

ABSTRACT
Animal models are important in understanding both the pathogenesis of and immunity to tuberculosis (TB). Unfortunately, we are beginning to understand that no animal model perfectly recapitulates the human TB syndrome, which encompasses numerous different stages. Furthermore, Mycobacterium tuberculosis infection is a very heterogeneous event at both the levels of pathogenesis and immunity. This review seeks to establish the current understanding of TB pathogenesis and immunity, as validated in the animal models of TB in active use today. We especially focus on the use of modern genomic approaches in these models to determine the mechanism and the role of specific molecular pathways. Animal models have significantly enhanced our understanding of TB. Incorporation of contemporary technologies such as single cell transcriptomics, high-parameter flow cytometric immune profiling, proteomics, proteomic flow cytometry and immunocytometry into the animal models in use will further enhance our understanding of TB and facilitate the development of treatment and vaccination strategies.
Keywords: Mycobacterium tuberculosis, animal models, translation
This review discusses the current state of the important animal models and techniques being used to study Mycobacterium tuberculosis infection and tuberculosis disease as it occurs in humans.
INTRODUCTION
Tuberculosis (TB) is a widespread infectious disease, latently infecting up to one third of the world's population and causing ~1.3 million deaths in 2016 (WHO 2017). Many of the world's regions with high disease prevalence are rife with malnutrition, have limited access to medical resources and are hotbeds for non-compliance with medical interventions, leading to multidrug resistant and extensively drug-resistant strains (Gandhi et al. 2006; Migliori et al. 2012).
Mycobacterium tuberculosis (Mtb) is a pathogen that is spread by aerosol droplets to uninfected individuals when infected patients cough( Figure 1). In the lungs, Mtb comes into contact with innate immune cells that attempt to eliminate the pathogen. When Mtb comes into contact with alveolar macrophages, it can infect the cells and persist following phagocytosis as an intracellular pathogen. During the innate immune phase, inflammatory cells, including dendritic cells, neutrophils and macrophages, traffic to the lungs and attempt to control the infection (Nunes-Alves et al. 2014). Dissemination of Mtb to the lymph nodes occurs, allowing dendritic cells to present bacterial antigens to T cells and prime them (Chackerian et al. 2002; Wolf et al. 2008). Priming of ESAT6 in particular occurs ~10 days post-infection in the mediastinal lymph nodes and is followed later by generation of effector T cells (Reiley et al. 2008). The adaptive immune response that follows T cell priming, particularly the generation of effector T cells and Th1 CD4+ T cells, leads to the formation of granulomas. Granulomas are organized structures where T cells and B cells encapsulate innate immune cells, such as activated neutrophils and macrophages, with a fibrotic capsule to create a gradient of hypoxia, with the greatest concentration of hypoxia formed in its center to isolate and prevent the growth of Mtb (Kumar et al. 2011). Granulomas caseate and fail to contain Mtb when host-specific responses are insufficient to prevent bacterial replication, Mtb-infected macrophages necrotize and bacilli escape the hypoxic core to disseminate into uninfected lung tissues or secondary sites of infection including the liver, spleen and brain. Mtb has evolved numerous mechanisms of host resistance including its ability to alter T cell cytokines in infected cells, such as interleukin-1β produced by dendritic cells, to induce Th2 immune responses and inhibit protective Th1 host responses (Dwivedi et al. 2012).
Figure 1.
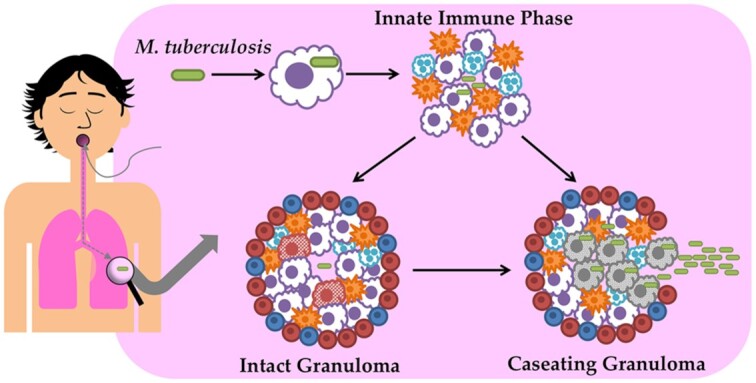
Mycobacterium tuberculosis (Mtb) is spread via aerosols and is harbored primarily in the lungs. When macrophages encounter and engulf the pathogens, it may be eliminated or may persist due to compensatory mechanisms. During the innate immune phase, macrophages, neutrophils and dendritic cells are recruited to the site of infection. After severeal week, T and B cells migrate to the site of infection and form granulomas that encapsulate the innate immune cells and bacilli in a fibrotic capsule that creates a gradient of hypoxia. If the immune cells are not able to kill the bacilli and prevent its growth, granulomas caseate and Mtbbacilli disseminate to other organs including the spleen, liver and brain.
When patients are infected with Mtb, there are multiple clinical outcomes that they can experience. Many patients will control the infection and are considered latently infected (LTBI). These individuals typically possess just a few intact granulomas and are positive on the Mantoux tuberculin skin test (TST). About 10% of all patients with TB will progress to active infection over the course of their lifetimes, some with early progression (Kwan and Ernst 2011). Comorbidities can also induce progression to active disease in latently infected patients, termed reactivation, including, but not limited to, type 2 diabetes (T2D) and human immunodeficiency virus (HIV) (Kwan and Ernst 2011; Ai et al. 2016). HIV alone increases the rate of activation from 10% over a patient's lifetime to 10% per year (Kwan and Ernst 2011). Due to the wide variety of potential states and outcomes, we currently lack the knowledge and tools to distinguish which individuals have cleared or will control Mtb and which will progress to active TB disease. The lack of information on responses that are associated with subsequent progression to active TB impedes identification of individuals at the highest risk of progression to active TB and who would benefit most from preventative therapy in resource-poor, high TB-prevalent regions. Additionally, the lack of information on favorable immune responses that lead to containment of infection, as well as their distinctions from those associated with progression to active disease, is a significant impediment to the design and evaluation of efficacious TB vaccines. Although it is generally understood that Th1 responses, such as IFN-γ that are generated in response to mycobacterial antigens, are protective, Th1 responses during active disease are unable to sterilize the host, complicating our ability to measure successful vaccination (Nunes-Alves et al. 2014).
As many mechanisms of pathogenesis and host response to infection are poorly understood, it is imperative to utilize animal models in order to investigate them. Animal models of TB infection and TB/HIV co-infection have the potential to yield insights about the host's responses to different strains, to different therapeutics and vaccines. They also have the ability to faithfully replicate comorbidities, particularly in the use of non-human primates (NHPs) with TB that are co-infected with simian immunodeficiency virus (SIV). The model used for studies, however, should always be carefully considered to maximize homology to human disease or symptomatology. For example, zebrafish infected with M. marinum have been a vital host to study caseous pathology within granulomas (Swaim et al. 2006; Cronan and Tobin 2014) despite being a non-mammalian organism. Balancing the ethical costs of the model, as well as the knowledge gap under investigation, are essential when choosing the proper animal model for studying TB infection. Here we discuss the most prominently used animal models of Mtb infection and their specific role in the identification of molecular determinants of TB pathology and immunity. We also describe the use of the various contemporary genome-wide approaches in the animal models of TB.
ANIMAL MODELS OF TB
Numerous animal models have been utilized for mechanistic and vaccine studies for TB. These range from more accurate, but more expensive, NHPs, to small and non-mammalian organisms like zebrafish. While NHPs best capitulate the human immune response and susceptibility to TB, often smaller animal models like mice, rats, guinea pigs, rabbits and zebrafish are better utilized for investigating more narrow aspects of the immune response to mycobacteria, such as granuloma formation, susceptibility to different strains, or pre-clinical vaccine responses. A summary of the characteristics of each model is shown in Table 1.
Table 1.
Comparison of biosafety safety level 2 (BSL-2) and BSL-3 animal models of Mtbinfection.
| Non-human primates | Mouse | Rabbit | Guinea pig | Rat | Zebrafish | |
|---|---|---|---|---|---|---|
| Susceptible to human clinical strains of Mtb | Yes | Yes | Yes | Yes | Yes | No |
| Potential for PK studies | Yes, but significant cost | No, differs from humans | Yes | No, differs from humans | Yes | No |
| BSL-2 model | No | No | No | No | No | Yes |
| BSL-3 model | Yes | Yes | Yes | Yes | Yes | No |
| Easy to house | No | Yes | No | No | Yes | Yes |
| Pulmonary pathology | Yes | Yes | Yes | Yes | Yes | No |
| Caseous pathology | Yes | No, except C3HeB/FeJ mouse | Yes | Yes | Yes | Yes |
| Cavitary lesions | Yes | No | Yes | No | No | Yes |
| Dissemination | Yes | Yes | Yes | Yes | Yes | Yes |
| Latency | Yes | No | Yes | No | Yes | Yes |
| Ready availability of immunological reagents | Yes, human cross-reactivity | Yes | No | No | No | No |
| CD1-cross reactivity | CD1a, CD1b, CD1c, CD1d | CD1d only | CD1a, CD1b, CD1d | CD1b, CD1c | CD1d only | No |
NHPS
NHPs have been actively used primarily in the national primate research centers. The seven national primate research centers possess specialized personnel, large facilities and a wealth of expertise and experience that allows complex NHP research to be conducted safely and effectively in a BSL3 setting. In addition, several other research centers and universities in the USA and around the world have developed facilities to leverage NHP models in addition to smaller animal models such as mice, rabbits, guinea pigs and rats. The use of NHPs, particularly macaques, as models of human disease has significantly increased recently. Of the >15 000 publications in PubMed that utilized macaque models of human disease, 60% were published in the last 10 years. These systems have also been significantly utilized in the modeling of infectious diseases, particularly HIV (∼4500 publications) (Hulskotte, Geretti and Osterhaus 1998; Veazey et al. 1998; Douek et al. 2002; Franchini et al. 2002; Brenchley et al. 2004; Hatziioannou et al. 2009; Weed et al. 2012; Kimata 2014), HSV (Peretti et al. 2005; Crostarosa et al. 2009; Calenda et al. 2017), influenza (Brown et al. 2010; Kitano et al. 2010; McDermott et al. 2011), Streptococcus pneumoniae (Philipp et al. 2012), Neisseria gonorrhoeae (Weyand et al. 2013), non-tuberculous mycobacteria (Winthrop et al. 2010; Henkle and Winthrop 2015) and malaria (Lombardini et al. 2015; Phares et al. 2017). These models have not only investigated the pathogenesis of infection, but have also related tissue pathology, vaccine and drug development and, of late, host-directed or immunotherapeutics (Kaushal et al. 2015).
Macaques have been extensively used in TB research (∼150 publications) (Kaushal and Mehra 2012; Kaushal et al. 2012; Foreman et al. 2017). In fact, macaques were extensively used for studying experimental Mtb infections in the early and middle parts of the 20th century, but as the advent of antibiotics and the widespread use of the Bacillus Calmette-Guérin (BCG) vaccine resulted in reduced TB rates, interest waned. Barclay led key studies five decades ago where Indian rhesus macaques were infected via the aerosol route with Mtb.(Barclay et al. 1970). Barclay et al. tested both BCG and Mtb cell wall as effective vaccines against Mtb challenge in this species (Ribi et al. 1971; Anacker et al. 1972), even demonstrating the feasibility of utilizing aerosol BCG vaccination to protect against TB challenge (Barclay et al. 1973). As TB disease resurged in the developed world in the 1980s and 1990s, this led to renewed interest in its research and it was at once clear that the model systems available to study TB at that time were insufficient. Walsh led the study of Mtb infection in the modern era in cynomolgus macaques and showed that it was possible to generate human-like LTBI in that species (Walsh et al. 1996). The cynomolgus macaque was then extensively used, primarily by Flynn and colleagues, to study aspects of Mtb infection (Capuano et al. 2003; Lin et al. 2009), especially early infection (Lin et al. 2006), reactivation due to SIV co-infection (Diedrich et al. 2010) and for vaccine evaluation (Lin et al. 2012; Mehra et al. 2013). The work of several other groups simultaneously led to a renewed interest in the rhesus macaque as a model for TB pathogenesis (Gormus et al. 2004; Lewinsohn et al. 2006; Qiu et al. 2008; Chen et al. 2009; Sharpe et al. 2009; Verreck et al. 2009, Sharpe et al. 2010). Mtb-infected rhesus macaques have since been utilized extensively to study the role of bacterial determinants of pathogenesis (Dutta et al. 2010), e.g. stress-response transcription factors (Mehra et al. 2012, 2015). These studies generated results different to those obtained in the typical mouse model and extended our knowledge of how Mtb has evolved to manage stress in its attempt to persist. This model has been invaluable to replicate the immune responses that lead to human granuloma formation and their failure because NHPs are able to produce granulomas that are comparable with those found in humans (Figure 1) (Mehra et al. 2010, 2013; Slight et al. 2013). The NHP model has been vital for testing vaccines and vaccine-induced responses (Kaushal et al. 2015; Foreman et al. 2017), in elucidating Mtb/HIV co-infections using SIV as a surrogate (Diedrich et al. 2010; Sestak et al. 2011; Foreman et al. 2016) and in ascertaining the role of individual cell types (Kuroda et al. 2018) and immune/metabolic pathways/molecules (Diedrich, Mattila and Flynn 2013; Cheng et al. 2017).
