

Abstract
Chemical probing experiments, coupled with empirically-determined free energy change relationships, can enable accurate modeling of the secondary structures of diverse and complex RNAs. A current frontier lies in modeling large and structurally heterogeneous transcripts, including complex eukaryotic RNAs. To validate and improve on experimentally driven approaches for modeling large transcripts, we obtained high-quality SHAPE data for the protein-free human 18S and 28S ribosomal RNAs (rRNAs). To our surprise, SHAPE-directed structure models for the human rRNAs poorly matched accepted structures. Analysis of predicted rRNA structures based on low-SHAPE and low-entropy (lowSS) metrics revealed that, whereas about 75% of E. coli rRNA sequences form well-determined lowSS secondary structure, only about 40% of the human rRNAs do. Critically, regions of the human rRNAs that specifically fold into well-determined lowSS structures were modeled to high accuracy using SHAPE data. This work reveals that eukaryotic rRNAs are more unfolded than are those of prokaryotic rRNAs, and indeed are largely unfolded overall, likely reflecting increased protein dependence for eukaryotic ribosome structure. Further, those regions and sub-structures that are well-determined can be identified de novo and successfully modeled by SHAPE-directed folding.
Graphical Abstract
INTRODUCTION
RNA is a central carrier of information in biological systems, and this information is encoded in both the primary sequence of the RNA and in higher-order structures that form when the RNA folds. Both highly stable, base-paired elements that populate single structures and also unpaired or less stable structures play important biological roles, and the extent of structure can modulate RNA function by forming, making accessible, or sequestering interaction sites for proteins, other RNAs, or small-molecule ligands1,2,3,4. Chemical probing technologies, especially the SHAPE (selective 2′-hydroxyl acylation analyzed by primer extension) strategy5,6, have proven to be powerful tools for characterizing local nucleotide flexibility in an experimentally concise and accurate way. These nucleotide flexibility data can be parameterized7,8 and incorporated into a standard RNA folding algorithm. This melded SHAPE-directed approach, applied to many short and medium sized RNAs and to long bacterial rRNAs, results in models that show good to outstanding accuracy when compared to accepted structures defined by comparative sequence analysis or high-resolution approaches7,8,9. SHAPE-directed structure modeling can also accurately model the structures of individual functional elements in viral, bacterial, and human RNAs and has identified novel functional motifs in these long RNAs10,11,12,13.
A current frontier in experimentally-directed RNA secondary structure modeling lies in modeling complex RNAs that contain a mixture of regions with persistent stable structure intermixed with regions containing conformationally dynamic elements14. Although it is clear that conformational dynamics are critical for function of many RNAs, chemical probing-driven approaches have been largely validated by analysis of RNAs with very stable and well-defined structures8,15. These test case RNAs are typically among the most highly structured RNAs known and include bacterial rRNAs and those RNAs that can be successfully crystalized7,9,16. In fact, RNAs with such well-defined structures are highly unusual and likely represent outliers in the RNA world. Moreover, widely reported “whole transcriptome” structure probing experiments involving complex and time-intensive protocols often include minimal validation, especially as applied to large RNAs. Indeed, the RNAs or RNA motifs used to validate many “transcriptome-wide” probing experiments span 200 nts or fewer, whereas the typical transcript in a eukaryotic cell exceeds 2,000 nucleotides.
As part of an effort to validate and improve SHAPE data constraints and parameters used for analysis of long and complex RNAs and of transcriptome-wide experiments, we used the SHAPE-MaP (mutational profiling) chemical probing strategy to examine the structure of full-length human 18S and 28S rRNAs, which are 1869 and 5070 nts in length, respectively. SHAPE-directed structure modeling of protein-free eukaryotic rRNAs in solution consistently showed poor overall accuracy (Fig. 1A) compared to the accepted structures, taken to be base pairs visualized in crystallographic studies17. This discrepancy prompted us to investigate whether the inability to model full-length human rRNAs reflects a limitation of SHAPE-directed structure modeling or represents a fundamental structural difference between deproteinized prokaryotic and eukaryotic rRNAs. Previous studies have shown that it is possible to identify well-structured regions in RNA genomes10,13, in long non-coding RNAs1, and in bacterial mRNAs12 using low SHAPE and low Shannon entropy metrics (lowSS regions). Using this metric, we discovered that, when examined in protein-free solution, human rRNAs are structurally much less well-determined, and are more unfolded, than are bacterial rRNAs under comparable conditions. The subset of regions within the human rRNAs that passed lowSS filters likely do have stable persistent structure and could indeed be modeled accurately by SHAPE-directed folding.
Figure 1. Accuracy of rRNA structure modeling.

