.38–2.01 | <0.0001 | 1.53 | 1.33–1.76 | <0.0001 |
| Type of HIV transmission | |||||||||
| MSM | Ref. | Ref. | Ref. | ||||||
| Heterosexual | 1.22 | 0.87–1.71 | 0.2 | 0.98 | 0.72–1.34 | 0.9 | 1.09 | 0.87–1.37 | 0.5 |
| Intravenous drug use | 1.00 | 0.61–1.63 | 1.0 | 0.72 | 0.49–1.04 | 0.08 | 0.85 | 0.63–1.14 | 0.3 |
| Other | 1.64 | 0.92–2.91 | 0.09 | 1.27 | 0.77–2.11 | 0.3 | 1.39 | 0.96–2.03 | 0.09 |
| Cohort entry (years) | |||||||||
| <1995 | Ref. | Ref. | Ref. | ||||||
| 1995–2000 | 0.98 | 0.70–1.38 | 0.9 | 0.83 | 0.60–1.14 | 0.3 | 0.90 | 0.71–1.13 | 0.4 |
| 2001–2005 | 0.77 | 0.49–1.20 | 0.2 | 0.92 | 0.66–1.27 | 0.6 | 0.90 | 0.70–1.15 | 0.4 |
| >2005 | 0.32 | 0.04–2.63 | 0.3 | 0.20 | 0.06–0.69 | 0.01 | 0.25 | 0.09–0.69 | 0.008 |
| PJP prophylaxisd | 0.44 | 0.32–0.60 | <0.0001 | 0.31 | 0.24–0.40 | <0.0001 | 0.36 | 0.30–0.44 | <0.0001 |
| ART/HAARTe | 0.79 | 0.75–0.83 | <0.0001 | 0.75 | 0.71–0.80 | <0.0001 | 0.77 | 0.74–0.80 | <0.0001 |
| HCV coinfectione | 0.93 | 0.87–1.00 | 0.05 | 0.94 | 0.89–0.99 | 0.01 | 0.93 | 0.90–0.97 | 0.001 |
| Tobacco smokingf | 0.77 | 0.57–1.03 | 0.08 | 0.89 | 0.68–1.15 | 0.4 | 0.83 | 0.68–1.01 | 0.06 |
| IL-4 rs2243250 TT/TC versus CCg | 1.43 | 1.07–1.92 | 0.02 | 1.42 | 1.08–1.85 | 0.01 | 1.42 | 1.17–1.73 | 0.0004 |
ART, antiretroviral treatment; CI, confidence interval; HCV, hepatitis C virus; OR, odds ratio; PJP, Pneumocystis jirovecii pneumonia; SHR, subhazard ratio (competing risk regression).
aVariables potentially associated with Pneumocystis carinii pneumonia (cut-off P < 0.1 by univariate testing, Supplemental Table 2) were entered into the multivariate analysis, with age and sex forced into the model. The number of patients is slightly lower than the number of patients included in the studies because some covariables are missing for some patients (refer to Table 1 for details).
bAt estimated HIV infection date (refer to Methods section); SHR is calculated per 1 additional year of age.
cRate of CD4+ depletion before HAART (refer to Methods section); Note: similar results were found when CD4+ were accounted for as a time-dependent covariates (refer to Supplemental Table 3).
dAt any time during follow-up.
eTime-dependent covariates.
fAt cohort entry: more than 10 U packet-year.
gGenetic associations are for the dominant mode of inheritance (patients homozygous and heterozygous for the rare allele are compared with the other). Because some genotypes were missing for rs2243250 in the discovery study, the association was also run for rs2070874, which is in strong LD with rs2243250 (R2 = 0.96): OR = 1.37, 95% CI 1.05–1.80, P = 0.02 (model including 1629 patients).
The association with rs4252125 in plasminogen tended to be associated after corrections for multiple tests (21 tests, P = 0.1) but was not replicated.