Old World NHPs originating from Africa and Asia, including macaques, are prevalent as an NHP model in TB research. New World NHPs, originating from Central and South America as well as Mexico, e.g. marmosets, have also been used for TB research. A challenge of the marmoset model remains that even low-dose infection has been shown to yield intensely pathological outcomes, making it difficult to faithfully model TB latency (Via et al. 2013; Cadena et al. 2016). Marmosets remain a captivating model organism, however, due to the ability to standardize experiments using twins, particularly for comparisons of strain virulence and disease progression (Via et al. 2013). Marmosets have also been a useful model for investigating the effect of different drug regimens because they produce fully formed granulomas that can be collected and evaluated to determine if a drug has fully sterilized a granuloma (Via et al. 2015). This is a method that has been evaluated additionally in cynomolgus macaques (Gideon et al. 2015). There is limited work on TB in baboons, although that species of monkeys is also susceptible to Mtb under experimental conditions (Martino, Hubbard and Schlabritz-Loutsevitch 2007). It may be useful to look at the effectiveness of the baboon as a model of TB co-infection because extensive work has been done in baboons to study T2D (Rincon-Choles et al. 2012). T2D is known to promote Mtb infection and TB disease (Gauld and Lyall 1947; Kapur and Harries 2013). Treatment strategies for T2D have exhibited promise as anti-TB host-directed therapeutics (Singhal et al. 2014).
In terms of determining which species to use as a model for TB, an understanding of what one wishes to accomplish must be reached, considering both the strain of Mtb being used as well as what effect is being investigated. In side-by-side comparisons of rhesus macaques and cynomolgus macaques, the former have been found to be more susceptible to developing active TB infections when infected with a virulent strain, Mtb Erdman (Langermans et al. 2001; Sharpe et al. 2009; Maiello et al. 2017). The lowest dose used of Mtb Erdman by Sharpe et al., however, was ∼30 CFU, with an upper limit of 500 CFU (Sharpe et al. 2009). In a later study, where a retained aerosol exposure was calculated to be as low as 3 CFU of Mtb Erdman, Sharpe et al. still found that rhesus macaques were overall more likely to progress to active disease and that cynomolgus macaques possessed a reduced disease burden (Sharpe et al. 2016). When a less virulent strain is used, such as CDC1551, it is possible to investigate latency even in rhesus macaques (Sestak et al. 2011; Foreman et al. 2016).
VALIDATION OF MOLECULAR DETERMINANTS OF INFECTION, DISEASE AND PROTECTION IN THE NHP MODEL
Over the last decade, important discoveries have been made using innovative approaches, including novel genomic techniques, to validate molecular determinants of infection and disease in NHPs. In 2007, Huang et al. established and validated a real-time quantitation system that identified 78 immune genes that were induced in rhesus macaques that were vaccinated/challenged with BCG (Huang et al. 2007). The first application of a system-wide approach in the NHP model of TB infection showed that gene expression in recently formed lung granulomas is characterized by a massive cytokine storm (Mehra et al. 2010). However, with the establishment of the chronic state of infection, this intense expression of pro-inflammatory cytokines was rapidly inhibited in later granulomas (Mehra et al. 2010). This study provided significant insights into the functioning of the primate granuloma. The initial study, conducted in rhesus, was then validated in cynomolgus macaques, where the effect of BCG vaccination on the progression of granuloma gene expression was also studied (Mehra et al. 2013). These studies indicated that primate lung TB granulomas were characterized by a high degree of expression of markers of adaptive immune dysfunction, such as indoleamine 2,3-dioxygenase (IDO), Arg1, LAG-3 and PD-1 (Mehra et al. 2013). Many of these observations have since been validated (Mattila et al. 2013; Phillips et al. 2015; Gautam et al. 2017). These results suggest that either immune exhaustion or suppression events may be common in granulomas. In support of this, T cells derived from NHP TB granulomas appear to be dysfunctional (Gideon et al. 2015), although it has recently been suggested that exhaustion may not be the main mechanism for this (Wong et al. 2018). Inhibition of IDO activity relieves the inhibition on T cells (Gautam et al. 2017) and it may soon be possible to test the impact of inhibiting other immune checkpoints, e.g. LAG-3 and PD-1 in this model, using either an antibody/depletion or inhibitor/antagonism approach. Numerous other cellular pathways that have been identified to be involved in TB pathogenesis using murine models of TB can be validated using NHPs to assess the feasibility of designing host-directed therapies directed against these pathways. These include the C-Abl pathway (Napier et al. 2011, 2015), the SIRT pathway (Cheng et al. 2017) and the AMPK pathways (Singhal et al. 2014). The C-Abl pathway can be modulated by the inhibitor Imatinib, a tyrosine kinase inhibitor (TKI). Inhibition of the C-Abl pathway by Imatinib correlates with better control of TB via myelopoesis (Napier et al. 2011, 2015). The SIRT pathway can be induced using agonists like pterostilbene (PTS) and its induction correlates with better control of TB due to enhanced macrophage function (Cheng et al. 2017). The AMPK pathway can be induced by the antidiabetic drug metformin (Singhal et al. 2014). Several of these pathways suggest the interface between immunity and cellular metabolism during Mtb infection. Mtb is already well described to preferentially alter the host's cellular metabolism, in addition to its own metabolism. Furthermore, these pathways also suggest mechanisms by which T2D and other metabolic syndromes could affect the immunity to TB in individual patients.
System-wide transcriptomic approaches in NHPs infected with Mtb have also been used to identify correlates of risk of TB development in the blood (Gideon et al. 2016) as well as lung tissue (Kaushal et al. 2015). These results show that transcriptional changes in this model translate well to the human situation and can be used for novel discoveries. In studies of macaques infected with specific Mtb mutants, the global host response has identified correlates of lack of disease despite infection (these responses may or may not translate to correlates of protection) (Mehra et al. 2012). By investigating global responses in the lungs of rhesus macaques infected with Mtb:ΔdosR mutants which are deficient in responding to hypoxia, we have found that Mtb actively subverts the recruitment of Th1 cells into the early granuloma (Mehra et al. 2015). System-wide measurements have also been applied to the granuloma level in both rhesus (Mehra et al. 2010) and cynomolgus (Mehra et al. 2013) macaques. In both models, we conclude that early lesions are characterized by a proinflammatory cytokine storm but later lesions are reprogrammed to suppress such responses (Mehra et al. 2010). It has also been found that a balance of pro- and anti-inflammatory cytokines, including IL-10 and IL-17, are associated with sterilization (Gideon et al. 2015). We have since successfully used lung and bronchoalveolar lavage (BAL) responses to study vaccine-induced protection (Kaushal et al. 2015), as well as the effect of host-directed therapeutics (Gautam et al. 2017).
Some of the recent advances in the NHP model have resulted from the use of high-definition live imaging using [18F]2-fluoro-deoxy-D-glucose ([18F]FDG) positron emission tomography–computed tomography (PET/CT) scanning (Lin et al. 2013; Coleman et al. 2014; Lin et al. 2014; Martin et al. 2017; White et al. 2017). PET has proven particularly useful in determining the inflammatory status of individual lesions. Generally, these studies suggest that PET-hot granulomas are prevalent in early time points and later time points are characterized by increased PET-cold lesions (Coleman et al. 2014). These studies confirm previous transcriptomics results that showed a cooling of proinflammatory signatures in granulomas over the course of time (Mehra et al. 2010, 2013). The most important message from these studies is the extensive heterogeneity in granuloma phenotype. Each granuloma appears to be an individual entity and factors governing their inflammatory status are not well understood. Recent cutting-edge molecular studies in this model include the use of digitally barcoded strains (Martin et al. 2017) to identify the founder bacilli for individual lesions and blockade of individual immune pathways and cells (Diedrich, Mattila and Flynn 2013; Gautam et al. 2017). Some of the recent advances in anti-TB vaccine discovery have involved the use of isogenic Mtb mutants (Kaushal et al. 2015; Foreman et al. 2017) or specific cytomegalovirus-based viral vectors in this model.
MICE
Rodent, and primarily murine, research is ubiquitous in science and is utilized in the study of nearly every human disease as ~99% of all mouse genes possess a human homolog (Consortium 2002). Mice also have the benefit of posing a low cost to laboratories and having a wide variety of available genetic knockouts for immunological studies. Cost is of particular importance to TB research since the requirement for specialized containment facilities leads to increased expenses to begin with. In terms of an animal model for TB, mice are susceptible to disseminating infection and are typically incapable of controlling the infection (Kramnik, Demant and Bloom 1998). Most strains do not form classical granulomas like those found in humans (see Figure 1) and instead form diffused, non-caseating structures, such as the C57BL/6 strain that forms non-necrotic lesions primarily comprised of neutrophils (Flynn 2006; Kramnik and Beamer 2016). Thus, while the mouse model of TB has been extremely useful in identifying key immune mechanisms responsible for the control of TB (Orme 2003), as well as for the assessment of drugs, therapeutics and regimens, its utility in effectively modeling the progression of Mtb infection, its pathology and the host-pathogen interactions that lead to either finite control or progressive TB disease, has been questioned. It has been suggested that the microenvironment within human TB lesions so profoundly impacts the physiology and the metabolism of the pathogen that the study of TB in systems that do not recapitulate these specific stress conditions (e.g. hypoxia) present within human TB lesions can be fundamentally misleading (Kaufmann et al. 2005). Recently, however, murine systems have been developed that recapitulate specific conditions. For example, granulomas formed in C3HeB/FeJ (Kramnik) mice infected with Mtb develop caseating necrosis and become centrally hypoxic (Driver et al. 2012). B6.C3Hsst1 mice provide stable hypoxic lesions to study (Kramnik 2008) and CBA/J IL-10 KO are able to model mature fibrotic granulomas (Cyktor et al. 2013). Of particular interest also are mouse models of Mtb/HIV co-infection that utilize humanized mice. HuMouse develop a progressive, disseminating Mtb infection producing caseous granulomatous inflammation (Calderon et al. 2013). Endsley and colleagues showed that increased Mtb replication (≥1-log) and larger and diffused lesions are observed in these mice following HIV/Mtb co-infection (Nusbaum et al. 2016).
Studies built on the comparison of inbred strains of mice, with varying degrees of susceptibility to Mtb infection (Fortin et al. 2007), are vital for deciphering the role of singular changes to the immune response that may have diverse effects on the overall host. These models have been used for multimodal in vivo and ex vivo imaging to assess necrosis, fibrosis, RNAseq of whole granulomas, macrophage recruitment and death, as well as IFN and ISR pathway activation for comprehensive quantitative evaluation of the dynamics of TB granulomas.
Recently, two additional mouse models have captured the imagination of the field, largely due to their ability to offer maximal allelic variation within a mouse model that more closely resembles human population dynamics. One, known as the Collaborative Cross (CC) model, is a large panel of new inbred mouse strains derived from an eight-way cross using a set of Jackson Labs mice that included three wild-derived strains. CC mice display a broad range of susceptibility to Mtb infection, which are expectedly heritable (Smith et al. 2016). The other, known as the Diversity Outbred (DO) model, represents a far more diverse underlying genotype and was produced by a novel outbreeding strategy that maintained a balanced mixture of the founder genomes and avoided allelic loss and inbreeding (Churchill et al. 2012). Each DO mouse is genetically unique and, unlike inbred strains, DO mice have normal levels of heterozygosity, recapitulating that of humans (Svenson et al. 2012). The Jackson Labs DO model was generated using 160 breeding lines as founders, some which were also used to construct the CC mice (Churchill et al. 2012). These DO mice have since been established as the models of choice to study qualitatively different outcomes in behavior (Logan et al. 2013), cancer (Winter et al. 2017) and infectious diseases (McHugh et al. 2013). DO mice display a wide range of pathologic phenotypes following influenza virus or bacterial co-infection (McHugh et al. 2013). These results offer a glimpse of the powerful potential that these newer mouse models, especially the DO outbred model, provide in the study of complex human diseases with varied phenotypes. TB is one such disease, with Mtb infection leading to varied outcomes over the course of time. Genetics has been strongly implicated in this diversity of phenotypes. When DO mice were infected with standard doses of Mtb via aerosol, super-susceptible, susceptible and resistant phenotypes were readily observed (Niazi et al. 2015). In addition to experiencing highly variable outcomes, DO mice exhibited a spectrum of necrotic (human-like) lesions including visible necrosis of individual macrophages, necrotizing granulomas with a fibrotic capsule, tuberculous pneumonia with intra-alveolar neutrophilic exudates and thrombosis of the lung capillaries (Niazi et al. 2015; Kramnik and Beamer 2016).