(A) Sensitivity (sens) and positive predictive value (ppv) for E. coli and human rRNA structures modeled with no SHAPE data, with restraints from experimental SHAPE data, and with idealized constraints (assigning SHAPE reactivities of zero and one, respectively, to base paired and single stranded nucleotides in the accepted structure). Values for sens and ppv, shown for the entire sequence and for lowSS regions only, are colored from low (red) to high (green). (B) Structure models for the human 28S rRNA, positions 160–275. Nucleotides are colored by SHAPE reactivity constraints used for modeling. This example emphasizes that portions of this RNA, in its protein-free form, fold differently than the accepted structure.
METHODS
Reference structures
Structures were obtained from Ribovision18 curated models, revised from crystallographic and cryo-EM structures of complete ribosomes corresponding to Protein Data Bank entries 3R8S, 4GD1, 3J3A, 3J3B, 3J3D, and 3J3F17,19,20
E. coli and mammalian cell lysis and protein digestion
A 25-mL aliquot of E. coli cells at an OD600 of 0.5 were pelleted at 8000 ×g, 4 °C for 10 min. Cells were lysed in 16.5 mL E. coli lysis buffer [15 mM Tris, pH 8, 450 mM sucrose, 8 mM EDTA, 0.4 mg/mL lysozyme] for 5 min at 23 °C, then 10 min at 0 °C. Protoplasts were collected at 5000 ×g and then resuspended in 2 mL proteinase K buffer [50 mM HEPES, pH 8, 200 mM NaCl, 5 mM MgCl2, 1.5% SDS, 0.2 mg/mL proteinase K], vortexed for 10 s, then incubated at 23 °C for 5 min and 0 °C for 10 min. For human rRNA data, total RNA from HEK293 cells (80% confluency) was extracted under conditions designed to maintain the underlying RNA structure21. Cells were lysed in cytoplasmic lysis buffer (40 mM Tris pH 8.0, 40 mM NaCl, 6 mM MgCl2, 1 mM CaCl2, 256 mM sucrose, 0.5% Triton X-100, 250 units/mL RNase inhibitor, 450 units/mL DNase I) for 5 minutes at 4 °C; nuclei were pelleted away; and SDS and proteinase K were added to 1.5% and 500 µg/mL final concentrations, respectively. Cytoplasmic lysates were incubated at room temperature for 45 min. RNA from E. coli or human cell lysates was extracted twice with one volume of phenol:chloroform:Isoamyl alcohol (25:24:1), pre-equilibrated in 1.1× RNA Folding Buffer (110 mM HEPES, pH 8.0, 110 mM NaCl, 5.55 mM MgCl2), and twice with one volume of chloroform.
SHAPE probing of cell-extracted RNA
E. coli and human RNA was exchanged into fresh 1.1× RNA Folding Buffer, incubated at 37 °C for 20 minutes, and split into two equal aliquots. One aliquot was treated with 1/9 volume 5.2 mg/100 µL 5-nitroisatoic anhydride22 and the second was treated with the same volume of DMSO (serving as unmodified control). After incubation for 10 minutes at 37 °C, RNA was precipitated by adding 1/10 volume 2 M ammonium acetate and 1 volume isopropanol, and incubating for 10 minutes at room temperature. Precipitated RNA was pelleted by centrifugation for 10 min at 12,000 g at 4 °C. The supernatant was removed, and the RNA pellet was washed with 1 volume 75% ethanol and centrifuged at 7,500 g for 5 min at 4 °C. The samples were resuspended in water before being treated with DNase (TURBO DNase, Thermo Fisher) for 1 hour at 37 °C and affinity purified (SPRI RNA beads, RNAClean XP, Beckman Coulter).
Reverse transcription and library preparation of ribosomal RNA
Modified and unmodified control RNA was subjected to reverse transcription using random nonamer primers as described10,21 with the addition of an initial 5 min 90 °C denaturation step for human rRNA. Reverse transcription (SuperScript II) was performed in the presence of 6 mM MnCl2 and 1 M betaine. The resulting cDNA was purified by size exclusion chromatography (G50 column, GE Healthcare). The cDNA was subjected to second-strand synthesis and double-stranded DNA was purified (AMPure XP beads in a 1:1.2 ratio, Agencourt). Sequencing libraries were prepared (NexteraXT; Illumina) from 1 µg of DNA. After size selection (with 1:0.8 AMPure XP beads) and quantification (QuBit high sensitivity dsDNA assay and Agilent Bioanalyzer 2100), libraries were sequenced (Illumina MiSeq 600 kits).