Discussion
In this study, we show for the first time an association between a SNP in the IL-4 gene and susceptibility to PJP. This association discovered in a study of 1645 patients was validated in a replication cohort of 1861 individuals. It was still present in multivariate analyses accounting for potential confounding factors such as CD4+ T-cell decline over time. It is further supported by several lines of evidence for a key role of IL-4, a cytokine, in the adaptive immune responses against P. jirovecii.
The IL-4 gene located on chromosome 5q31.1 encodes IL-4, a polyfunctional cytokine produced by activated T cells, type 2 innate lymphoid cells and mast cells, which is involved in adaptive immunity [33]. Its biological activity is mediated through a heterodimeric structured receptor (IL-4R) consisting of IL-4Rα together with either a γ chain (type1 receptor) or a IL13R-α-1 (type2 receptor) molecule (reviewed in [34,35]). IL-4 promotes the differentiation of CD4+ T cells into the Th2 phenotype (also mediated by IL-13 and IL-10), leading to B-cell activation and production of neutralizing antibodies such as IgE and IgG1 [34]. It also counterbalances the Th1 phenotype (mediated by IFNγ and TNFα) and subsequent activation of cell-mediated immunity and phagocytic activity [36].
A number of studies have shown that immunity against Pneumocystis spp. is mediated by both Th1 and Th2 responses [37]. Inhibition of the Th1 response by using anti-TNFα antibodies induced decreased [36] or delayed [38] pathogen clearance in two different mice models of PCP. Reversely, stimulation of Th1 responses by using an adenoviral vector encoding IFNγ protected T cells depleted mice from PCP [39] and recombinant IFNγ increased survival in a rat model of PCP. Inhibition of B cells in mice by using antibodies targeting CD20 also leads to increased susceptibility for PJP [40]. The risk of Pneumocystis spp. infections in humans is increased in patients with primary immune deficiencies, such as X-linked hyper-IgM syndrome [41], as well as in patients treated with monoclonal antibodies against the CD20+ antigen on B cells (rituximab or obinutuzumab [42,43]), the CD52 antigen on B and T cells (alemtuzumab [44]), or with Bruton's tyrosine kinase inhibitor (ibrutinib [45]).
Several studies suggested that the presence of the -590T allele in rs2243250 is associated with increased serum or plasma IL-4 levels [46–49], although this was not universally confirmed [50,51]. Higher IL-4 gene expression may result from a new binding site for nuclear factor of activated T cells, the main transcription factor for the IL-4 expression, at the nucleotide position -590 (Supplemental Fig. S2) [52]. Conversely, the -590T allele was associated with reduced IFNγ and TNFα expression and/or production by human immune cells stimulated with phorbol myristate acetate/Ionomycin (including neutrophils, monocytes and lymphocytes), suggesting that higher IL-4 production could counteract Th1 responses, leading to decreased Pneumocystis spp. clearance [46].
Altogether, this data suggest that increased IL-4 levels in -590T allele carriers result in increased susceptibility to infections mainly as a results from reduced Th1 responses, and that this defect cannot be adequately compensated by a concomittant or subsequent increase in Th2 responses. Consistent with this hypothesis, the -590T allele was associated with an increased risk of vulvo-vaginal candidiasis, as well as increased vaginal IL-4 levels, in a cohort of 85 Latvian women [53] and a higher risk of paracoccidioidomycosis in a cohort of 81 Brazilian individuals [51]. In a cohort of adult leukemia patients, the -590T allele was protective for hepatosplenic candidiasis, as a possible result of diminished immune reconstitution after neutropenia [20]. In addition, numerous studies associated the -590T variant with susceptibility to pathogens other than fungi, such as respiratory syncytial virus (RSV) [54–57], Plasmodium falciparum[58], Brucella spp. [59], Clostridium difficile[60] and bacteria causing periodontitis [61–65].