More recently, the DO mouse collection has been used to measure the impact of host-directed therapeutics (specifically vitamin D) on the progression of Mtb infection (Bhatt et al. 2016). In these studies, changes in host-specific immune responses, e.g. cellular recruitment and phenotype, its impact on the granuloma structure and changes in bacterial burden could be readily measured (Bhatt et al. 2016). DO mice exhibit a range of bacterial burdens following aerosol infection with Mtb, enabling DO mice to act as an improved mouse co-infection model (Monin et al. 2015). The DO mouse model has been able to recapitulate complex phenotypes related to Mtb infection, such as helminth co-infections (Monin et al. 2015), as well as vaccine efficacy (Griffiths et al. 2016). Furthermore, several genes in a TB susceptibility signature identified in an adolescent cohort study of human household contacts (Zak et al. 2016) that predicted progression to TB far earlier than when clinical signs were apparent can be detected in the lungs of Mtb-infected DO mice, but not conventional B6 mice, by RNAseq (our unpublished data).
In the process of testing other toll-like receptor (TLR) agonists to similarly activate innate DC pathways in mice, we identified monophosphoryl lipid A (MPL) as a safe adjuvant for pulmonary delivery (Ahmed et al. 2017). We next tested whether the delivery of MPL and CD40 agonist FGK 4.5 in DO mice improves Mtb control, as seen in inbred B6 mice (Ahmed et al. 2017). Accordingly, individual DO mice were vaccinated with BCG, and at the time of Mtb infection received MPL, FGK4.5 along with IL-10R neutralizing antibody. IL-10 was identified as a key anti-inflammatory cytokine that limits T cell responses (Griffiths et al. 2016). These new data show that DO mice that received the DC activation therapy resulted in improved Mtb control and formed enhanced B cell follicles, which serve as a correlate of improved Mtb control in both murine and NHP animal models (Slight et al.2013 ; Kaushal et al. 2015; Foreman et al. 2016). DO mice mimic the spectrum of disease seen in human TB and, similar to humans, exhibit variability in control upon Mtb challenge (Gopal et al. 2013). These model systems are, therefore, likely to advance our understanding of TB pathogenesis and immunity in the years to come. Furthermore, these will serve as initial screening tools for therapeutic agents in the setting of human-like genetic variability.
Mice are immensely valuable for their ability to provide a clear analysis of protection in vaccine studies and in mechanistic studies using genetic knockouts. This latter attribute is particularly important since the interaction of the Mtb bacillus with the host is complex and multi-parametric. Numerous cell types of the immune system and several immune pathways are involved, giving rise to variability and heterogeneity in phenotypes, which mechanistic approaches can better control.
VALIDATION OF MOLECULAR DETERMINANTS OF INFECTION, DISEASE AND PROTECTION IN THE MURINE MODEL
Seminal murine studies have established principles of Mtb infection progression and TB disease in the murine model and identified bacterial determinants of disease and host contributions into both protection from and pathology of TB. The current iteration of the murine model of TB was optimized in the early 1990s with the advent of the ability to infect mice with aerosols of Mtb. These studies showed that Mtb was more virulent when the exposure route was aerosol instead of the previously used intravenous route (North 1995). This led to the adoption of the aerosol route of murine infection as the model of choice by the field. Further experimentation with this route of infection showed that, unlike systemic infection with Mtb, adaptive immune responses are not initially elicited following Mtb infection via the aerosol route (Cooper 2014), a result which has since been confirmed in other experimental models (Mehra et al. 2010).
Using a mouse model where the route of infection is comparable to humans was a defining achievement. Simultaneously, landmark studies by Jacobs, Bloom and others allowed the development of genetic systems for the manipulation and the investigation of Mtb (Jacobs et al. 1989, 1990; Cirillo et al. 1991; Jacobs et al. 1991; Balasubramanian et al. 1996). These systems allowed the generation of isogenic mutants of Mtb for testing in murine virulence experiments. Eventually, genome-wide libraries of Mtb transposon-interrupted mutants were generated which allowed a system-wide investigation of the bacterial genes required for the survival of the pathogen in murine lungs (Sassetti and Rubin 2003; Lamichhane, Tyagi and Bishai 2005). Successful execution of these projects has since allowed a limited number of similar investigations to be performed in guinea pigs and macaques (Jain et al. 2007; Dutta et al. 2010). One approach was to generate Mariner-transposon mutants of Mtb with lesions in individual genes and to pool them for use in in vitro or in vivo studies, known as Designer Arrays for Define Mutant Analysis (DeADMAn). This allowed >1/3rd of all Mtb genes to be mutated and individual mutants included in the library (Lamichhane et al. 2003). Bayesian statistical analyses of this library revealed that at least a third of the Mtb genes may be essential and it may not be possible to interrupt these. The DeADMAn approach has been used to study molecules required for bacterial survival and persistence in mice (Lamichhane, Tyagi and Bishai 2005). It was found that 6% of the Mtb mutants were essential for growth in the mouse model, including those encoding for mycobacterial membrane large and small protein family (mmpL/S), as well as genes involved in the synthesis of (p)ppGpp, the alarmone that signals stringent response due to stress and amino acid starvation (Haseltine and Block 1973). Most of these observations have since been confirmed using specific isogenic mutants and the molecules implicated in these murine screens have been validated to be critical for TB disease progression (Varela et al. 2012; Weiss and Stallings 2013; Degiacomi et al. 2017). Another approach that was used to test the consequences of genome-wide mutagenesis of Mtb genes in the mouse model was Transposon Site Hybridization (TraSH). This approach combined high-density insertional mutagenesis with either microarray- or TnSeq-based mapping of mutant pools enriched or diminished post-infection (Sassetti, Boyd and Rubin 2001; Sassetti and Rubin 2003; Griffin et al. 2011, 2012; DeJesus et al. 2017). These studies identified a core group of ∼200 genes (5% of the genome, a number that compares favorably to the one arrived at by the DeADMAn approach), which were required for the survival of Mtb in mice. Many of these genes were unique to mycobacteria and the majority were of hitherto unknown functions (Sassetti and Rubin 2003). Many of the core genes required for virulence in the mouse model have since been individually validated to encode molecules important for in vivo survival and/or persistence of the bacillus.
Since the first draft of the Mtb genome sequence became available, it has become abundantly clear that this remarkable pathogen has evolved to respond to a variety of stress conditions during its in vivo life cycle (Cole et al. 1998). Numerous stress conditions that could be biologically relevant in vivo have been identified, e.g. damage to Mtb cell wall, or DNA, acidic pH in the phagosomal environment, nitrosative and oxidative stresses, nutritional starvation including phosphate, and hypoxia (Stallings and Glickman 2010). These stressors do not work in isolation but may in fact be interlinked. Furthermore, Mtb genes and pathways have been implicated to be involved in defense against such stress conditions and specific knockout of molecules or pathways analyzed for phenotype relative to wild-type Mtb in the mouse model of aerosol infection. Thus, a mutant in the kasB gene, which encodes an enzyme in the mycolic acid synthesis pathway, was susceptible to treatment with antimicrobial peptides that damage cell wall and found to be highly susceptible to killing in the murine model. These results showed a connection between the specific molecular function of an Mtb protein and its ability to encounter and respond to in vivo relevant stress. Numerous other mutants in genes involved in cell wall component synthesis, maintenance and modification (e.g. phthiocerol dimycocerosate, mycolic acids and cyclopropanes) have been observed to be deficient for survival in murine lungs (Glickman, Cox and Jacobs 2000; Rao et al. 2005). Similarly, mutants in Mtb genes that protect against or repair DNA damage also have significant defects for growth in the murine model. For example, Rv1633, which encodes for UvrB, forms a critical part of the nucleotide excision repair machinery (Darwin et al. 2003). Interestingly, there was a significant defect in the killing of mice by a strain unable to synthesize UvrB, but the difference in tissue bacterial burdens was minimal (Darwin et al. 2003). These results suggest that while the murine model may be good for replicating several stress conditions that Mtb faces in human lungs, it is possible that specific mechanisms that restrict the replication of the bacillus in human lungs may be somewhat differently invoked in murine lungs. Mtb mutants in serine protease genes Rv3671c and Rv2224c were unable to survive in murine lungs, validating their role in protecting the bacillus from acidic pH by preserving intrabacillary pH (Vandal et al. 2008; Vandal, Nathan and Ehrt 2009). Mtb uses a multitude of pathways to counter oxidative burst of the phagosome, indicating the importance of being able to survive in the wake of this stress. These include a catalase gene katG (Pym, Saint-Joanis and Cole 2002) and two superoxide dismutase genes, sodA (Edwards et al. 2001) and sodC (Piddington et al. 2001). Mutants in katG have strong defects in murine growth (Pym, Saint-Joanis and Cole 2002) and sodA is an essential gene. When the expression of sodA was perturbed by antisense RNA approach, the resulting strain survived poorly in mice (Edwards et al. 2001). Mtb also experiences oxidoreductive stress and has extensive means to respond to it. Mtb encodes a stress response network orchestrated by an alternate, extracytoplasmic factor response sigma factor sigH, which is induced in response to reductive, acidic pH, heat shock and a variety of other stresses (Fernandes et al. 1999; Manganelli et al. 1999). SigH orchestrates an antioxidative response by inducing the expression of thioredoxin/thioredoxin reductase proteins (Bell et al. 2016). Surprisingly, a mutant in the sigH gene, while being susceptible to oxidoreductive stress, could grow in mice to levels comparable with wild-type Mtb despite evidence of lesser pathology (Kaushal et al. 2002).
The murine model has perhaps been most beneficial in establishing the key role of various components of the immune system in protection from TB. Thus, seminal studies by Flynn and Bloom, and Cooper and Orme, established the importance of CD4+ T cells in protecting from TB, especially those expressing IFN-γ and IL-12 and those involved in TNF-α induction (Cooper et al. 1993; Flynn et al. 1993a, 1993b, 1995a, 1995b; Cooper et al. 1997). Since then, mouse studies have revealed a relatively smaller role of CD8+ T cells in protection from Mtb infection (Lin and Flynn 2015) and an even more miniscule contribution of B cells and humoral immunity (Chan et al. 2014). Work by Khader and Cooper has since revealed an interesting role for the Th17 branch of immunity in TB (Khader et al. 2005, 2007). More recent work suggests that the role of some immune pathways, particularly Th17, may be governed by the strain of Mtb used and its relative pathogenicity (Gopal et al. 2014). Similarly, Turner and colleagues have established the negative role of IL-10 signaling in the control of Mtb infection (Beamer et al. 2008). These studies have since allowed us to focus on the function of specific lymphocytes. Thus, it has been postulated that murine granuloma T cells may have compromised cytokine section and functional activity (Egen et al. 2008; Bold et al. 2011; Egen et al. 2011).
RABBITS
Early on, rabbits of different origins were shown to have differential susceptibility to TB (Lurie, Zappasodi and Tickner 1955). Rabbits have commonly been infected via the aerosol route (Manabe et al. 2008), however human strains of Mtb are not fully virulent in rabbits (Dannenberg and Collins 2001). During infection with M. bovis, rabbits are able to form pulmonary cavitation, which has been considered to resemble Mtb-induced granulomas in humans more faithfully than those formed in mice and guinea pigs (Dannenberg 2006). Most of the species available today are resistant to Mtb as Lurie's susceptible breed had died out (Dharmadhikari and Nardell 2008). Seminal studies by Dannenberg et al. showed that alveolar macrophages are the target cell for Mtb and are required for resistance to TB (Henderson, Dannenberg and Lurie 1963; Rojas-Espinosa et al. 1974). Resistance to TB required proteolytic enzymes functioning in the endocytic pathway (Henderson, Dannenberg and Lurie 1963; Rojas-Espinosa et al. 1974). Dannenberg also illuminated the phenomenon of delayed type hypersensitivity in the rabbit model (Dannenberg 1991). More recently, the model has been employed to study mechanisms of Mtb persistence and reactivation (Geiman et al. 2004; Kesavan et al. 2009), cavitary dissemination (Nedeltchev et al. 2009), response of Mtb to host-mediated hypoxia in the granuloma (Converse et al. 2009), correlates of immune-mediated latency (Subbian et al. 2013a, 2013b) and both the environment of failed immunity and its correction by host-directed therapeutics (Subbian et al. 2011a, 2011b, 2016). Manabe et al. have developed the only animal model of TB-related immune reconstitution inflammatory syndrome (IRIS) in rabbits to study the progression and severity of the immune reconstitution inflammatory syndrome (Manabe et al. 2008). The model has now been employed for testing drug effectiveness, particularly pharmacokinetics and pharmacodynamics, to estimate drug penetration into different types of lesions in experimentally infected rabbits (Rifat et al. 2018). Rabbits also form caseating, necrotic granulomas (Figure 1) which, under the correct conditions, may liquefy and cavitate (Nedeltchev et al. 2009). Bishai and colleagues have developed a reliable model of cavitary disease in rabbits and confirmed pathological and molecular correlates of human disease. This model permits in vivo imaging to observe and quantify the events leading to cavitation (Kubler et al. 2015) and thus has tremendous potential in the study of granuloma breakdown.