Reverse transcription and library preparation of U1 RNA
Reverse transcription was conducted on total human RNA from HEK293 cells using a gene specific primer for U1 RNA (5’-CAGGG GAAAG CGCGA A). The cDNA was purified; amplification and library preparation for sequencing were performed using a two-step PCR reaction; PCR products were purified; and libraries were sequenced (Illumina MiSeq instrument) as described22.
Structure modeling
Sequencing data were processed using ShapeMapper 2 software23 with a minimum required read depth of 1000. Superfold6 was used to model base pairs and pairing probabilities. RNA structures were modeled using constraints from experimental SHAPE reactivities, idealized data (1 for unpaired nucleotides and 0 for base-paired nucleotides based on the accepted structure), or no reactivity data (all reactivities set to −999). rRNAs were modeled with maximum pairing distances of 600 and 800 nucleotides for the small and large subunits, respectively. Structures were scored by comparing predicted minimum free energy structures to accepted structures17 Scoring allowed for 1-base-pair offset7. The sens value was calculated as the number of correctly predicted base pairs in the model divided by the number of canonical base pairs in the accepted structure, excluding pairs for which SHAPE data was not present at both positions. The ppv value was calculated as the number of correctly predicted base pairs divided by the number of predicted base pairs in the model, excluding no-data pairs.
Direct reactivity comparison
For direct comparison of SHAPE reactivities across experiments performed under identical conditions, reactivity profiles (Fig. 2) were computed as ln(rateM / rateU), where rateM, rateU refer to MaP rates in SHAPE-modified and untreated samples, respectively. Positions with read depth below 1000 or untreated MaP rate above 0.05 were excluded. Kernel density estimates (Fig. 2A) were calculated using the seaborn python package (https://seaborn.pydata.org) with default bandwidth settings. Kernel density estimates can be conceptualized as smooth histograms, and are calculated by treating each data point as a gaussian curve with a fixed width and summing all curves over the range of values.
Figure 2. Comparison of extent of folding in E. coli versus human rRNAs in regions with conserved sequence and structure.

(A) Raw (unnormalized) background-corrected chemical-adduct induced mutational profiling (MaP) rates for E. coli and human ribosomes probed with 5NIA. Reactivities calculated as ln(rateM / rateU), where rateM, rateU refer to mutation rates in SHAPE-modified and untreated samples, respectively. Box plots span the central 50% of the data, the interquartile range [IQR, from quartile 1 (Q1) to quartile 3 (Q3)]; center line indicates median; whiskers indicate the most extreme points within (Q1 – 1.5 ✕ IQR) to (Q2 + 1.5 ✕ IQR). **, Student’s t-test p-values less than 10-9. (B) RNA region that is well and similarly structured in both E. coli and human rRNAs. Adduct reactivity rates shown as step plots. Accepted, crystal-structure defined, base pairs shown as arcs. Base pairs present in both E. coli and human rRNA are black; pairs present only in E. coli or human rRNAs are colored blue or orange, respectively. ρ, Spearman correlation coefficient between profiles. (C) Selected regions in which human rRNA is notably less well-folded than the homologous E. coli region. Note consistently higher SHAPE reactivity rates for human rRNA than for E. coli.
Identifying low-SHAPE/low-entropy regions
Low-SHAPE/low-entropy regions were identified using a combination of SHAPE reactivity and Shannon entropy, using calculated base pairing probabilities12. Median SHAPE reactivity and Shannon entropy were calculated over 51-nt centered windows. Regions longer than 25 nucleotides with windowed SHAPE reactivities below 0.3 and windowed Shannon entropies below 0.08 were defined as lowSS regions. Regions were expanded to include nested base pairs that had base pairing probabilities greater than 90%. The selected SHAPE and entropy thresholds employed here balance region detection with model accuracy, identifying many well-structured regions in accurately modeled structures. Stricter (lower) thresholds increase structure modeling accuracy at the expense of detection rate, and less stringent (higher) thresholds recover more and longer lowSS regions but result in less accurate structures (Fig. S1).
RESULTS
Structure modeling of full-length rRNAs
As an initial point of comparison, the secondary structures and pairing probabilities of the E. coli 16S and 23S rRNAs were modeled using SuperFold6 with no SHAPE constraints or using experimental SHAPE data obtained for this study for RNAs gently extracted from bacterial cells (at 5 mM Mg2+ concentration) while maintaining the overall structure of the rRNA7,12 (see Methods). In the absence of SHAPE constraints, the structures of the E. coli 16S and 23S rRNA are modeled with low accuracy in agreement with previous analyses of these and of other RNAs8,9. The observed sensitivities (sens, percent accepted base pairs modeled correctly) and positive predictive values (ppv, percent modeled base pairs in the accepted structure) are in the 50–60% range (Fig. 1A, top; see entire rRNA columns). As expected7,8, structure modeling accuracy notably increased with the use of SHAPE data as a pseudo-free energy change restraint to achieve a sensitivity of about 74% for both subunits (Fig. 1A, top). A sensitivity greater than 90% for modeling of the 16S rRNA is obtained when the RNA is probed at 10 mM Mg2+ and regions not locally compatible with SHAPE data are omitted7. For simplicity, this latter adjustment was not applied in this study, due to the complexity of defining omitted regions for the human rRNAs. True sens and ppv values are roughly 10% higher than the values reported here.