Also consistent with this hypothesis, animal studies showed that IL-4 deficiency is associated with protection against fungal, mycobacterial and parasitic infections. In a cyclophosphamide-induced mice model of invasive aspergillosis, mice deficient in IL-4 had increased survival [66] and increased broncho-alveolar lavage IFNγ levels, compared to WT mice. In a mouse model of tuberculosis, IL-4-deficient mice had decreased disease severity and increased TNFα lung expression compared to WT mice [67]. In a mouse model of RSV infection, overexpression of IL-4 was associated with decreased viral clearance and neutralization of IL-4 with a reduced illness score [68,69]. In a murine model of Leishmania major infection, parasite clearance was positively correlated with the production of IFNγ (Th1) and negatively correlated with that of IL-4, IL-5 and IL-13 (Th2) [70].
Like other genetic association studies, our study has some limitations. The date of HIV-1 infection was estimated by using a joint back calculation model in seroprevalent patients [31]. Although it is by far the largest association study for PJP infection, our study may have failed to detect associations with rare variants, such as those in Dectin-1 (MAF = 0.08), Toll-like receptor 1 (TLR1) (MAF = 0.08) or TLR4 (MAF = 0.05), which have been associated with susceptibility to infections due to other fungi. Our study did not replicate a previously reported association with MBL2 low expression haplotypes [21], despite reasonable power to do so (>80% power to detect an association with hazard ratio = 1.5; Supplemental Table S1). Despite substantial evidence for a role for rs2243250 on IL-4 production, baseline IL-4 levels have not been measured in study patients to further support genetic associations. In addition, while the SHCS is a well established longitudinal cohort with robust follow-up, patients management strategies including prophylaxis and antiretroviral treatment have been evolving over year. Yet, despite the limitations, association with IL-4 SNP was still significant in multivariate models accounting for prophylaxis and different periods of cohort entry.
In conclusion, this data demonstrates an association between PJP and the presence of the interleukin-4-590T/C polymorphism in a large cohort of HIV patients. This SNP may influence the Th2/Th1 responses required for appropriate immunity against Pneumocystis spp. and increase susceptibility to infection in HIV-positive patients with low level of CD4+ T cells.
Acknowledgements
We thank all the study nurses and SHCS members who were engaged in the data collection and provided care for the patients as well as technical assistants and all the other laboratory members that were in charge for sample shipment and DNA extraction.
Members of the Swiss HIV Cohort Study: Anagnostopoulos A, Battegay M, Bernasconi E, Böni J, Braun DL, Bucher HC, Calmy A, Cavassini M, Ciuffi A, Dollenmaier G, Egger M, Elzi L, Fehr J, Fellay J, Furrer H (Chairman of the Clinical and Laboratory Committee), Fux CA, Günthard HF (President of the SHCS), Haerry D (deputy of ‘Positive Council’), Hasse B, Hirsch HH, Hoffmann M, Hösli I, Huber M, Kahlert C, Kaiser L, Keiser O, Klimkait T, Kouyos RD, Kovari H, Ledergerber B, Martinetti G, Martinez de Tejada B, Marzolini C, Metzner KJ, Müller N, Nicca D, Paioni P, Pantaleo G, Perreau M, Rauch A (Chairman of the Scientific Board), Rudin C (Chairman of the Mother & Child Substudy), Scherrer AU (Head of Data Centre), Schmid P, Speck R, Stöckle M, Tarr P, Trkola A, Vernazza P, Wandeler G, Weber R, Yerly S.
The study has been financed within the framework of the Swiss HIV Cohort Study, supported by the Swiss National Science Foundation (grant no. 177499), by SHCS project no. 803 and by the SHCS research Foundation. This work was supported by research funding from the Leenaards Foundation, the Santos-Suarez Foundation and a Mérieux Research Grant (MRG). PYB is recipient of grants from the Swiss National Science Foundation (32003B-127613, 320030–144054 and 33IC30_179636) and the European Union's Seventh Framework Program (FP7/2007–2013) under grant agreement no. HEALTH-2010–260338 (ALLFUN).
Conflicts of interest
There are no conflicts of interest.
Supplementary Material
Footnotes
Matthias Cavassini and Pierre-Yves Bochud contributed equally to the article.
Members of the Swiss HIV Cohort Study are indicated in Acknowledgments section.