VALIDATION OF MOLECULAR DETERMINANTS OF INFECTION, DISEASE AND PROTECTION IN THE RABBIT MODEL
As discussed above, Subbian et al. have characterized various outcomes of Mtb infection in the rabbit model (Subbian et al. 2011a, 2012,2013b; Tsenova et al. 2014). They also applied whole genome transcriptomic evaluation to better understand universal host responses as they relate to the progression of infection (Subbian et al. 2012). Rabbits infected with low doses of low-virulence strain CDC1551, which elicits strong immune responses, initially exhibited bacterial growth and pathology, but eventually controlled infection in a latent form (Subbian et al. 2012). Genome-wide transcriptional analysis supported this notion, with the number of genes involved in both inflammatory responses and in lung remodeling (required for granuloma formation) peaking 2 weeks after infection in rabbit lungs and then progressively reducing in extent (Subbian et al. 2012). Similar results were earlier obtained in both the rhesus (Mehra et al. 2010) and the cynomolgus (Mehra et al. 2013) macaque models, which is discussed subsequently. The use of PET/CT imaging was first employed in the rabbit model of TB to study the dynamics of infection. These studies also confirmed earlier transcriptomic outcomes that low-dose experimental infection in this model involved an initial inflammatory phase followed by a chronic phase with reduced granuloma-associated inflammatory signal (Via et al. 2012). Thus, the use of state-of-the-art molecular approaches in animal models have extended our understanding of the granuloma formation/maturation kinetics in a manner not possible in humans. Circulatory human cytokine/chemokine profile-based molecular analytes are now being used to successfully study Mtb infection in rabbits (Dehnad et al. 2016).
Transcriptomics has also been employed in the rabbit model to successfully evaluate responses to therapy, including host-directed therapy (Subbian et al. 2011a). However, the most striking use of global ‘omics’ approach in this model has been to compare infection with a high-immunogenic vs. a high-virulence strain of Mtb (Subbian et al. 2013b). According to these reports, Mtb CDC1551, and perhaps other hyper-immunogenic strains, express early lung transcriptional profiles of infection that result in reduced STAT1 induction and polymorphonuclear leukocyte (PMN) recruitment and inflammation. Hence, the usual outcome of infection in rabbits with this strain is LTBI (Bishai et al. 1999; Subbian et al. 2012, 2013b). On the contrary, STAT1 and inflammatory signals were highly expressed following comparable infection with the hypervirulent HN878 strain and led to high levels of PMN recruitment and cavitary disease (Subbian et al.2013b). While further research across experimental models is necessary, these results provide a potential mechanism for how hyper-virulent strains may have acquired the ability to elicit lung pathology as a means of overcoming the ability of the large majority of human beings to restrict Mtb infection as LTBI.
GUINEA PIGS
Guinea pigs are susceptible to Mtb and form granulomas, so they have been useful in immunohistopathological stages of pulmonary granuloma formation (Dannenberg and Collins 2001; Turner, Basaraba and Orme 2003). Guinea pigs have also been useful in investigations of responses to antigenic lipopeptides due to their possession of type I CD1 presentation molecules (Kaufmann et al. 2016). Initially considered the gold standard for vaccine testing against TB, the guinea pig model is considered highly susceptible (Clark, Hall and Williams 2014) and has suffered from a lack of immunological tools such as guinea pig-specific antibodies for immune analysis in the past. It is expected, however, that more tools will be available in the future. Thus, a genome-wide interrogation of transcriptional responses to Mtb infection in this model is now possible (Jain, Dey and Tyagi 2012). An immune-gene specific array platform has been available for over a decade now and has been used to study Mtb infection in this model (Tree et al. 2006). This reagent has validated that Mtb infection of guinea pigs elicits strong and immediate Th1 responses (Tree et al. 2006), indicating its ability to recapitulate aspects of the human immune response to Mtb.
VALIDATION OF MOLECULAR DETERMINANTS OF INFECTION, DISEASE AND PROTECTION IN THE GUINEA PIG MODEL
Guinea pigs have been successfully leveraged to study Mtb-specific metabolic fate. With the increasing understanding that the immune system interacts with metabolism with influential consequences on the fate of various infections, interest in the guinea pig model of TB has also increased. Recently, important studies have investigated persisters in lung granulomas of this animal model (Lenaerts et al. 2007). Basaraba et al. have recently been successful in developing a model of TB/T2D in these guinea pigs (Podell et al. 2014, 2017). This is an exceptionally significant advance. T2D is a major cause of morbidity and mortality worldwide amongst non-infectious causes and accounts for ∼90% of the entire diabetic burden (Deshpande, Harris-Hayes and Schootman 2008). Furthermore, the rates of T2D have been significantly increasing both globally as well as in areas of the world where TB is endemic. Thus, globally, it is predicted that the number of T2D patients will double between 2010 and 2030 (Rowley et al. 2017). It has been proposed that T2D is a comorbidity that is fueling the TB pandemic in certain parts of the world at levels comparable with HIV co-infection, especially in regions where TB is already endemic (Dooley and Chaisson 2009). When T2D was induced in guinea pigs, Mtb infection caused a significantly higher degree of TB disease with reduced survival and severe pathological presentation than controls (Podell et al. 2014). This coincided with uncontrolled inflammatory responses. Thus, the guinea pig model allows an in-depth study of T2D/TB comorbidity, including the testing of novel approaches for therapeutic control.
The guinea pig model has also been used in mutant infection screening experiments to better understand the bacillary determinants of pathogenesis (Jain et al. 2007). When compared with mice infected with the identical mutant library, guinea pigs permitted an accelerated detection of genes required for the in vivo survival of Mtb (Jain et al. 2007). These results suggest that TB-related pathology in guinea pig lungs results in relatively greater immune pressure on the bacillus than the corresponding murine pathology.
RATS
Rats remain a more recent and less developed model of TB infection out of all mammalian models. Recently, Lewis rats (Sugawara, Yamada and Mizuno 2004a), American cotton rats (Elwood et al. 2007), Wistar rats (Gaonkar et al. 2010; Cardona et al. 2011; Singhal et al. 2011) and diabetic rat strains (Sugawara, Yamada and Mizuno 2004b; Sugawara and Mizuno 2008) have been used to develop the rat model of TB infection. They have also been used to demonstrate Th1-shifts in immunity following BCG vaccination (Cardona et al. 2011). Unlike mice, rats have been found to form well-organized granulomas and accumulate foamy macrophages (Figure 1), which have been associated with pulmonary lesion resolution (Singhal et al. 2011). Rats have been used successfully as an animal model for testing drug efficacy (Gaonkar et al. 2010; Foo et al. 2011) and antigen-specific T cell responses (Cardona et al. 2011; Foo et al. 2011; Singhal et al. 2011) and have been a valuable animal model for pharamacokinetic and toxicology studies by pharmaceutical companies (Gumbo et al. 2015). Similar to NHP models of TB infection, Wistar rats exposed to a low dose infection were found to control or clear bacilli by 4 months post-infection, depending on the strain used (Singhal et al. 2011), indicating that they have the potential to model latency. As a co-infection model, rats would be of benefit in the exploration of diabetes as Goto Kakizaki T2D rats (McGillivray et al. 2014) and Komeda diabetes-prone rats (Sugawara and Mizuno 2008) have been shown to have a higher susceptibility to TB compared with wild-type rats. Like mice, rats are simple to house, however they can withstand repeated blood collections and could be used during long-term infection studies.
ZEBRAFISH
Despite the fact that zebrafish do not possess lungs, researchers have been successful in utilizing their infection with M. marinum to uncover the mechanisms of microgranuloma formation prior to antigen-specific responses (Volkman et al. 2010; Ramakrishnan 2013). This model is useful in the determination of elementary mycobacterial pathogenesis and treatment. Many seminal recent discoveries about granuloma induction and the role of various mycobacterial virulence factors in this process have been initiated in this model (Volkman et al. 2010; Cambier et al. 2014). Zebrafish were vital in showing that macrophage death spreads bacilli to adjacent macrophages encapsulated within the same granuloma (Volkman et al. 2004). Many of these observations were extended from the fish model and validated in clinical human samples (Tobin et al. 2010). Recently, investigations in this system have identified specific mechanisms by which granulomas mature, with specialized phagocyte morphology contributing to the pathology of the lesion and promoting bacillary replication and persistence (Cronan et al. 2016). The zebrafish embryo model was used to show innate immune mechanisms that lead to granuloma formation, including induction of innate immune aggregates (Figure 1) and activation of M. marinum granuloma-specific genes (Davis et al. 2002). Zebrafish are small, easy to image (especially in the larval stage where the lesions can be directly visualized) and offer the advantage of significantly powered group sizes while posing minimal increases in overall cost (Cronan and Tobin 2014). The lack of host homology, however, limits the ability of zebrafish to completely model human infection, particularly latency. Special facilities are required for studies utilizing the fish model, however these studies have an additional benefit of being conducted at BSL2 since most studies utilize the strain M. marinum, which rarely infects humans, rather than Mtb.
IMPORTANCE OF ANIMAL MODELS FOR DRUG AND VACCINE EFFICACY
Animal models are important for evaluating new drug regimens prior to introduction in humans. With humans, the threat of developing drug or multidrug resistant strains prevents rapid introduction and animals, particularly mice, provide a niche where drug regimens can be used quickly to determine if new regimens are efficacious in treating and eliminating TB. Mice were useful in showing that rifapentine and moxifloxacin could be used to eliminate TB within 2 months, rather than the standard 6 months with rifampin (R), isoniazid (H) and pyrazinamide (Z) (Rosenthal et al. 2007). The use of different strains is vital to the evaluation of drug efficacy for later human use. Dual and triple drug combinations of H, R and Z have shown that different challenge strains yielded different in vivo efficacy in BALB/c mice (De Groote et al. 2012), leading to the need to develop improved drug regimens for eventual human use. Animal models allow an avenue to explore these new drugs prior to human use. Different quinolones (Lenaerts et al. 2008) and nitroimidazo-oxazines (Nuermberger et al. 2008) have recently been investigated to replace the current three-drug regimen. Recently, a subclass of quinolones called 2-pyridones has been investigated, including KRQ-10 018, and was found to be similar in in vivo efficacy to isoniazid and have less activity than moxifloxacin (Lenaerts et al. 2008). When the nitroimidazo-oxazine PA-824 was used with moxifloxacin and pyrazinamide regimen as a substitute for rifapentine, it cured mice more quickly than the standard regimen, but not within 3 months of treatment (Nuermberger et al. 2008).
As early as 1997, it was suggested that vaccine efficacy is altered based on the number of passages that the BCG strain has undergone (Behr and Small 1997). Behr et al. found that significant genetic changes had accumulated in many of the BCG strains, including the loss of region of difference 2 (RD2) in BCG-Denmark, BCG-Tice and BCG-Glaxo (Behr and Small 1999). In mice, it was found that the Prague and Japanese strains of BCG had lower recovery rates compared with three other BCG strains and were unable to protect against a second challenge with Mtb (Lagranderie et al. 1996). These studies highlight that, despite the presence of a vaccine that has been accepted for a long period, proper maintenance of the vaccine strain is vital in ensuring its efficacy. Experiments in animal models are thus necessary to ensure that efficacy is maintained over time to protect vulnerable human populations.
It has been observed for some time that BCG is primarily efficacious in the prevention of disseminated TB in children, so studies in other animal models are vital in investigating improvements that must be made to improve protection. Animal models have supported the lack of widespread protection produced by the BCG vaccine. Using Mtb Erdman and HN878 challenge to evaluate parenteral BCG vaccination efficacy, Verreck et al. showed that protection widely varies in rhesus macaques, as is the case in humans (Verreck et al. 2017). Until we possess a vaccine that prevents adult pulmonary TB, it will be impossible to eliminate the disease. Due to the existence of multidrug and extremely drug-resistant TB, treatment improvements alone will not be sufficient to contain the infection. Improved developments in vaccines, treatments and patient support for improved compliance will be necessary to ultimately eliminate TB.