Next, we chemically probed human 18S and 28S rRNAs (the small and large subunit RNAs, respectively) in a similar cell-free state; the rRNAs were gently extracted from HEK293 cells. As expected, structure models were inaccurate without SHAPE constraints, with sens and ppv values in the 20–40% range, as compared to the accepted models17 (Fig. 1A, bottom; entire rRNA columns). However, in contrast to the E. coli rRNA, the addition of SHAPE data restraints did not yield accurate secondary structure models for the full-length human rRNAs. The human 18S and 28S rRNAs were modeled with sensitivities of only 32% and 61%, respectively, meaning that the SHAPE-directed structures still deviated substantially from the accepted models (Fig. 1A, bottom; 1B).
Modeling rRNA structure with idealized data
The SHAPE-directed pseudo-free energy change strategy was developed primarily using prokaryotic RNAs with compact structures7,9, and one formal possibility was that the resulting parameters are not appropriate for modeling human rRNA. We therefore examined whether idealized restraints based on the accepted model of the human rRNAs would allow accurate folding. To create idealized datasets, we assigned a SHAPE reactivity of zero to nucleotides that are base paired and a SHAPE reactivity of 1 to nucleotides that are single stranded in the accepted secondary structures of the rRNAs. Models constrained by the idealized SHAPE data resulted in secondary structure models for the E. coli rRNAs with sens and ppv values in the 87–90% range (Fig. 1A, top). The idealized data thus yielded a significant increase in modeling accuracy.
The idealized restraints also notably improved modeling of the human rRNAs with sens values of 65% and 90% for the 18S and 28S rRNAs, respectively; some regions were modeled with very high accuracy (Fig. 1B). These accuracies are not quite as high as those of E. coli rRNA models but are sufficiently high to strongly suggest that the observed disagreements between SHAPE-directed models and accepted structures are not due to a general limitation of SHAPE-constrained structure modeling but, instead, reflect fundamental biological structural differences between protein-free bacterial and human rRNAs. Indeed, there are many regions in the human rRNAs in which the observed experimental SHAPE data are simply incompatible with the accepted structure; these regions can be accurately modeled into the accepted structure when guided by idealized data, however (Fig. 1B).
Direct comparison of SHAPE reactivities between human and E. coli rRNA
As a second approach to establish whether the E. coli and human rRNAs show fundamentally different levels of folding, distinct from secondary structure modeling, we directly compared SHAPE reactivities between the two species. We examined SHAPE reactivity profiles over the full rRNA lengths and over selected regions in the small subunit with locally conserved sequence and structure, using raw background-corrected SHAPE adduct-induced mutation rates from probing experiments performed under identical conditions. Global SHAPE reactivity profiles for human rRNA are substantially shifted to higher modification rates as compared to E. coli rRNA (Fig. 2A). The human rRNAs are therefore overall intrinsically more reactive to SHAPE than are the E. coli rRNAs.
We also focused on reactivity profiles for four regions showing clear sequence and structure conservation between the human and E. coli small subunit rRNAs, defined as gap-free alignments over at least 15 nts, with at least 50% sequence identity and one shared helix longer than two base pairs. In E. coli numbering, these regions were helix 8 (residues 144–178), helix 18 (500–545), helix 23 (673–717), and helix 24 (765–820), all of which have well-described roles in ribosome function. Helix 18 is a component of the mRNA entry site latch24; and helices 8, 23 and 24 all form important bridging interactions with the large subunit25.
Helix 23 is fully or nearly fully folded in both E. coli and human rRNAs, as evidenced by shared low reactivities over putative base paired positions and substantially similar reactivity profiles (Fig. 2B). In contrast, the other three regions are folded in E. coli rRNA but strikingly unfolded in human rRNA, even though the expected secondary structures are similar (Fig. 2C). Thus, isolated human rRNAs adopt notably different structures than their E. coli counterparts, even in regions with conserved sequences and functions.