Contributor Information
Collaborators: the Swiss HIV Cohort Study
References
- 1.Thomas CF, Jr, Limper AH. Pneumocystis pneumonia. N Engl J Med 2004; 350:2487–2498. [DOI] [PubMed] [Google Scholar]
- 2.Llibre JM, Revollo B, Vanegas S, Lopez-Nunez JJ, Ornelas A, Marin JM, et al. Pneumocystis jirovecii pneumonia in HIV-1-infected patients in the late-HAART era in developed countries. Scand J Infect Dis 2013; 45:635–644. [DOI] [PubMed] [Google Scholar]
- 3.Siegel M, Masur H, Kovacs J. Pneumocystis jirovecii pneumonia in human immunodeficiency virus infection. Semin Respir Crit Care Med 2016; 37:243–256. [DOI] [PubMed] [Google Scholar]
- 4.Lee SH, Kim KH, Lee SG, Chen DH, Jung DS, Moon CS, et al. Trends of mortality and cause of death among HIV-infected patients in Korea, 1990–2011. J Korean Med Sci 2013; 28:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mocroft A, Reiss P, Kirk O, Mussini C, Girardi E, Morlat P, et al. Opportunistic Infections Project Team of the Collaboration of Observational HIV Epidemiological Research in Europe (COHERE). Is it safe to discontinue primary Pneumocystis jirovecii pneumonia prophylaxis in patients with virologically suppressed HIV infection and a CD4 cell count <200 cells/microL?. Clin Infect Dis 2010; 51:611–619. [DOI] [PubMed] [Google Scholar]
- 6.Stansell JD, Osmond DH, Charlebois E, LaVange L, Wallace JM, Alexander BV, et al. Predictors of Pneumocystis carinii pneumonia in HIV-infected persons. Pulmonary complications of HIV Infection Study Group. Am J Respir Crit Care Med 1997; 155:60–66. [DOI] [PubMed] [Google Scholar]
- 7.Kaplan JE, Hanson DL, Navin TR, Jones JL. Risk factors for primary Pneumocystis carinii pneumonia in human immunodeficiency virus-infected adolescents and adults in the United States: reassessment of indications for chemoprophylaxis. J Infect Dis 1998; 178:1126–1132. [DOI] [PubMed] [Google Scholar]
- 8.Morris A, Lundgren JD, Masur H, Walzer PD, Hanson DL, Frederick T, et al. Current epidemiology of Pneumocystis pneumonia. Emerg Infect Dis 2004; 10:1713–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wojtowicz A, Lecompte TD, Bibert S, Manuel O, Rueger S, Berger C, et al. PTX3 polymorphisms and invasive mold infections after solid organ transplant. Clin Infect Dis 2015; 61:619–622. [DOI] [PubMed] [Google Scholar]
- 10.Cunha C, Aversa F, Lacerda JF, Busca A, Kurzai O, Grube M, et al. Genetic PTX3 deficiency and aspergillosis in stem-cell transplantation. N Engl J Med 2014; 370:421–432. [DOI] [PubMed] [Google Scholar]
- 11.Brunel AS, Wojtowicz A, Lamoth F, Spertini O, Neofytos D, Calandra T, et al. Pentraxin-3 polymorphisms and invasive mold infections in acute leukemia patients with intensive chemotherapy. Haematologica 2018; 103:e527–e530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cunha C, Di Ianni M, Bozza S, Giovannini G, Zagarella S, Zelante T, et al. Dectin-1 Y238X polymorphism associates with susceptibility to invasive aspergillosis in hematopoietic transplantation through impairment of both recipient- and donor-dependent mechanisms of antifungal immunity. Blood 2010; 116:5394–5402. [DOI] [PubMed] [Google Scholar]