IMPORTANCE OF STRAIN-STRAIN COMPARISON IN ANIMAL MODELS
It has been estimated that humans can be infected with Mtb with an infectious dose as low as 10 CFU. Humans have a 10% lifetime risk of progressing to active disease once they have acquired TB (Society AT 2000). As a result, using an animal model with a higher susceptibility rate would make sense in replicating the natural, human response to infection. As humans have different susceptibilities to different strains of Mtb, it is ideal to compare the susceptibilities of each animal model with different strains of Mtb, as well as using multiple strains of Mtb to model treatment efficacy (De Groote et al. 2012). Several animal models have been used for side-by-side susceptibility comparisons of different strains such as Mtb Erdman, H37Rv and CDC1551 including mice (Kelley and Collins 1999; Manca et al. 1999; Ordway et al. 2007; De Groote et al. 2012), rabbits (Manabe et al. 2003; Tsenova et al. 2005), rats (Singhal et al. 2011) and NHPs (Cadena et al. 2016). In early comparisons over the course of 3 months, mouse studies failed to show differential susceptibilities among different strains of Mtb in intravenous or aerosol challenge (Kelley and Collins 1999). A study involving comparisons of rabbits aerosol-challenged with Mtb Erdman, CDC1551 and H37Rv found that Erdman was the most virulent strain and caused a broader spectrum of disease within 16–20 weeks post-challenge (Manabe et al. 2003). Several studies of strain-strain comparisons have been conducted within the NHP model as well. Despite marmosets having high susceptibility, they have been used to reveal that strain and infectious dose can lead to heterogenous responses by comparing low-dose Erdman and CDC1551 infection ranging from 1–12 CFU (Cadena et al. 2016).
The use of animal models in strain-strain comparison are beneficial in that they enable one to investigate both differences in the host's responses to the individual strains as well as differential virulence mechanisms of the strains themselves. Using New Zealand white rabbits, Tsenova et al. found that phenolic glycolipid (PGL) biosynthesis is critical in subverting the host's ability to produce protective TNF-α by comparing rabbits infected with CDC1551, HN878 and HN878pks1–15::hyg (Tsenova et al. 2005). Using CDC1551 and HN878 infection, Subbian et al. showed that differential leukocyte activation and recruitment within the first 3 h of infection led to vastly different outcomes despite identical infectious doses being used (Subbian et al.2013b). HN878-infected rabbits that developed cavitary disease had increased expression of pro-inflammatory genes that are associated with extravasation and activation of PMNs in the lungs, including those producing TNF-α, IL-8, IL-15, MCP-1 and CXCL10. Other studies have found that CDC1551 induces a more rapid and robust response in mice, with granulomas containing high levels of TNF-α, IL-6, IL-10, IL-12 and IFN-γ mRNA (Manca et al. 1999). Mice were also vital in showing that some of HN878 hypervirulence is due to the induction of stronger Th1 responses and rapid Treg emergence compared with other strains that result in their decreased survival (Ordway et al. 2007). Studies utilizing multiple challenge strains are also vital in evaluating the protection offered by different vaccines. BCG has shown efficacy in protecting mice from various strains of Mtb, however these differ incredibly from clinical observations in humans (Jeon et al. 2008).
ADDITIONAL GENOMIC APPROACHES IN THE ANIMAL MODELS OF TB
The availability of genome sequences in the past two decades has spawned the sciences of systems biology, where the understanding of the entire genome is leveraged to understand the responses of both the pathogen and the host on a ‘system-wide’ level. Initial studies focused on a simplistic design where the response of Mtb to various in vitro conditions directly or indirectly corresponding to stresses that the pathogen might be subjected to in vivo were measured. These experiments clearly illuminated that Mtb has developed an intricate network of transcriptional networks to fine-tune responses to different stresses. Key molecules in this network included sigma factors, which serve to guide the RNA polymerase to specifically initiate gene expression. The Mtb genome encodes about a dozen stress- or condition-specific sigma factors whose expression are often intricately controlled at multiple levels (Gomez, Chen and Bishai 1997). Thus, it was shown that sigE responds to stress that weakens the Mtb cell wall and, unlike wild-type Mtb, ΔsigE mutants are effectively killed by phagocytes (Manganelli et al. 2001). SigE is one of the few molecules known to be expressed in Mtb-infected macrophages by selective capture of transcribed sequences (SCOTS), an early genome-wide RNA measurement technique. Since those early days, the animal models of TB have progressed tremendously, and state-of-the art techniques are now routinely used in these to study both the pathogenesis of Mtb as well as the evaluation of host responses involved in immunity as well as pathology. These include comparative genomics, post-genomic mutations, transcriptomics (beginning with DNA microarrays, but now including RNAseq and single cell RNAseq), proteomics (including to detect proteins with modifications such as phosphorylation, etc.), lipidomics, metabolomics and whole animal live imaging by magnetic resonance imaging (MRI) and PET/CT.
CONCLUSIONS
No one animal model recapitulates the breadth of heterogeneity in human TB. However, several new mouse models can portray specific events in the granuloma such as hypoxia or fibrosis. Similarly, guinea pigs, zebrafish and rabbit infection models have significantly contributed to our understanding of Mtb infection and the resulting immune response. Perhaps the best recapitulation of Mtb-specific pathogenesis events and immunity occurs in NHPs. Mtb-infected macaques have been used for transcriptome-wide immune response evaluation in peripheral blood to back-translate key findings for humans. These studies have been extended to evaluate BAL and lung samples. State-of-the-art molecular imaging tools such as PET/CT have been successfully employed in this model. The use of bar-coded strains and isogenic mutants to better understand TB pathogenesis has revealed interesting information from this model. Finally, the use of the NHP model is informing vaccine and therapeutic discovery critical to the future control of the TB pandemic. Overall, the use of animal models improves our understanding of Mtb disease pathogenesis and provides us with models that recapitulate aspects of human disease for the testing of new vaccine and treatment regimens that are vital to ultimately eliminate TB.
FUNDING
Support for this work came from NIH awards to DK (AI111943, AI135726, AI134240), DK/SAK (AI111914, AI123780) and SM (AI134245) and institutional NIH awards (OD011133, OD011104).
Conflict of interest. None declared.
REFERENCES
- Ahmed M, Jiao H, Domingo-Gonzalez Ret al.. Rationalized design of a mucosal vaccine protects against Mycobacterium tuberculosis challenge in mice. J Leukoc Biol. 2017;101:1373–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai JW, Ruan QL, Liu QHet al.. Updates on the risk factors for latent tuberculosis reactivation and their managements. Emerg Microbes Infect. 2016;5:e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anacker RL, Brehmer W, Barclay WRet al.. Superiority of intravenously administered BCG and BCG cell walls in protecting rhesus monkeys (Macaca mulatta) against airborne tuberculosis. Z Immunitatsforsch Exp Klin Immunol. 1972;143:363–76. [PubMed] [Google Scholar]
- Balasubramanian V, Pavelka MS Jr., Bardarov SSet al.. Allelic exchange in Mycobacterium tuberculosis with long linear recombination substrates. J Bacteriol. 1996;178:273–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay WR, Anacker RL, Brehmer Wet al.. Aerosol-induced tuberculosis in subhuman primates and the course of the disease after intravenous BCG vaccination. Infect Immun. 1970;2:574–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay WR, Busey WM, Dalgard DWet al.. Protection of monkeys against airborne tuberculosis by aerosol vaccination with bacillus Calmette-Guerin. Am Rev Respir Dis. 1973;107:351–8. [DOI] [PubMed] [Google Scholar]
- Beamer GL, Flaherty DK, Assogba BDet al.. Interleukin-10 promotes Mycobacterium tuberculosis disease progression in CBA/J mice. J Immunol. 2008;181:5545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr MA, Small PM. Has BCG attenuated to impotence?. Nature. 1997;389:133–4. [DOI] [PubMed] [Google Scholar]
- Behr MA, Small PM. A historical and molecular phylogeny of BCG strains. Vaccine. 1999;17:915–22. [DOI] [PubMed] [Google Scholar]
- Bell LC, Pollara G, Pascoe Met al.. In vivo molecular dissection of the effects of HIV-1 in active tuberculosis. PLoS Pathog. 2016;12:e1005469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt K, Rafi W, Shah Net al.. 1,25 (OH)2D3 treatment alters the granulomatous response in M. tuberculosis infected mice. Sci Rep. 2016;6:34469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishai WD, Parrish AM Jr., Ruiz Net al.. Virulence of Mycobacterium tuberculosis CDC1551 and H37Rv in rabbits evaluated by Lurie's pulmonary tubercle count method. Infect Immun. 1999;67:4931–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bold TD, Banaei N, Wolf AJet al.. Suboptimal activation of antigen-specific CD4+ effector cells enables persistence of M. tuberculosis in vivo. PLoS Pathog. 2011;7:e1002063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley JM, Schacker TW, Ruff LEet al.. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. 2004;200:749–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JN, Palermo RE, Baskin CRet al.. Macaque proteome response to highly pathogenic avian influenza and 1918 reassortant influenza virus infections. J Virol. 2010;84: 12058–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadena AM, Klein EC, White AGet al.. Low dose Mycobacterium tuberculosis yield diverse host outcomes in common marmosets (Callithrix jacchus). Comp Med. 2016;66:412–9. [PMC free article] [PubMed] [Google Scholar]
- Calderon VE, Valbuena G, Goez Yet al.. A humanized mouse model of tuberculosis. PLoS One. 2013;8:e63331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calenda G, Villegas G, Barnable Pet al.. MZC gel inhibits SHIV-RT and HSV-2 in macaque vaginal mucosa and SHIV-RT in rectal mucosa. J Acquir Immune Defic Syndr. 2017;74:e67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambier CJ, Takaki KK, Larson RPet al.. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature. 2014;505:218–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuano SV 3rd, Croix DA, Pawar Set al.. Experimental Mycobacterium tuberculosis infection of cynomolgus macaques closely resembles the various manifestations of human M. tuberculosis infection. Infect Immun. 2003;71:5831–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona P-J, Mathys V, Kiass Met al.. BCG induces protection against Mycobacterium tuberculosis infection in the Wistar rat model. PLoS One. 2011;6:e28082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chackerian AA, Alt JM, Perera TVet al.. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infect Immun. 2002;70:4501–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J, Mehta S, Bharrhan Set al.. The role of B cells and humoral immunity in Mycobacterium tuberculosis infection. Semin Immunol. 2014;26:588–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Huang D, Wang RCet al.. A critical role for CD8 T cells in a nonhuman primate model of tuberculosis. PLoS Pathog. 2009;5:e1000392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Gutierrez NM, Marzuki MBet al.. Host sirtuin 1 regulates mycobacterial immunopathogenesis and represents a therapeutic target against tuberculosis. Sci Immunol. 2017;2:eaaj1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA, Gatti DM, Munger SCet al.. The diversity outbred mouse population. Mamm Genome. 2012;23:713–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo JD, Barletta RG, Bloom BRet al.. A novel transposon trap for mycobacteria: isolation and characterization of IS1096. J Bacteriol. 1991;173:7772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark S, Hall Y, Williams A. Animal models of tuberculosis: guinea pigs. Cold Spring Harb Perspect Med. 2014;5:a018572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill Jet al.. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. [DOI] [PubMed] [Google Scholar]
- Coleman MT, Chen RY, Lee Met al.. PET/CT imaging reveals a therapeutic response to oxazolidinones in macaques and humans with tuberculosis. Sci Transl Med. 2014;6:265ra167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium MGS. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–62. [DOI] [PubMed] [Google Scholar]