Identification of well-structured (lowSS) RNA regions
Prior work has emphasized that regions that are highly structured (as detected by a low local SHAPE reactivity) and that have well-determined structures (supported by a low Shannon entropy) tend to be strongly correlated with function10,11,12. These regions might comprise well-folded elements within the human rRNAs where structure can be more accurately predicted by SHAPE-directed structure modeling. Local regions of low SHAPE/low Shannon entropy (lowSS) within the E. coli and human rRNAs were identified using a combination of SHAPE data and SHAPE-informed Shannon entropy, calculated over smoothed windows across the primary sequence10,12,6. Although SHAPE reactivity influences the calculation of Shannon entropy, the SHAPE and entropy metrics provide orthogonal information, and the combination of the two terms identifies regions whose secondary structures are modeled with the highest accuracy (Fig. S1). Reactivity and entropy thresholds were chosen to maximize the overall number of nucleotides modeled while maintaining high secondary structure modeling accuracy.
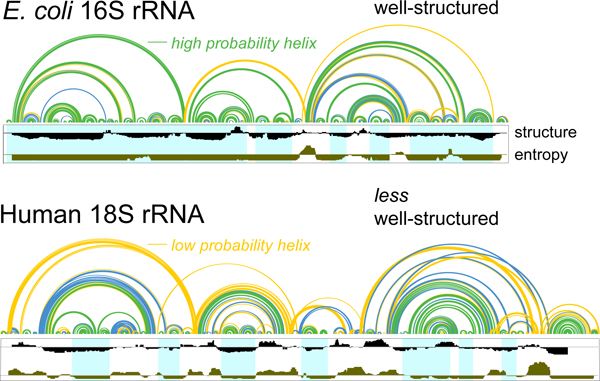
We identified well-structured regions in both the E. coli and human rRNAs by the lowSS criteria and then modeled each using SHAPE restraints. For the E. coli rRNAs, focusing on the lowSS regions had a small, but positive, effect on the already high model accuracy (Fig. 1A, top). lowSS regions comprise 81 and 69% of the E. coli 16S and 23S rRNAs, respectively, and these extensive regions of well-defined structure allow both RNAs to be modeled with high accuracy, even outside of the lowSS regions. In contrast, models of the human rRNAs spanning only the lowSS regions, 34 and 43% of 18S and 28S rRNAs, respectively, were substantially more accurate than models of the full-length rRNA sequences. In the human 18S rRNA, seven well-structured regions were identified and these were modeled with base pair sens of 76%, a dramatic improvement over the 32% sensitivity for the full-length rRNA (Fig. 1A, 3). The human 28S rRNA model contained 14 lowSS regions and these were modeled with 77% sens (Fig. 1, 4).
Figure 3. Structural characterization of the human 18S rRNA.

(A) (top) Model of the full-length 18S rRNA with arcs representing correct, incorrect, and missing base pairs relative to the accepted structure. (middle) SHAPE reactivity (black) and entropy (brown) shown as medians over 51-nt centered windows along the full-length 18S rRNA with the axes crossing at 0.3 and 0.08, respectively. (bottom) SHAPE-directed structure models for lowSS regions modeled as independent elements. (B) Nucleotide-level models of well-structured regions of the 18S rRNA. Nucleotides are colored by SHAPE reactivity (legend in Fig. 1). Missed and incorrect base pairs are shown with red and purple arcs or lines, respectively; in general, these base pairs are supported by the experimental SHAPE reactivities, but are not consistent with the accepted structure.
Figure 4. Structural characterization of the human 28S rRNA.

(A) (top) Model of the full-length 28S rRNA with arcs representing correct, incorrect, and missing base pairs relative to the accepted structure. (middle) SHAPE reactivity (black) and entropy (brown) shown as medians over 51-nt centered windows along the full-length 18S rRNA with the axes crossing at 0.3 and 0.08, respectively. (bottom) SHAPE-directed structure models for lowSS regions modeled as independent elements. (B) Nucleotide-level models of selected regions, highlighting plausible SHAPE-supported local refolding. Local structures that are clearly not compatible with the accepted structure are boxed (defined as nucleotides with high reactivity in base paired regions or low reactivity in consecutive unpaired regions). Nucleotides are colored by SHAPE reactivity (legend in Fig. 1).
This analysis reveals that, although SHAPE data alone are ill-suited for recovering the accepted structures of full-length human rRNAs, the low-SHAPE/low-entropy strategy robustly identifies local well-defined secondary structures. Well-structured regions that pass the lowSS filters in human rRNA involve important functional elements such as the L1 stalk, GTPase associated center, A-site finger, and an element of the 18S rRNA that directly interacts with mRNA in the E-site26–30 (Fig. S2).