- 13.Chai LY, de Boer MG, van der Velden WJ, Plantinga TS, van Spriel AB, Jacobs C, et al. The Y238X stop codon polymorphism in the human beta-glucan receptor dectin-1 and susceptibility to invasive aspergillosis. J Infect Dis 2011; 203:736–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sainz J, Lupianez CB, Segura-Catena J, Vazquez L, Rios R, Oyonarte S, et al. Dectin-1 and DC-SIGN polymorphisms associated with invasive pulmonary aspergillosis infection. PLoS One 2012; 7:e32273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wojtowicz A, Bochud PY. Host genetics of invasive Aspergillus and Candida infections. Semin Immunopathol 2015; 37:173–186. [DOI] [PubMed] [Google Scholar]
- 16.Wojtowicz A, Bochud PY. Risk stratification and immunogenetic risk for infections following stem cell transplantation. Virulence 2016; 7:917–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wojtowicz A, Gresnigt MS, Lecompte T, Bibert S, Manuel O, Joosten LA, et al. IL1B and DEFB1 polymorphisms increase susceptibility to invasive mold infection after solid-organ transplantation. J Infect Dis 2015; 211:1646–1657. [DOI] [PubMed] [Google Scholar]
- 18.Sainz J, Perez E, Gomez-Lopera S, Jurado M. IL1 gene cluster polymorphisms and its haplotypes may predict the risk to develop invasive pulmonary aspergillosis and modulate C-reactive protein level. J Clin Immunol 2008; 28:473–485. [DOI] [PubMed] [Google Scholar]
- 19.Wojtowicz A, Tissot F, Lamoth F, Orasch C, Eggimann P, Siegemund M, et al. Polymorphisms in tumor necrosis factor-alpha increase susceptibility to intra-abdominal Candida infection in high-risk surgical ICU patients. Crit Care Med 2014; 42:e304–e308. [DOI] [PubMed] [Google Scholar]
- 20.Choi EH, Foster CB, Taylor JG, Erichsen HC, Chen RA, Walsh TJ, et al. Association between chronic disseminated candidiasis in adult acute leukemia and common IL4 promoter haplotypes. J Infect Dis 2003; 187:1153–1156. [DOI] [PubMed] [Google Scholar]
- 21.Yanagisawa K, Ogawa Y, Uchiumi H, Gohda F, Mawatari M, Ishizaki T, et al. Gene polymorphisms of mannose-binding lectin confer susceptibility to Pneumocystis pneumonia in HIV-infected patients. J Infect Chemother 2015; 21:769–775. [DOI] [PubMed] [Google Scholar]
- 22.An P, Li R, Wang JM, Yoshimura T, Takahashi M, Samudralal R, et al. Role of exonic variation in chemokine receptor genes on AIDS: CCRL2 F167Y association with pneumocystis pneumonia. PLoS Genet 2011; 7:e1002328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.An P, Penugonda S, Thorball CW, Bartha I, Goedert JJ, Donfield S, et al. Role of APOBEC3F gene variation in HIV-1 disease progression and pneumocystis pneumonia. PLoS Genet 2016; 12:e1005921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forthal DN, Landucci G, Bream J, Jacobson LP, Phan TB, Montoya B. FcgammaRIIa genotype predicts progression of HIV infection. J Immunol 2007; 179:7916–7923. [DOI] [PubMed] [Google Scholar]
- 25.Ledergerber B, von Overbeck J, Egger M, Luthy R. The Swiss HIV Cohort Study: rationale, organization and selected baseline characteristics. Soz Praventivmed 1994; 39:387–394. [DOI] [PubMed] [Google Scholar]
- 26.Sudre P, Rickenbach M, Taffe P, Janin P, Volkart AC, Francioli P, et al. Clinical epidemiology and research on HIV infection in Switzerland: the Swiss HIV Cohort Study 1988–2000. Schweiz Med Wochenschr 2000; 130:1493–1500. [PubMed] [Google Scholar]
- 27.Centers for Disease Control and Prevention. 1993 Classification system for HIV infection and expanded surveillance case definition for acquired immunodeficiency syndrome (AIDS) among adolescents and adults. MMWR Morb Mortal Wkly Rep 1992; 41:1–19. [PubMed] [Google Scholar]
