

Abstract
TRPM2 is a Ca2+-permeable, nonselective cation channel that plays a role in oxidant-induced cell death, insulin secretion, and cytokine release. Few TRPM2 inhibitors have been reported, which hampers the validation of TRPM2 as a drug target. While screening our in-house marine-derived chemical library, we identified scalaradial and 12-deacetylscalaradial as the active components within an extract of an undescribed species of Cacospongia (class Demospongiae, family Thorectidae) that strongly inhibited TRPM2-mediated Ca2+ influx in TRPM2-overexpressing HEK293 cells. In whole-cell patch-clamp experiments, scalaradial (and similarly 12-deacetylscalaradial) inhibited TRPM2-mediated currents in a concentration- and time-dependent manner (~20 min to full onset; IC50 210 nM). Scalaradial inhibited TRPM7 with less potency (IC50 760 nM) but failed to inhibit CRAC, TRPM4, and TRPV1 currents in whole-cell patch clamp experiments. Scalaradial’s effect on TRPM2 channels was shown to be independent of its well-known ability to inhibit secreted phospholipase A2 (sPLA2) and its reported effects on extracellular signal-regulated kinases (ERK) and Akt pathways. In addition, scalaradial was shown to inhibit endogenous TRPM2 currents in a rat insulinoma cell line (IC50 330 nM). Based on its potency and emerging specificity profile, scalaradial is an important addition to the small number of known TRPM2 inhibitors.
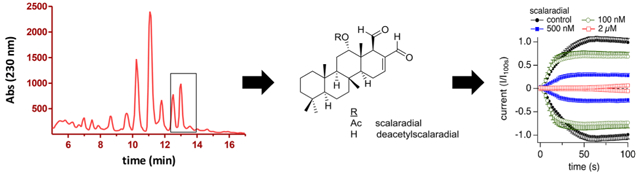
Graphical Abstract
We have focused our marine natural products screening program on the melastatin-like transient receptor potential (TRPM) group of cation channels, including the second member, TRPM2. The TRPM ion channels are an eight-membered family structurally characterized by an N-terminus calmodulin binding IQ-like motif, six trans-membrane segments, a pore-forming loop domain between the fifth and sixth transmembrane helices, and for three of the members—TRPM2, TRPM6, and TRPM7—an enzyme domain in the C-terminus.1,2 Because of this unique dual ion channel and enzymatic function, these three proteins are sometimes called “chanzymes”. Overall, the TRPM protein family represents a diverse profile of permeability to different mono- and divalent cations, as well as different mechanisms of modulation.
TRPM2 forms a voltage-insensitive, nonselective cation channel and possesses an adenosine diphosphate ribose (ADPR) pyrophosphatase domain in its C-terminus (Nudix-like domain NUDT9-H).3 TRPM2 channel activity is primarily activated by binding of ADPR to the NUDT9-H, allowing the permeation of Na+, K+, and Ca2+ into the cytosol for an unusually long period of several seconds.3 Furthermore, elevation of intracellular Ca2+ increases the channel’s sensitivity to ADPR, thereby providing a positive feedback mechanism for Ca2+ influx.4 TRPM2 is also activated by events downstream of cellular exposure to reactive oxygen species that involve the enzymes poly-ADP-ribose polymerase (PARP) and poly-ADP-ribose glycohydrolase (PARG).5 It is hypothesized that intracellular ADPR concentration increases following the activation of PARP, an enzyme involved in the repair of reactive oxygen species (ROS)-mediated DNA damage. ROS stimulates TRPM2 activity, which contributes to cellular Ca2+ overload, suggesting a role for TRPM2 in apoptosis and as a sensor of intracellular oxidants.6
TRPM2 is most notably expressed in the central nervous system with broad, yet regional variability throughout both neuronal and non-neuronal cells in the hippocampus, cerebral cortex, thalamus, and midbrain.1 Expression of TRPM2 mRNA has also been detected in a diverse range of tissues and cell types, including bone marrow, spleen, heart, liver, lung tissue, and gastrointestinal tissues. TRPM2-mediated currents have also been recorded in endothelial, immune cells, Jurkat T cells, microglia, cardio myocytes, and insulinoma cells.1 The protein can be localized in both the plasma and lysosomal membranes, although the mechanism in which this distribution is regulated is not yet understood.7
TRPM2 is recognized as a potential therapeutic target for oxidative-stress-related pathologies, neurodegenerative diseases, and chronic inflammation. Specifically, it is implicated in stroke,8 Parkinson’s disease,9 amyloid-β-induced neurovascular dysfunction,10 cardiac remodeling following myocardial in-farction,11 cell damage following traumatic brain injury,12 diabetes,13 irritable bowel syndrome,14 prostate cancer cell proliferation,15 and decreased susceptibility of breast cancer cells to chemotherapeutics.16 Mice knockout studies that have substantiated a therapeutic potential for TRPM2 inhibition include reduction of neutrophil infiltration and ulceration in a colitis inflammation model,14 reduction of bleomycin-induced lung inflammation,17 and protection against β-amyloid neuronal toxicity.18
There are several known inhibitors of TRPM2, including flufenamic acid,19 clotrimazole,20 2-aminoethyl diphenylborinate (2-APB),21 anthranilic acid,22 and curcumin.23 However, most of these compounds lack either specificity and/or potency. Despite these disadvantages, these types of inhibitors continue to progress the current understanding of TRPM2 and remain an important resource. In the present study we have identified an organic extract of a Cacospongia species that strongly inhibits TRPM2-mediated currents in a time- and concentration-dependent manner. We describe the bioassay-linked fractionation of the active sponge extract, which led to the discovery of the known sesterterpenes scalaradial and 12-deacetylscalaradial as potent TRPM2 inhibitors. We also present evidence that scalaradial inhibits TRPM2 independent of its known secreted phospholipase A2 (sPLA2) and Akt inhibitory activities and demonstrates a specificity profile that is unique from other TRPM2 inhibitors.