Local rRNA refolding
The lowSS regions in the human rRNAs were generally modeled to good agreement with the accepted structures. However, SHAPE reactivities in some regions, including some lowSS regions, are clearly not consistent with the accepted structures in regions of both the human 18S and 28S rRNAs (Figs. 1B, 4B, boxed residues). We interpret these data as indicating that, for RNAs chemically probed in a cell-free (and protein-free) state, models for lowSS regions are better representations of these regions than are the accepted structures, likely because these regions locally refold when RNA is removed from the cellular (and native protein) environment.
DISCUSSION
This work supports three overarching conclusions. First, folding of the protein-free bacterial and human rRNAs are fundamentally different: in solution, human rRNA structure is more unfolded and less well-determined than that of the bacterial rRNAs (Fig. 1, 2). Second, algorithms that couple chemical probing data with empirical free energy change relationships cannot recover the accepted structure of all RNAs with high accuracy. This inability can be attributed to RNAs that rarely populate the accepted structure, as notably observed here for the human rRNAs, and to small errors in thermodynamic and chemical reactivity parameters which especially limit the accuracy of modeling long range interactions31,32. Third, the low SHAPE reactivity/low Shannon entropy (lowSS) metric is a powerful approach for characterizing the well-determinedness of folding for large RNAs and for addressing the challenge presented by conclusion 2. The lowSS metric identifies the subset of regions in long RNAs that can be modeled accurately by SHAPE-directed folding (Fig. 1, 3, 4).
SHAPE provides empirical information on local nucleotide flexibility that can be used to model the secondary structure of short RNAs, bacterial rRNAs, and numerous other RNAs – which have stable and well-defined structures – with high accuracy7,8. However, SHAPE-directed folding alone can produce misleading or inaccurate models for long RNAs that contain poorly structured regions or regions capable of adopting multiple conformations. This challenge in de novo modeling of complex RNAs can be addressed, in part, by focusing on those regions within a large RNA that do fold to form a well-defined structure. The lowSS filter appears robust for both short and long RNAs, and across RNAs with different levels of structure. For example, the majority of nucleotides in the highly structured human U1 snRNA (78%) meet the lowSS criterion (Fig. 5A). Both the E. coli 16S and 23S rRNAs are well-structured with 81% and 69% of nucleotides in lowSS regions (Fig. 5B). Based on these three RNAs, both short and long RNAs can have extensive lowSS regions. In contrast, the human 18S and 28S rRNAs, with only 34% and 43% of nucleotides in lowSS regions, respectively, have extensive poorly-structured regions (Fig. 5C). Many regions of human rRNA are simply unfolded, despite sharing conserved sequence and structure with E. coli rRNA (Fig. 2).
Figure 5. Extent of well-determined structures in small and large RNAs.

(A) human U1 snRNA, (B) E. coli 16S and 23S rRNA, and (C) human 18S and 28S rRNA. Arcs represent modeled base pairing probabilities (colors defined in key). Windowed SHAPE reactivities and entropy are shown in black and brown, respectively. Well-structured regions are highlighted in light blue. Percentages of nucleotides in lowSS regions exclude positions with no SHAPE data (primarily located near the 5’ and 3’ ends of each RNA, and one central section of the 28S rRNA).
Our data emphasize that human rRNA is intrinsically less structured than E. coli rRNA under the conditions probed here. This study also suggests that the human rRNAs may not be well suited for use in validating modeling accuracies for transcriptome-wide studies. Eukaryotic rRNAs likely require more support from proteins for full structure formation than do bacterial rRNAs, consistent with models of ribosome evolution that show the accretion of a protein shell in eukaryotic lineages33,34. Eukaryotic ribosome structure, assembly, and regulation are all more complex as compared to prokaryotic ribosomes35. Other aspects of the eukaryotic cellular environment, such as local ion concentrations or differences in macromolecular crowding, might play larger roles for eukaryotic ribosome assembly and folding than for assembly of the bacterial translation machinery. Unlike the relatively stable “RNA rocks” of prokaryotic ribosomes, only a small fraction of eukaryotic rRNA appears to be well folded in isolation.
Eukaryotic rRNAs, multi-kilobase mRNAs12, viral RNAs10,11, and non-coding RNAs1 appear to be less structured overall than well-understood short structured RNAs like bacterial riboswitches8,36 but do have regions of stable functional structures. In such cases, lowSS regions provide a starting point for locating well-folded and potentially functional structural elements. Indeed, lowSS regions identify the vast majority of well-characterized functional motifs in viral genomic RNAs10,11,13, and the lowSS metric enabled de novo identification and validation of multiple novel regulatory elements in the E. coli transcriptome12. Similarly, well-structured lowSS regions in the human rRNAs include important functional elements. The lowSS metric will likely prove a broadly useful tool for future modeling and functional studies of diverse classes of long RNA transcripts.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the U.S. National Institutes of Health (R35 GM122532 to K.M.W.). C.A.G. was supported in part by a training grant in Molecular Biophysics (T32GM008570). C.A.W. is an American Cancer Society Postdoctoral Fellow (ACS 130845-RSG-17-114-01-RMC).