- 28.Zellweger C, Opravil M, Bernasconi E, Cavassini M, Bucher HC, Schiffer V, et al. Long-term safety of discontinuation of secondary prophylaxis against Pneumocystis pneumonia: prospective multicentre study. AIDS 2004; 18:2047–2053. [DOI] [PubMed] [Google Scholar]
- 29.Ebner L, Walti LN, Rauch A, Furrer H, Cusini A, Meyer AM, et al. Clinical course, radiological manifestations, and outcome of Pneumocystis jirovecii pneumonia in HIV patients and renal transplant recipients. PLoS One 2016; 11:e0164320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bochud PY, Hersberger M, Taffe P, Bochud M, Stein CM, Rodrigues SD, et al. Polymorphisms in Toll-like receptor 9 influence the clinical course of HIV-1 infection. AIDS 2007; 21:441–446. [DOI] [PubMed] [Google Scholar]
- 31.Taffe P, May M. Swiss HIV Cohort Study. A joint back calculation model for the imputation of the date of HIV infection in a prevalent cohort. Stat Med 2008; 27:4835–4853. [DOI] [PubMed] [Google Scholar]
- 32.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005; 21:263–265. [DOI] [PubMed] [Google Scholar]
- 33.Arai N, Nomura D, Villaret D, DeWaal Malefijt R, Seiki M, Yoshida M, et al. Complete nucleotide sequence of the chromosomal gene for human IL-4 and its expression. J Immunol 1989; 142:274–282. [PubMed] [Google Scholar]
- 34.Paul WE. History of interleukin-4. Cytokine 2015; 75:3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol 1999; 17:701–738. [DOI] [PubMed] [Google Scholar]
- 36.Chen W, Havell EA, Harmsen AG. Importance of endogenous tumor necrosis factor alpha and gamma interferon in host resistance against Pneumocystis carinii infection. Infect Immun 1992; 60:1279–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steele C, Shellito JE, Kolls JK. Immunity against the opportunistic fungal pathogen Pneumocystis. Med Mycol 2005; 43:1–19. [DOI] [PubMed] [Google Scholar]
- 38.Kolls JK, Lei D, Vazquez C, Odom G, Summer WR, Nelson S, et al. Exacerbation of murine Pneumocystis carinii infection by adenoviral-mediated gene transfer of a TNF inhibitor. Am J Respir Cell Mol Biol 1997; 16:112–118. [DOI] [PubMed] [Google Scholar]
- 39.Kolls JK, Habetz S, Shean MK, Vazquez C, Brown JA, Lei D, et al. IFN-gamma and CD8+ T cells restore host defenses against Pneumocystis carinii in mice depleted of CD4+ T cells. J Immunol 1999; 162:2890–2894. [PubMed] [Google Scholar]
- 40.Elsegeiny W, Eddens T, Chen K, Kolls JK. Anti-CD20 antibody therapy and susceptibility to Pneumocystis pneumonia. Infect Immun 2015; 83:2043–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Milledge J, Kakakios A, Gillis J, Fitzgerald DA. Pneumocystis carinii pneumonia as a presenting feature of X-linked hyper-IgM syndrome. J Paediatr Child Health 2003; 39:704–706. [DOI] [PubMed] [Google Scholar]
- 42.Martin-Garrido I, Carmona EM, Specks U, Limper AH. Pneumocystis pneumonia in patients treated with rituximab. Chest 2013; 144:258–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Venhuizen AC, Hustinx WN, van Houte AJ, Veth G, van der Griend R. Three cases of Pneumocystis jirovecii pneumonia (PCP) during first-line treatment with rituximab in combination with CHOP-14 for aggressive B-cell non-Hodgkin's lymphoma. Eur J Haematol 2008; 80:275–276. [DOI] [PubMed] [Google Scholar]
- 44.Kim SJ, Moon JH, Kim H, Kim JS, Hwang YY, Intragumtornchai T, et al. Nonbacterial infections in Asian patients treated with alemtuzumab: a retrospective study of the Asian Lymphoma Study Group. Leuk Lymphoma 2012; 53:1515–1524. [DOI] [PubMed] [Google Scholar]