- Converse PJ, Karakousis PC, Klinkenberg LGet al.. Role of the dosR-dosS two-component regulatory system in Mycobacterium tuberculosis virulence in three animal models. Infect Immun. 2009;77:1230–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AM. Mouse model of tuberculosis. Cold Spring Harb Perspect Med. 2014;5:a018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AM, Dalton DK, Stewart TAet al.. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med. 1993;178:2243–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AM, Magram J, Ferrante Jet al.. Interleukin 12 (IL-12) is crucial to the development of protective immunity in mice intravenously infected with mycobacterium tuberculosis. J Exp Med. 1997;186:39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronan MR, Beerman RW, Rosenberg AFet al.. Macrophage epithelial reprogramming underlies mycobacterial granuloma formation and promotes infection. Immunity. 2016;45:861–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronan MR, Tobin DM. Fit for consumption: zebrafish as a model for tuberculosis. Dis Model Mech. 2014;7:777–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crostarosa F, Aravantinou M, Akpogheneta OJet al.. A macaque model to study vaginal HSV-2/immunodeficiency virus co-infection and the impact of HSV-2 on microbicide efficacy. PLoS One. 2009;4:e8060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyktor JC, Carruthers B, Kominsky RAet al.. IL-10 inhibits mature fibrotic granuloma formation during Mycobacterium tuberculosis infection. J Immunol. 2013;190:2778–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannenberg AM Jr. Delayed-type hypersensitivity and cell-mediated immunity in the pathogenesis of tuberculosis. Immunol Today. 1991;12:228–33. [DOI] [PubMed] [Google Scholar]
- Dannenberg AM Jr. Pathogenesis of Human Pulmonary Tuberculosis: Insights from the Rabbit Model. 2006, Washington, DC: ASM Press. [Google Scholar]
- Dannenberg AM Jr., Collins FM. Progressive pulmonary tuberculosis is not due to increasing numbers of viable bacilli in rabbits, mice and guinea pigs, but is due to a continuous host response to mycobacterial products. Tuberculosis. 2001;81:229–42. [DOI] [PubMed] [Google Scholar]
- Darwin KH, Ehrt S, Gutierrez-Ramos JCet al.. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science. 2003;302:1963–6. [DOI] [PubMed] [Google Scholar]
- Davis JM, Clay H, Lewis JLet al.. Real-time visualization of mycobacterium-macrophage interactions leading to initiation of granuloma formation in zebrafish embryos. Immunity. 2002;17:693–702. [DOI] [PubMed] [Google Scholar]
- De Groote MA, Gruppo V, Woolhiser LKet al.. Importance of confirming data on the in vivo efficacy of novel antibacterial drug regimens against various strains of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2012;56:731–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degiacomi G, Benjak A, Madacki Jet al.. Essentiality of mmpL3 and impact of its silencing on Mycobacterium tuberculosis gene expression. Sci Rep. 2017;7:43495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehnad A, Ravindran R, Subbian Set al.. Development of immune-biomarkers of pulmonary tuberculosis in a rabbit model. Tuberculosis (Edinb). 2016;101:1–7. [DOI] [PubMed] [Google Scholar]
- DeJesus MA, Gerrick ER, Xu Wet al.. Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. MBio. 2017;8:e02133-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande AD, Harris-Hayes M, Schootman M. Epidemiology of diabetes and diabetes-related complications. Phys Ther. 2008;88:1254–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmadhikari AS, Nardell EA. What animal models teach humans about tuberculosis. Am J Respir Cell Mol Biol. 2008;39:503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diedrich CR, Mattila JT, Flynn JL. Monocyte-derived IL-5 reduces TNF production by Mycobacterium tuberculosis-specific CD4 T cells during SIV/M. tuberculosis coinfection. J Immunol. 2013;190:6320–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diedrich CR, Mattila JT, Klein Eet al.. Reactivation of latent tuberculosis in cynomolgus macaques infected with SIV is associated with early peripheral T cell depletion and not virus load. PLoS One. 2010;5:e9611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley KE, Chaisson RE. Tuberculosis and diabetes mellitus: convergence of two epidemics. Lancet Infect Dis. 2009;9:737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douek DC, Brenchley JM, Betts MRet al.. HIV preferentially infects HIV-specific CD4+ T cells. Nature. 2002;417:95–8. [DOI] [PubMed] [Google Scholar]
- Driver ER, Ryan GJ, Hoff DRet al.. Evaluation of a mouse model of necrotic granuloma formation using C3HeB/FeJ mice for testing of drugs against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2012;56:3181–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta NK, Mehra S, Didier PJet al.. Genetic requirements for the survival of tubercle bacilli in primates. J Infect Dis. 2010;201:1743–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi VP, Bhattacharya D, Chatterjee Set al.. Mycobacterium tuberculosis directs T helper 2 cell differentiation by inducing interleukin-1beta production in dendritic cells. J Biol Chem. 2012;287:33656–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards KM, Cynamon MH, Voladri RKet al.. Iron-cofactored superoxide dismutase inhibits host responses to Mycobacterium tuberculosis. Am J Respir Crit Care Med. 2001;164:2213–9. [DOI] [PubMed] [Google Scholar]
- Egen JG, Rothfuchs AG, Feng CGet al.. Macrophage and T cell dynamics during the development and disintegration of mycobacterial granulomas. Immunity. 2008;28:271–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egen JG, Rothfuchs AG, Feng CGet al.. Intravital imaging reveals limited antigen presentation and T cell effector function in mycobacterial granulomas. Immunity. 2011;34:807–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwood RL, Wilson S, Blanco JCet al.. The American cotton rat: a novel model for pulmonary tuberculosis. Tuberculosis (Edinb). 2007;87:145–54. [DOI] [PubMed] [Google Scholar]
- Fernandes NW, Kong QL, Puyang Det al.. A mycobacterial extracytoplasmic sigma factor involved in survival following heat shock and oxidative stress. J Bacteriol. 1999;181:4266–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JL. Lessons from experimental Mycobacterium tuberculosis infections. Microbes Infect. 2006;8:1179–88. [DOI] [PubMed] [Google Scholar]
- Flynn JL, Chan J, Triebold KJet al.. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med. 1993a;178:2249–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JL, Goldstein MM, Chan Jet al.. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995a;2:561–72. [DOI] [PubMed] [Google Scholar]
- Flynn JL, Goldstein MM, Triebold KJet al.. Major histocompatibility complex class I-restricted T cells are necessary for protection against M. tuberculosis in mice. Infect Agents Dis. 1993b;2:259–62. [PubMed] [Google Scholar]
- Flynn JL, Goldstein MM, Triebold KJet al.. IL-12 increases resistance of BALB/c mice to Mycobacterium tuberculosis infection. J Immunol. 1995b;155:2515–24. [PubMed] [Google Scholar]
- Foo DG, Tay HC, Siew JYet al.. T cell monitoring of chemotherapy in experimental rat tuberculosis. Antimicrob Agents Chemother. 2011;55:3677–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foreman TW, Mehra S, Lackner AAet al.. Translational research in the nonhuman primate model of tuberculosis. ILAR J. 2017;58:151–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foreman TW, Mehra S, LoBato DNet al.. CD4+ T-cell-independent mechanisms suppress reactivation of latent tuberculosis in a macaque model of HIV coinfection. Proc Natl Acad Sci USA. 2016;113:E5636–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foreman TW, Veatch AV, LoBato DNet al.. Nonpathologic infection of macaques by an attenuated mycobacterial vaccine is not reactivated in the setting of HIV co-infection. Am J Pathol. 2017;187:2811–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin A, Abel L, Casanova JLet al.. Host genetics of mycobacterial diseases in mice and men: forward genetic studies of BCG-osis and tuberculosis. Annu Rev Genomics Hum Genet. 2007;8:163–92. [DOI] [PubMed] [Google Scholar]
- Franchini G, Nacsa J, Hel Zet al.. Immune intervention strategies for HIV-1 infection of humans in the SIV macaque model. Vaccine. 2002;20 Suppl 4:A52–60. [DOI] [PubMed] [Google Scholar]
- Gandhi NR, Moll A, Sturm AWet al.. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet North Am Ed. 2006;368:1575–80. [DOI] [PubMed] [Google Scholar]
- Gaonkar S, Bharath S, Kumar Net al.. Aerosol infection model of tuberculosis in Wistar rats. Int J Microbiol. 2010;2010:426035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauld WR, Lyall A. Tuberculosis as a complication of diabetes. Br Med J. 1947;1:677–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam US, Foreman TW, Bucsan ANet al.. In vivo inhibition of tryptophan catabolism reorganizes the tuberculoma and augments immune-mediated control of Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2017;115:E62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiman DE, Kaushal D, Ko Cet al.. Attenuation of late-stage disease in mice infected by the Mycobacterium tuberculosis mutant lacking the SigF alternate sigma factor and identification of SigF-dependent genes by microarray analysis. Infect Immun. 2004;72:1733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gideon HP, Phuah J, Myers AJet al.. Variability in tuberculosis granuloma T cell responses exists, but a balance of pro- and anti-inflammatory cytokines is associated with sterilization. PLoS Pathog. 2015;11:e1004603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gideon HP, Skinner JA, Baldwin Net al.. Early whole blood transcriptional signatures are associated with severity of lung inflammation in cynomolgus macaques with Mycobacterium tuberculosis infection. J Immunol. 2016;197:4817–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman MS, Cox JS, Jacobs WR Jr.. A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis. Mol Cell. 2000;5:717–27. [DOI] [PubMed] [Google Scholar]
- Gomez JE, Chen JM, Bishai WR. Sigma factors of Mycobacterium tuberculosis. Tuber Lung Dis. 1997;78:175–83. [DOI] [PubMed] [Google Scholar]
- Gopal R, Monin L, Slight Set al.. Unexpected role for IL-17 in protective immunity against hypervirulent Mycobacterium tuberculosis HN878 infection. PLoS Pathog. 2014;10:e1004099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal R, Monin L, Torres Det al.. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med. 2013;188:1137–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gormus BJ, Blanchard JL, Alvarez XHet al.. Evidence for a rhesus monkey model of asymptomatic tuberculosis. J Med Primatol. 2004;33:134–45. [DOI] [PubMed] [Google Scholar]
- Griffin JE, Gawronski JD, Dejesus MAet al.. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7:e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JE, Pandey AK, Gilmore SAet al.. Cholesterol catabolism by Mycobacterium tuberculosis requires transcriptional and metabolic adaptations. Chem Biol. 2012;19:218–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths KL, Ahmed M, Das Set al.. Targeting dendritic cells to accelerate T-cell activation overcomes a bottleneck in tuberculosis vaccine efficacy. Nat Commun. 2016;7:13894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbo T, Lenaerts AJ, Hanna Det al.. Nonclinical models for antituberculosis drug development: a landscape analysis. J Infect Dis. 2015;211 Suppl 3:S83–95. [DOI] [PubMed] [Google Scholar]
- Haseltine WA, Block R. Synthesis of guanosine tetra- and pentaphosphate requires the presence of a codon-specific, uncharged transfer ribonucleic acid in the acceptor site of ribosomes. Proc Natl Acad Sci USA. 1973;70:1564–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatziioannou T, Ambrose Z, Chung NPet al.. A macaque model of HIV-1 infection. Proc Natl Acad Sci USA. 2009;106:4425–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson HJ, Dannenberg AM Jr., Lurie MB. Phagocytosis of tubercle bacilli by rabbit pulmonary alveolar macrophages and its relation to native resistance to tuberculosis. J Immunol. 1963;91:553–6. [PubMed] [Google Scholar]
- Henkle E, Winthrop KL. Nontuberculous mycobacteria infections in immunosuppressed hosts. Clin Chest Med. 2015;36:91–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Qiu L, Wang Ret al.. Immune gene networks of mycobacterial vaccine–elicited cellular responses and immunity. J Infect Dis. 2007;195:55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulskotte EG, Geretti AM, Osterhaus AD. Towards an HIV-1 vaccine: lessons from studies in macaque models. Vaccine. 1998;16:904–15. [DOI] [PubMed] [Google Scholar]
- Jacobs WR Jr., Kalpana GV, Cirillo JDet al.. Genetic systems for mycobacteria. Methods Enzymol. 1991;204:537–55. [DOI] [PubMed] [Google Scholar]
- Jacobs WR Jr., Snapper SB, Lugosi Let al.. Development of genetic systems for the mycobacteria. Acta Leprol. 1989;7 Suppl 1:203–7. [PubMed] [Google Scholar]
- Jacobs WR Jr., Snapper SB, Lugosi Let al.. Development of BCG as a recombinant vaccine vehicle. Curr Top Microbiol Immunol. 1990;155:153–60. [DOI] [PubMed] [Google Scholar]
- Jain R, Dey B, Tyagi AK. Development of the first oligonucleotide microarray for global gene expression profiling in guinea pigs: defining the transcription signature of infectious diseases. BMC Genomics. 2012;13:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain SK, Hernandez-Abanto SM, Cheng QJet al.. Accelerated detection of Mycobacterium tuberculosis genes essential for bacterial survival in guinea pigs, compared with mice. J Infect Dis. 2007;195:1634–42. [DOI] [PubMed] [Google Scholar]
- Jeon BY, Derrick SC, Lim JHet al.. Mycobacterium bovis BCG immunization induces protective immunity against nine different Mycobacterium tuberculosis strains in mice. Infect Immun. 2008;76:5173–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur A, Harries AD. The double burden of diabetes and tuberculosis - public health implications. Diabetes Res Clin Pract. 2013;101:10–19. [DOI] [PubMed] [Google Scholar]