Footnotes
SUPPORTING INFORMATION
Two figures, plus tables giving the full SHAPE data for the E. coli and human rRNAs.
DISLOSURE
K.M.W. is an advisor to and holds equity in Ribometrix, to which mutational profiling (MaP) technologies have been licensed.
REFERENCES
- (1).Smola MJ, Christy TW, Inoue K, Nicholson CO, Friedersdorf M, Keene JD, Lee DM, Calabrese JM, and Weeks KM (2016) SHAPE reveals transcript-wide interactions, complex structural domains, and protein interactions across the Xist lncRNA in living cells. Proc. Natl. Acad. Sci 113, 10322–10327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Mustoe AM, Corley M, Laederach A, and Weeks KM (2018) Messenger RNA Structure Regulates Translation Initiation: A Mechanism Exploited from Bacteria to Humans. Biochemistry 57, 3537–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Cech TR, and Steitz JA (2014) The Noncoding RNA Revolution—Trashing Old Rules to Forge New Ones. Cell 157, 77–94. [DOI] [PubMed] [Google Scholar]
- (4).Lewis CJT, Pan T, and Kalsotra A (2017) RNA modifications and structures cooperate to guide RNA–protein interactions. Nat. Rev. Mol. Cell Biol 18, 202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Weeks KM, and Mauger DM (2011) Exploring RNA Structural Codes with SHAPE Chemistry. Acc. Chem. Res 44, 1280–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Smola MJ, Rice GM, Busan S, Siegfried NA, and Weeks KM (2015) Selective 2’-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nat. Protoc 10, 1643–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Deigan KE, Li TW, Mathews DH, and Weeks KM (2009) Accurate SHAPE-directed RNA structure determination. Proc. Natl. Acad. Sci. U. S. A 106, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hajdin CE, Bellaousov S, Huggins W, Leonard CW, Mathews DH, and Weeks KM (2013) Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots. Proc. Natl. Acad. Sci. U. S. A 110, 5498–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Rice GM, Leonard CW, and Weeks KM (2014) RNA secondary structure modeling at consistent high accuracy using differential SHAPE. RNA 20, 846–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Siegfried NA, Busan S, Rice GM, Nelson JAE, and Weeks KM (2014) RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat. Methods 11, 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mauger DM, Golden M, Yamane D, Williford S, Lemon SM, Martin DP, and Weeks KM (2015) Functionally conserved architecture of hepatitis C virus RNA genomes. Proc. Natl. Acad. Sci. U. S. A 112, 3692–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mustoe AM, Busan S, Rice GM, Hajdin CE, Peterson BK, Ruda VM, Kubica N, Nutiu R, Baryza JL, and Weeks KM (2018) Pervasive Regulatory Functions of mRNA Structure Revealed by High-Resolution SHAPE Probing. Cell 173, 181–195.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dethoff EA, Boerneke MA, Gokhale NS, Muhire BM, Martin DP, Sacco MT, McFadden MJ, Weinstein JB, Messer WB, Horner SM, and Weeks KM (2018) Pervasive tertiary structure in the dengue virus RNA genome. Proc. Natl. Acad. Sci. U. S. A 115, 11513–11518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Blanco MR, Martin JS, Kahlscheuer ML, Krishnan R, Abelson J, Laederach A, and Walter NG (2015) Single Molecule Cluster Analysis dissects splicing pathway conformational dynamics. Nat. Methods 12, 1077–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Al-Hashimi HM, and Walter NG (2008) RNA dynamics: it is about time. Curr. Opin. Struct. Biol 18, 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lavender CA, Lorenz R, Zhang G, Tamayo R, Hofacker IL, and Weeks KM (2015) Model-Free RNA Sequence and Structure Alignment Informed by SHAPE Probing Reveals a Conserved Alternate Secondary Structure for 16S rRNA. PLOS Comput. Biol (Wilke CO, Ed.) 11, e1004126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Petrov AS, Bernier CR, Gulen B, Waterbury CC, Hershkovits E, Hsiao C, Harvey SC, Hud NV, Fox GE, Wartell RM, and Williams LD (2014) Secondary Structures of rRNAs from All Three Domains of Life. PLoS One (Barash D, Ed.) 9, e88222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Bernier CR, Petrov AS, Waterbury CC, Jett J, Li F, Freil LE, Xiong X, Wang L, Migliozzi BLR, Hershkovits E, Xue Y, Hsiao C, Bowman JC, Harvey SC, Grover MA, Wartell ZJ, and Williams LD (2014) RiboVision suite for visualization and analysis of ribosomes. Faraday Discuss 169, 195–207. [DOI] [PubMed] [Google Scholar]