- 45.Ahn IE, Jerussi T, Farooqui M, Tian X, Wiestner A, Gea-Banacloche J. Atypical Pneumocystis jirovecii pneumonia in previously untreated patients with CLL on single-agent ibrutinib. Blood 2016; 128:1940–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anovazzi G, Medeiros MC, Pigossi SC, Finoti LS, Souza Moreira TM, Mayer MP, et al. Functionality and opposite roles of two interleukin 4 haplotypes in immune cells. Genes Immun 2017; 18:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Imran M, Laddha NC, Dwivedi M, Mansuri MS, Singh J, Rani R, et al. Interleukin-4 genetic variants correlate with its transcript and protein levels in patients with vitiligo. Br J Dermatol 2012; 167:314–323. [DOI] [PubMed] [Google Scholar]
- 48.Li J, Lin LH, Wang J, Peng X, Dai HR, Xiao H, et al. Interleukin-4 and interleukin-13 pathway genetics affect disease susceptibility, serum immunoglobulin E levels, and gene expression in asthma. Ann Allergy Asthma Immunol 2014; 113:173–179.e1. [DOI] [PubMed] [Google Scholar]
- 49.Akkad DA, Arning L, Ibrahim SM, Epplen JT. Sex specifically associated promoter polymorphism in multiple sclerosis affects interleukin 4 expression levels. Genes Immun 2007; 8:703–706. [DOI] [PubMed] [Google Scholar]
- 50.Hussein YM, El-Shal AS, Rezk NA, Abdel Galil SM, Alzahrani SS. Influence of interleukin-4 gene polymorphisms and interleukin-4 serum level on susceptibility and severity of rheumatoid arthritis in Egyptian population. Cytokine 2013; 61:849–855. [DOI] [PubMed] [Google Scholar]
- 51.Bozzi A, Reis BS, Pereira PP, Pedroso EP, Goes AM. Interferon-gamma and interleukin-4 single nucleotide gene polymorphisms in paracoccidioidomycosis. Cytokine 2009; 48:212–217. [DOI] [PubMed] [Google Scholar]
- 52.Kim BS, Park SM, Uhm TG, Kang JH, Park JS, Jang AS, et al. Effect of single nucleotide polymorphisms within the interleukin-4 promoter on aspirin intolerance in asthmatics and interleukin-4 promoter activity. Pharmacogenet Genomics 2010; 20:748–758. [DOI] [PubMed] [Google Scholar]
- 53.Babula O, Lazdane G, Kroica J, Linhares IM, Ledger WJ, Witkin SS. Frequency of interleukin-4 (IL-4) -589 gene polymorphism and vaginal concentrations of IL-4, nitric oxide, and mannose-binding lectin in women with recurrent vulvovaginal candidiasis. Clin Infect Dis 2005; 40:1258–1262. [DOI] [PubMed] [Google Scholar]
- 54.Choi EH, Lee HJ, Yoo T, Chanock SJ. A common haplotype of interleukin-4 gene IL4 is associated with severe respiratory syncytial virus disease in Korean children. J Infect Dis 2002; 186:1207–1211. [DOI] [PubMed] [Google Scholar]
- 55.Forton JT, Rowlands K, Rockett K, Hanchard N, Herbert M, Kwiatkowski DP, et al. Genetic association study for RSV bronchiolitis in infancy at the 5q31 cytokine cluster. Thorax 2009; 64:345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoebee B, Rietveld E, Bont L, Oosten M, Hodemaekers HM, Nagelkerke NJ, et al. Association of severe respiratory syncytial virus bronchiolitis with interleukin-4 and interleukin-4 receptor alpha polymorphisms. J Infect Dis 2003; 187:2–11. [DOI] [PubMed] [Google Scholar]
- 57.Zhang M, Lu Y, Zhang X, Lu A, Wang L, Chen C. Interleukin-4 polymorphism is associated with severity of respiratory syncytial virus infection. J Paediatr Child Health 2016; 52:25–29. [DOI] [PubMed] [Google Scholar]