- Kaufmann E, Spohr C, Battenfeld Set al.. BCG vaccination induces robust CD4+ T cell responses to Mycobacterium tuberculosis complex-specific lipopeptides in guinea pigs. J Immunol. 2016;196:2723–32. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH, Cole ST, Mizrahi Vet al.. Mycobacterium tuberculosis and the host response. J Exp Med. 2005;201:1693–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal D, Foreman TW, Gautam USet al.. Mucosal vaccination with attenuated Mycobacterium tuberculosis induces strong central memory responses and protects against tuberculosis. Nat Commun. 2015;6:8533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal D, Mehra S. Faithful experimental models of human infection. Mycobact Dis. 2012;2:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal D, Mehra S, Didier PJet al.. The non-human primate model of tuberculosis. J Med Primatol. 2012;41:191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal D, Schroeder BG, Tyagi Set al.. Reduced immunopathology and mortality despite tissue persistence in a Mycobacterium tuberculosis mutant lacking alternative sigma factor, SigH. Proc Natl Acad Sci USA. 2002;99:8330–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley CL, Collins FM. Growth of a highly virulent strain of Mycobacterium tuberculosis in mice of differing susceptibility to tuberculous challenge. Tuber Lung Dis. 1999;79:367–70. [DOI] [PubMed] [Google Scholar]
- Kesavan AK, Brooks M, Tufariello Jet al.. Tuberculosis genes expressed during persistence and reactivation in the resistant rabbit model. Tuberculosis (Edinb). 2009;89:17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khader SA, Bell GK, Pearl JEet al.. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–77. [DOI] [PubMed] [Google Scholar]
- Khader SA, Pearl JE, Sakamoto Ket al.. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J Immunol. 2005;175:788–95. [DOI] [PubMed] [Google Scholar]
- Kimata JT. Stepping toward a macaque model of HIV-1 induced AIDS. Viruses. 2014;6:3643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano M, Itoh Y, Kodama Met al.. Establishment of a cynomolgus macaque model of influenza B virus infection. Virology. 2010;407:178–84. [DOI] [PubMed] [Google Scholar]
- Kramnik I. Genetic dissection of host resistance to Mycobacterium tuberculosis: the sst1 locus and the Ipr1 gene. Curr Top Microbiol Immunol. 2008;321:123–48. [DOI] [PubMed] [Google Scholar]
- Kramnik I, Beamer G. Mouse models of human TB pathology: roles in the analysis of necrosis and the development of host-directed therapies. Semin Immunopathol. 2016;38:221–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramnik I, Demant P, Bloom BB. Susceptibility to tuberculosis as a complex genetic trait: analysis using recombinant congenic strains of mice. Novartis Found Symp. 1998;217:120–31. [DOI] [PubMed] [Google Scholar]
- Kubler A, Luna B, Larsson Cet al.. Mycobacterium tuberculosis dysregulates MMP/TIMP balance to drive rapid cavitation and unrestrained bacterial proliferation. J Pathol. 2015;235:431–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Farhana A, Guidry Let al.. Redox homeostasis in mycobacteria: the key to tuberculosis control?. Expert Rev Mol Med. 2011;13:e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda MJ, Sugimoto C, Cai Yet al.. High turnover of tissue macrophages contributes to tuberculosis reactivation in simian immunodeficiency virus-infected rhesus macaques. J Infect Dis. 2018;217:1865–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan CK, Ernst JD. HIV and tuberculosis: a deadly human syndemic. Clin Microbiol Rev. 2011;24:351–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagranderie MR, Balazuc AM, Deriaud Eet al.. Comparison of immune responses of mice immunized with five different Mycobacterium bovis BCG vaccine strains. Infect Immun. 1996;64:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhane G, Tyagi S, Bishai WR. Designer arrays for defined mutant analysis to detect genes essential for survival of Mycobacterium tuberculosis in mouse lungs. Infect Immun. 2005;73:2533–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhane G, Zignol M, Blades NJet al.. A postgenomic method for predicting essential genes at subsaturation levels of mutagenesis: application to Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2003;100:7213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langermans JA, Andersen P, van Soolingen Det al.. Divergent effect of bacillus Calmette-Guérin (BCG) vaccination on Mycobacterium tuberculosis infection in highly related macaque species: implications for primate models in tuberculosis vaccine research. Proc Natl Acad Sci USA. 2001;98:11497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaerts AJ, Bitting C, Woolhiser Let al.. Evaluation of a 2-pyridone, KRQ-10018, against Mycobacterium tuberculosis in vitro and in vivo. Antimicrob Agents Chemother. 2008;52:1513–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaerts AJ, Hoff D, Aly Set al.. Location of persisting mycobacteria in a guinea pig model of tuberculosis revealed by r207910. Antimicrob Agents Chemother. 2007;51:3338–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewinsohn DM, Tydeman IS, Frieder Met al.. High resolution radiographic and fine immunologic definition of TB disease progression in the rhesus macaque. Microbes Infect. 2006;8:2587–98. [DOI] [PubMed] [Google Scholar]
- Lin PL, Coleman T, Carney JPet al.. Radiologic responses in cynomolgous macaques for assessing tuberculosis chemotherapy regimens. Antimicrob Agents Chemother. 2013;57:4237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PL, Dietrich J, Tan Eet al.. The multistage vaccine H56 boosts the effects of BCG to protect cynomolgus macaques against active tuberculosis and reactivation of latent Mycobacterium tuberculosis infection. J Clin Invest. 2012;122:303–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PL, Flynn JL. CD8 T cells and Mycobacterium tuberculosis infection. Semin Immunopathol. 2015;37:239–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PL, Ford CB, Coleman MTet al.. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat Med. 2014;20:75–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PL, Pawar S, Myers Aet al.. Early events in Mycobacterium tuberculosis infection in cynomolgus macaques. Infect Immun. 2006;74:3790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PL, Rodgers M, Smith Let al.. Quantitative comparison of active and latent tuberculosis in the cynomolgus macaque model. Infect Immun. 2009;77:4631–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan RW, Robledo RF, Recla JMet al.. High-precision genetic mapping of behavioral traits in the diversity outbred mouse population. Genes Brain Behav. 2013;12:424–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardini ED, Gettayacamin M, Turner GDet al.. A review of plasmodium coatneyi-macaque models of severe malaria. Vet Pathol. 2015;52:998–1011. [DOI] [PubMed] [Google Scholar]
- Lurie MB, Zappasodi P, Tickner C. On the nature of genetic resistance to tuberculosis in the light of the host-parasite relationships in natively resistant and susceptible rabbits. Am Rev Tuberc. 1955;72:297–329. [DOI] [PubMed] [Google Scholar]
- Maiello P, DiFazio RM, Cadena AMet al.. Rhesus macaques are more susceptible to progressive tuberculosis than cynomolgus macaques: A quantitative comparison. Infect Immun. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe YC, Dannenberg AM Jr., Tyagi SKet al.. Different strains of Mycobacterium tuberculosis cause various spectrums of disease in the rabbit model of tuberculosis. Infect Immun. 2003;71:6004–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe YC, Kesavan AK, Lopez-Molina Jet al.. The aerosol rabbit model of TB latency, reactivation and immune reconstitution inflammatory syndrome. Tuberculosis (Edinb). 2008;88:187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manca C, Tsenova L, Barry CE 3rdet al.. Mycobacterium tuberculosis CDC1551 induces a more vigorous host response in vivo and in vitro, but is not more virulent than other clinical isolates. J Immunol. 1999;162:6740–6. [PubMed] [Google Scholar]
- Manganelli R, Dubnau E, Tyagi Set al.. Differential expression of 10 sigma factor genes in Mycobacterium tuberculosis. Mol Microbiol. 1999;31:715–24. [DOI] [PubMed] [Google Scholar]
- Manganelli R, Voskuil MI, Schoolnik GKet al.. The Mycobacterium tuberculosis ECF sigma factor sigmaE: role in global gene expression and survival in macrophages. Mol Microbiol. 2001;41:423–37. [DOI] [PubMed] [Google Scholar]
- Martin CJ, Cadena AM, Leung VWet al.. Digitally barcoding mycobacterium tuberculosis reveals in vivo infection dynamics in the macaque model of tuberculosis. MBio. 2017;8:e00312–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino M, Hubbard GB, Schlabritz-Loutsevitch N. Tuberculosis (Mycobacterium tuberculosis) in a pregnant baboon (Papio cynocephalus). J Med Primatol. 2007;36:108–12. [DOI] [PubMed] [Google Scholar]
- Mattila JT, Ojo OO, Kepka-Lenhart Det al.. Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J Immunol. 2013;191:773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott JE, Shankaran H, Eisfeld AJet al.. Conserved host response to highly pathogenic avian influenza virus infection in human cell culture, mouse and macaque model systems. BMC Syst Biol. 2011;5:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGillivray A, Golden NA, Gautam USet al.. The Mycobacterium tuberculosis Rv2745c plays an important role in responding to redox stress. PLoS One. 2014;9:e93604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh KJ, Mandalapu S, Kolls JKet al.. A novel outbred mouse model of 2009 pandemic influenza and bacterial co-infection severity. PLoS One. 2013;8:e82865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehra S, Alvarez X, Didier PJet al.. Granuloma correlates of protection against tuberculosis and mechanisms of immune modulation by Mycobacterium tuberculosis. J Infect Dis. 2013;207:1115–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehra S, Foreman TW, Didier PJet al.. The DosR regulon modulates adaptive immunity and is essential for M. tuberculosis persistence. Am J Respir Crit Care Med. 2015;191:1185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehra S, Golden NA, Stuckey Ket al.. The Mycobacterium tuberculosis stress response factor SigH is required for bacterial burden as well as immunopathology in primate lungs. J Infect Dis. 2012;205:1203–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehra S, Pahar B, Dutta NKet al.. Transcriptional reprogramming in nonhuman primate (rhesus macaque) tuberculosis granulomas. PLoS One. 2010;5:e12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliori GB, Centis R, D'Ambrosio Let al.. Totally drug-resistant and extremely drug-resistant tuberculosis: the same disease?. Clin Infect Dis. 2012;54:1379–80. [DOI] [PubMed] [Google Scholar]
- Monin L, Griffiths KL, Lam WYet al.. Helminth-induced arginase-1 exacerbates lung inflammation and disease severity in tuberculosis. J Clin Invest. 2015;125:4699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napier RJ, Norris BA, Swimm Aet al.. Low doses of imatinib induce myelopoiesis and enhance host anti-microbial immunity. PLoS Pathog. 2015;11:e1004770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napier RJ, Rafi W, Cheruvu Met al.. Imatinib-sensitive tyrosine kinases regulate mycobacterial pathogenesis and represent therapeutic targets against tuberculosis. Cell Host Microbe. 2011;10:475–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedeltchev GG, Raghunand TR, Jassal MSet al.. Extrapulmonary dissemination of Mycobacterium bovis but not Mycobacterium tuberculosis in a bronchoscopic rabbit model of cavitary tuberculosis. Infect Immun. 2009;77:598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niazi MK, Dhulekar N, Schmidt Det al.. Lung necrosis and neutrophils reflect common pathways of susceptibility to Mycobacterium tuberculosis in genetically diverse, immune-competent mice. Dis Model Mech. 2015;8:1141–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RJ. Mycobacterium tuberculosis is strikingly more virulent for mice when given via the respiratory than via the intravenous route. J Infect Dis. 1995;172:1550–3. [DOI] [PubMed] [Google Scholar]
- Nuermberger E, Tyagi S, Tasneen Ret al.. Powerful bactericidal and sterilizing activity of a regimen containing PA-824, moxifloxacin, and pyrazinamide in a murine model of tuberculosis. Antimicrob Agents Chemother. 2008;52:1522–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes-Alves C, Booty MG, Carpenter SMet al.. In search of a new paradigm for protective immunity to TB. Nat Rev Microbiol. 2014;12:289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusbaum RJ, Calderon VE, Huante MBet al.. Pulmonary tuberculosis in humanized mice infected with HIV-1. Sci Rep. 2016;6:21522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordway D, Henao-Tamayo M, Harton Met al.. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J Immunol. 2007;179:522–31. [DOI] [PubMed] [Google Scholar]
- Orme IM. The mouse as a useful model of tuberculosis. Tuberculosis (Edinb). 2003;83:112–5. [DOI] [PubMed] [Google Scholar]