- (19).Anger AM, Armache J-P, Berninghausen O, Habeck M, Subklewe M, Wilson DN, and Beckmann R (2013) Structures of the human and Drosophila 80S ribosome. Nature 497, 80–85. [DOI] [PubMed] [Google Scholar]
- (20).Dunkle JA, Wang L, Feldman MB, Pulk A, Chen VB, Kapral GJ, Noeske J, Richardson JS, Blanchard SC, and Cate JHD (2011) Structures of the bacterial ribosome in classical and hybrid states of tRNA binding. Science 332, 981–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Smola MJ, Calabrese JM, and Weeks KM (2015) Detection of RNA-Protein Interactions in Living Cells with SHAPE. Biochemistry 54, 6867–6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Busan S, Weidmann CA, Sengupta A, and Weeks KM (2019) Guidelines for SHAPE Reagent Choice and Detection Strategy for RNA Structure Probing Studies. Biochemistry 58, 2655–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Busan S, and Weeks KM (2018) Accurate detection of chemical modifications in RNA by mutational profiling (MaP) with ShapeMapper 2. RNA 24, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hussain T, Llácer JL, Wimberly BT, Kieft JS, and Ramakrishnan V (2016) Large-Scale Movements of IF3 and tRNA during Bacterial Translation Initiation. Cell 167, 133–144.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Liu Q, and Fredrick K (2016) Intersubunit Bridges of the Bacterial Ribosome. J. Mol. Biol 428, 2146–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Doris SM, Smith DR, Beamesderfer JN, Raphael BJ, Nathanson JA, and Gerbi SA (2015) Universal and domain-specific sequences in 23S-28S ribosomal RNA identified by computational phylogenetics. RNA 21, 1719–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Mohan S, and Noller HF (2017) Recurring RNA structural motifs underlie the mechanics of L1 stalk movement. Nat. Commun 8, 14285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Egebjerg J, Douthwaite SR, Liljas A, and Garrett RA (1990) Characterization of the binding sites of protein L11 and the L10.(L12)4 pentameric complex in the GTPase domain of 23 S ribosomal RNA from Escherichia coli. J. Mol. Biol 213, 275–288. [DOI] [PubMed] [Google Scholar]
- (29).Piekna-Przybylska D, Przybylski P, Baudin-Baillieu A, Rousset J-P, and Fournier MJ (2008) Ribosome performance is enhanced by a rich cluster of pseudouridines in the A-site finger region of the large subunit. J. Biol. Chem 283, 26026–26036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Pisarev AV, Kolupaeva VG, Yusupov MM, Hellen CUT, and Pestova TV (2008) Ribosomal position and contacts of mRNA in eukaryotic translation initiation complexes. EMBO J 27, 1609–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Doshi KJ, Cannone JJ, Cobaugh CW, and Gutell RR (2004) Evaluation of the suitability of free-energy minimization using nearest-neighbor energy parameters for RNA secondary structure prediction. BMC Bioinformatics 5, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Wu Y, Shi B, Ding X, Liu T, Hu X, Yip KY, Yang ZR, Mathews DH, and Lu ZJ (2015) Improved prediction of RNA secondary structure by integrating the free energy model with restraints derived from experimental probing data. Nucleic Acids Res 43, 7247–7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Petrov AS, Bernier CR, Hsiao C, Norris AM, Kovacs NA, Waterbury CC, Stepanov VG, Harvey SC, Fox GE, Wartell RM, Hud NV, and Williams LD (2014) Evolution of the ribosome at atomic resolution. Proc. Natl. Acad. Sci. U. S. A 111, 10251–10256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Melnikov S, Ben-Shem A, Garreau de Loubresse N, Jenner L, Yusupova G, and Yusupov M (2012) One core, two shells: bacterial and eukaryotic ribosomes. Nat. Struct. Mol. Biol 19, 560–567. [DOI] [PubMed] [Google Scholar]
- (35).Klinge S, Voigts-Hoffmann F, Leibundgut M, and Ban N (2012) Atomic structures of the eukaryotic ribosome. Trends Biochem. Sci 37, 189–198. [DOI] [PubMed] [Google Scholar]
- (36).McCown PJ, Corbino KA, Stav S, Sherlock ME, and Breaker RR (2017) Riboswitch diversity and distribution. RNA 23, 995–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.