- 58.Vafa M, Maiga B, Israelsson E, Dolo A, Doumbo OK, Troye-Blomberg M. Impact of the IL-4-590 C/T transition on the levels of Plasmodium falciparum specific IgE, IgG, IgG subclasses and total IgE in two sympatric ethnic groups living in Mali. Microbes Infect 2009; 11:779–784. [DOI] [PubMed] [Google Scholar]
- 59.Rasouli M, Kiany S. Association of interferon-gamma and interleukin-4 gene polymorphisms with susceptibility to brucellosis in Iranian patients. Cytokine 2007; 38:49–53. [DOI] [PubMed] [Google Scholar]
- 60.Connelly TM, Koltun WA, Sangster W, Berg AS, Hegarty JP, Harris L, 3rd, et al. An interleukin-4 polymorphism is associated with susceptibility to Clostridium difficile infection in patients with inflammatory bowel disease: results of a retrospective cohort study. Surgery 2014; 156:769–774. [DOI] [PubMed] [Google Scholar]
- 61.Anovazzi G, Kim YJ, Viana AC, Curtis KM, Orrico SR, Cirelli JA, et al. Polymorphisms and haplotypes in the interleukin-4 gene are associated with chronic periodontitis in a Brazilian population. J Periodontol 2010; 81:392–402. [DOI] [PubMed] [Google Scholar]
- 62.Gonzales JR, Kobayashi T, Michel J, Mann M, Yoshie H, Meyle J. Interleukin-4 gene polymorphisms in Japanese and Caucasian patients with aggressive periodontitis. J Clin Periodontol 2004; 31:384–389. [DOI] [PubMed] [Google Scholar]
- 63.Gonzales JR, Mann M, Stelzig J, Bodeker RH, Meyle J. Single-nucleotide polymorphisms in the IL-4 and IL-13 promoter region in aggressive periodontitis. J Clin Periodontol 2007; 34:473–479. [DOI] [PubMed] [Google Scholar]
- 64.Holla LI, Fassmann A, Augustin P, Halabala T, Znojil V, Vanek J. The association of interleukin-4 haplotypes with chronic periodontitis in a Czech population. J Periodontol 2008; 79:1927–1933. [DOI] [PubMed] [Google Scholar]
- 65.Loo WT, Fan CB, Bai LJ, Yue Y, Dou YD, Wang M, et al. Gene polymorphism and protein of human pro- and anti-inflammatory cytokines in Chinese healthy subjects and chronic periodontitis patients. J Transl Med 2012; 10 Suppl 1:S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cenci E, Mencacci A, Del Sero G, Bacci A, Montagnoli C, d’Ostiani CF, et al. Interleukin-4 causes susceptibility to invasive pulmonary aspergillosis through suppression of protective type I responses. J Infect Dis 1999; 180:1957–1968. [DOI] [PubMed] [Google Scholar]
- 67.Hernandez-Pando R, Aguilar D, Hernandez ML, Orozco H, Rook G. Pulmonary tuberculosis in BALB/c mice with nonfunctional IL-4 genes: changes in the inflammatory effects of TNF-alpha and in the regulation of fibrosis. Eur J Immunol 2004; 34:174–183. [DOI] [PubMed] [Google Scholar]
- 68.Fischer JE, Johnson JE, Kuli-Zade RK, Johnson TR, Aung S, Parker RA, et al. Overexpression of interleukin-4 delays virus clearance in mice infected with respiratory syncytial virus. J Virol 1997; 71:8672–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tang YW, Graham BS. Anti-IL-4 treatment at immunization modulates cytokine expression, reduces illness, and increases cytotoxic T lymphocyte activity in mice challenged with respiratory syncytial virus. J Clin Invest 1994; 94:1953–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Himmelrich H, Launois P, Maillard I, Biedermann T, Tacchini-Cottier F, Locksley RM, et al. In BALB/c mice, IL-4 production during the initial phase of infection with Leishmania major is necessary and sufficient to instruct Th2 cell development resulting in progressive disease. J Immunol 2000; 164:4819–4825. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.