- Peretti S, Shaw A, Blanchard Jet al.. Immunomodulatory effects of HSV-2 infection on immature macaque dendritic cells modify innate and adaptive responses. Blood. 2005;106:1305–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phares TW, May AD, Genito CJet al.. Rhesus macaque and mouse models for down-selecting circumsporozoite protein based malaria vaccines differ significantly in immunogenicity and functional outcomes. Malar J. 2017;16:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipp MT, Doyle LA, Martin DSet al.. A rhesus macaque model of Streptococcus pneumoniae carriage. J Med Primatol. 2012;41:60–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips BL, Mehra S, Ahsan MHet al.. LAG3 expression in active Mycobacterium tuberculosis infections. Am J Pathol. 2015;185:820–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piddington DL, Fang FC, Laessig Tet al.. Cu,Zn superoxide dismutase of Mycobacterium tuberculosis contributes to survival in activated macrophages that are generating an oxidative burst. Infect Immun. 2001;69:4980–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podell BK, Ackart DF, Obregon-Henao Aet al.. Increased severity of tuberculosis in guinea pigs with type 2 diabetes: a model of diabetes-tuberculosis comorbidity. Am J Pathol. 2014;184:1104–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podell BK, Ackart DF, Richardson MAet al.. A model of type 2 diabetes in the guinea pig using sequential diet-induced glucose intolerance and streptozotocin treatment. Dis Model Mech. 2017;10:151–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pym AS, Saint-Joanis B, Cole ST. Effect of katG mutations on the virulence of Mycobacterium tuberculosis and the implication for transmission in humans. Infect Immun. 2002;70:4955–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu L, Huang D, Chen CYet al.. Severe tuberculosis induces unbalanced up-regulation of gene networks and overexpression of IL-22, MIP-1alpha, CCL27, IP-10, CCR4, CCR5, CXCR3, PD1, PDL2, IL-3, IFN-beta, TIM1, and TLR2 but low antigen-specific cellular responses. J Infect Dis. 2008;198:1514–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan L. The zebrafish guide to tuberculosis immunity and treatment. Cold Spring Harb Symp Quant Biol. 2013;78:179–92. [DOI] [PubMed] [Google Scholar]
- Rao V, Fujiwara N, Porcelli SAet al.. Mycobacterium tuberculosis controls host innate immune activation through cyclopropane modification of a glycolipid effector molecule. J Exp Med. 2005;201:535–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiley WW, Calayag MD, Wittmer STet al.. ESAT-6-specific CD4 T cell responses to aerosol Mycobacterium tuberculosis infection are initiated in the mediastinal lymph nodes. Proc Natl Acad Sci USA. 2008;105:10961–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribi E, Anacker RL, Barclay WRet al.. Efficacy of mycobacterial cell walls as a vaccine against airborne tuberculosis in the Rheusus monkey. J Infect Dis. 1971;123:527–38. [DOI] [PubMed] [Google Scholar]
- Rifat D, Prideaux B, Savic RMet al.. Pharmacokinetics of rifapentine and rifampin in a rabbit model of tuberculosis and correlation with clinical trial data. Sci Transl Med. 2018;10:eaai7786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon-Choles H, Abboud HE, Lee Set al.. Renal histopathology of a baboon model with type 2 diabetes. Toxicol Pathol. 2012;40:1020–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas-Espinosa O, Dannenberg AM Jr., Sternberger LAet al.. The role of cathepsin D in the pathogenesis of tuberculosis. A histochemical study employing unlabeled antibodies and the peroxidase-antiperoxidase complex. Am J Pathol. 1974;74:1–17. [PMC free article] [PubMed] [Google Scholar]
- Rosenthal IM, Zhang M, Williams KNet al.. Daily dosing of rifapentine cures tuberculosis in three months or less in the murine model. PLoS Med. 2007;4:e344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley WR, Bezold C, Arikan Yet al.. Diabetes 2030: insights from yesterday, today, and future trends. Popul Health Manag. 2017;20:6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti CM, Boyd DH, Rubin EJ. Comprehensive identification of conditionally essential genes in mycobacteria. Proc Natl Acad Sci USA. 2001;98:12712–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci USA. 2003;100:12989–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sestak K, Conroy L, Aye PPet al.. Improved xenobiotic metabolism and reduced susceptibility to cancer in gluten-sensitive macaques upon introduction of a gluten-free diet. PLoS One. 2011;6:e18648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe S, White A, Gleeson Fet al.. Ultra low dose aerosol challenge with Mycobacterium tuberculosis leads to divergent outcomes in rhesus and cynomolgus macaques. Tuberculosis (Edinb). 2016;96:1–12. [DOI] [PubMed] [Google Scholar]
- Sharpe SA, Eschelbach E, Basaraba RJet al.. Determination of lesion volume by MRI and stereology in a macaque model of tuberculosis. Tuberculosis (Edinb). 2009;89:405–16. [DOI] [PubMed] [Google Scholar]
- Sharpe SA, McShane H, Dennis MJet al.. Establishment of an aerosol challenge model of tuberculosis in rhesus macaques and an evaluation of endpoints for vaccine testing. Clin Vaccine Immunol. 2010;17:1170–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal A, Aliouat el M, Hervé Met al.. Experimental tuberculosis in the Wistar rat: a model for protective immunity and control of infection. PLoS One. 2011;6:e18632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal A, Jie L, Kumar Pet al.. Metformin as adjunct antituberculosis therapy. Sci Transl Med. 2014;6:263ra159. [DOI] [PubMed] [Google Scholar]
- Slight SR, Rangel-Moreno J, Gopal Ret al.. CXCR5(+) T helper cells mediate protective immunity against tuberculosis. J Clin Invest. 2013;123:712–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CM, Proulx MK, Olive AJet al.. Tuberculosis susceptibility and vaccine protection are independently controlled by host genotype. MBio. 2016;7:e01516-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Society AT. Targeted tuberculin testing and treatment of latent tuberculosis infection. MMWR Recomm Rep. 2000;49(RR-6):1–51. [PubMed] [Google Scholar]
- Stallings CL, Glickman MS. Is Mycobacterium tuberculosis stressed out? A critical assessment of the genetic evidence. Microbes Infect. 2010;12:1091–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbian S, Bandyopadhyay N, Tsenova Let al.. Early innate immunity determines outcome of Mycobacterium tuberculosis pulmonary infection in rabbits. Cell Commun Signal. 2013a;11:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbian S, O'Brien P, Kushner NLet al.. Molecular immunologic correlates of spontaneous latency in a rabbit model of pulmonary tuberculosis. Cell Commun Signal. 2013b;11:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbian S, Tsenova L, Holloway Jet al.. Adjunctive phosphodiesterase-4 inhibitor therapy improves antibiotic response to pulmonary tuberculosis in a rabbit model. EBioMedicine. 2016;4:104–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbian S, Tsenova L, O'Brien Pet al.. Phosphodiesterase-4 inhibition alters gene expression and improves isoniazid-mediated clearance of Mycobacterium tuberculosis in rabbit lungs. PLoS Pathog. 2011a;7:e1002262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbian S, Tsenova L, O'Brien Pet al.. Spontaneous latency in a rabbit model of pulmonary tuberculosis. Am J Pathol. 2012;181:1711–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbian S, Tsenova L, Yang Get al.. Chronic pulmonary cavitary tuberculosis in rabbits: a failed host immune response. Open Biol. 2011b;1:110016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara I, Mizuno S. Higher susceptibility of type 1 diabetic rats to Mycobacterium tuberculosis infection. Tohoku J Exp Med. 2008;216:363–70. [DOI] [PubMed] [Google Scholar]
- Sugawara I, Yamada H, Mizuno S. Pathological and immunological profiles of rat tuberculosis. Int J Exp Pathol. 2004a;85:125–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara I, Yamada H, Mizuno S. Pulmonary tuberculosis in spontaneously diabetic goto kakizaki rats. Tohoku J Exp Med. 2004b;204:135–45. [DOI] [PubMed] [Google Scholar]
- Svenson KL, Gatti DM, Valdar Wet al.. High-resolution genetic mapping using the mouse diversity outbred population. Genetics. 2012;190:437–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaim LE, Connolly LE, Volkman HEet al.. Mycobacterium marinum infection of adult zebrafish causes caseating granulomatous tuberculosis and is moderated by adaptive immunity. Infect Immun. 2006;74:6108–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin DM, Vary JC Jr., Ray JPet al.. The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell. 2010;140:717–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tree JA, Elmore MJ, Javed Set al.. Development of a guinea pig immune response-related microarray and its use to define the host response following Mycobacterium bovis BCG vaccination. Infect Immun. 2006;74:1436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsenova L, Ellison E, Harbacheuski Ret al.. Virulence of selected Mycobacterium tuberculosis clinical isolates in the rabbit model of meningitis is dependent on phenolic glycolipid produced by the bacilli. J Infect Dis. 2005;192:98–106. [DOI] [PubMed] [Google Scholar]
- Tsenova L, O'Brien P, Holloway Jet al.. Etanercept exacerbates inflammation and pathology in a rabbit model of active pulmonary tuberculosis. J Interferon Cytokine Res. 2014;34: 716–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner OC, Basaraba RJ, Orme IM. Immunopathogenesis of pulmonary granulomas in the guinea pig after infection with Mycobacterium tuberculosis. Infect Immun. 2003;71:864–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandal OH, Nathan CF, Ehrt S. Acid resistance in Mycobacterium tuberculosis. J Bacteriol. 2009;191:4714–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandal OH, Pierini LM, Schnappinger Det al.. A membrane protein preserves intrabacterial pH in intraphagosomal Mycobacterium tuberculosis. Nat Med. 2008;14:849–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela C, Rittmann D, Singh Aet al.. MmpL genes are associated with mycolic acid metabolism in mycobacteria and corynebacteria. Chem Biol. 2012;19:498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veazey RS, DeMaria M, Chalifoux LVet al.. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science. 1998;280:427–31. [DOI] [PubMed] [Google Scholar]
- Verreck FA, Vervenne RA, Kondova Iet al.. MVA.85A boosting of BCG and an attenuated, phoP deficient M. tuberculosis vaccine both show protective efficacy against tuberculosis in rhesus macaques. PLoS One. 2009;4:e5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verreck FAW, Tchilian EZ, Vervenne RAWet al.. Variable BCG efficacy in rhesus populations: pulmonary BCG provides protection where standard intra-dermal vaccination fails. Tuberculosis (Edinb). 2017;104:46–57. [DOI] [PubMed] [Google Scholar]
- Via LE, England K, Weiner DMet al.. A sterilizing tuberculosis treatment regimen is associated with faster clearance of bacteria in cavitary lesions in marmosets. Antimicrob Agents Chemother. 2015;59:4181–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Via LE, Schimel D, Weiner DMet al.. Infection dynamics and response to chemotherapy in a rabbit model of tuberculosis using [(1)(8)F]2-fluoro-deoxy-D-glucose positron emission tomography and computed tomography. Antimicrob Agents Chemother. 2012;56:4391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Via LE, Weiner DM, Schimel Det al.. Differential virulence and disease progression following Mycobacterium tuberculosis complex infection of the common marmoset (Callithrix jacchus). Infect Immun. 2013;81:2909–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman HE, Clay H, Beery Det al.. Tuberculous granuloma formation is enhanced by a mycobacterium virulence determinant. PLoS Biol. 2004;2:e367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman HE, Pozos TC, Zheng Jet al.. Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science. 2010;327:466–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh GP, Tan EV, dela Cruz ECet al.. The Philippine cynomolgus monkey (Macaca fasicularis) provides a new nonhuman primate model of tuberculosis that resembles human disease. Nat Med. 1996;2:430–6. [DOI] [PubMed] [Google Scholar]
- Weed M, Adams RJ, Hienz RDet al.. SIV/macaque model of HIV infection in cocaine users: minimal effects of cocaine on behavior, virus replication, and CNS inflammation. J Neuroimmune Pharmacol. 2012;7:401–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Stallings CL. Essential roles for Mycobacterium tuberculosis Rel beyond the production of (p)ppGpp. J Bacteriol. 2013;195:5629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyand NJ, Wertheimer AM, Hobbs TRet al.. Neisseria infection of rhesus macaques as a model to study colonization, transmission, persistence, and horizontal gene transfer. Proc Natl Acad Sci USA. 2013;110:3059–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AG, Maiello P, Coleman MTet al.. Analysis of 18FDG PET/CT imaging as a tool for studying mycobacterium tuberculosis infection and treatment in non-human primates. J Vis Exp. 2017, DOI: 10.3791/56375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. Global Tuberculosis Report 2017. 2017, Geneva: World Heatlh Organization. [Google Scholar]
- Winter JM, Gildea DE, Andreas JPet al.. Mapping complex traits in a diversity outbred f1 mouse population identifies germline modifiers of metastasis in human prostate cancer. Cell Syst. 2017;4:31–45. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winthrop KL, McNelley E, Kendall Bet al.. Pulmonary nontuberculous mycobacterial disease prevalence and clinical features: an emerging public health disease. Am J Respir Crit Care Med. 2010;182:977–82. [DOI] [PubMed] [Google Scholar]
- Wolf AJ, Desvignes L, Linas Bet al.. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J Exp Med. 2008;205:105–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong EA, Joslyn L, Grant NLet al.. Low levels of T cell exhaustion in tuberculous lung granulomas. Infect Immun. 2018;86:e00426–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zak DE, Penn-Nicholson A, Scriba TJet al.. A blood RNA signature for tuberculosis disease risk: a prospective cohort study. Lancet. 2016;387:2312–22. [DOI] [PMC free article] [PubMed] [Google Scholar]