RESULTS AND DISCUSSION
Scalaradial-Containing Extracts and Fractions from an Undescribed Species of Cacospongia Inhibit TRPM2-Mediated Ca2+ Influx in HEK293 Cells.
In our screening program for TRPM2 inhibitors from marine organisms, an organic extract of specimens identified as belonging to the genus Cacospongia (class Demospongiae, family Thorectidae) was found to significantly suppress TRPM2-mediated Ca2+ influx in human TRPM2-overexpressing HEK293 cells (TRPM2-HEK293), following exposure to H2O2, as measured by intracellular fura-2 fluorescence (Figure 1A). The active extract was then chromatographed by analytical-scale reversed-phase HPLC while fractions were collected into a 96-well plate (Figure 1C). Assay of the plate indicated that fraction wells containing scalaradial and 12-deacetylscalaradial as their major components (tR 12.5–13.5 min) demonstrated reproducible inhibition in our TRPM2-HEK293 cell Ca2+ imaging assay (Figure 1B). The inhibition of TRPM2-mediated Ca2+ flux in the fluorescence-based assay was corroborated in whole-cell patch clamp experiments with TRPM2-HEK293 cells (Figure 1D), which motivated a scaled up isolation of scalaradial and 12-deacetylscalaradial from the extract of Cacospongia sp.
Figure 1.

Bioassay-linked fractionation of TRPM2-active organic extract of Cacospongia sp. (A, B) Hydrogen peroxide-induced Ca2+ entry in our calcium imaging assay using TRPM2-HEK293 cells following (A) treatment with vehicle control (closed circles, n = 15) versus an organic extract of Cacospongia sp. (open circles, n = 3) or (B)treatment with vehicle control (closed circles, n = 20) versus HPLC fractions of the extract eluting at 12.0–12.5 min (black trace, open circles, n = 2), 12.5–13.0 min (red trace, open circles, n = 2), 13.0–13.5 min (red trace, close circles, n = 2), or 13.5–14.0 min (black trace, closed squares, n = 2). H2O2 (250 μM) was added at 30 s. Fractions (approximately 17 μg/mL each) were incubated for 20–30 min. Cells were preincubated with fura-2 AM, and change in relative fluorescence units was measured at 510 nm after excitation at 340 nm.(C)Chromatogram of semipreparative reversed-phase HPLC of the organic extract residue. Eluent (2.0–30.0 min) was collected into 96-well plates in 30–60 s fractions and tested at proportional concentrations in the TRPM2 Ca2+ imaging assay. Components eluting between 12.5 and 13.5 min consistently concentrated TRPM2 inhibitory activity (grayed area) (see panel B). The major components eluting between 12.5 and 13.5 min were identified as the known scalaradial (SD) and 12-deacetylscalaradial (DSD), which were subsequently isolated and assayed. (D) Average normalized whole-cell current development of human TRPM2 overexpressed in HEK293 cells3 following treatment with vehicle control (closed circles, n = 4) versus HPLC fractions of the Cacospongia sp. extract eluting at 13.0–14.0 min (approximately 1 μg/mL, open circles, n = 4). Cells were incubated with the indicated fraction/control for 30–60 min in standard external Ringer’s solution before patching. Internal solution was unbuffered (no Ca2+ chelator) standard K-glutamate-based solution supplemented with 100 μM ADPR. Currents were elicited by voltage ramps from −100 mV to 100 mV over 50 ms at 0.5 Hz intervals. Current amplitudes were extracted at −80 mV, normalized to the current assessed at 100 s, averaged, and plotted versus time of the experiment. Induction time for tetracycline was 6–9 h. Error bars indicate SEM. For panels A, B, and D, data at the 100 s time point were statistically evaluated. Each of the following samples showed significance compared to control: (A) crude extract; (B) 12.5–13.0 and 13.0–13.5 min fractions; (D) 13.0–14.0 min fraction.
Scalaradial Inhibits TRPM2-Mediated Currents in a Concentration- and Time-Dependent Manner As Measured by Whole-Cell Patch-Clamp Analysis.
Isolated scalaradial and 12-deacetylscalaradial were tested for TRPM2 inhibitory activity in whole-cell patch clamp analysis using tetracycline-inducible HEK293 cells overexpressing human TRPM2 (TRPM2-HEK293).3 Cells were exposed to intracellular ADPR to measure TRPM2-mediated ion flux in the presence or absence of scalaradial or 12-deacetylscalaradial. The initial assays established these compounds as the active constituents in the Cacospongia extract fractions that inhibit the TRPM2-mediated Ca2+ influx. (12-Deacetylscalaradial demonstrated a similar activity and potency in patch clamp experiments compared with scalaradial and was not further characterized.) Following this, we sought to further evaluate the interaction of scalaradial with TRPM2 channels. Figure 2A shows that acute application of 2 μM scalaradial did not significantly affect overexpressed heterologous TRPM2 currents patched in TRPM2-HEK293 cells. On the other hand, Figure 2B, C, and F show a concentration-dependent inhibition of TRPM2 currents after 30–60 min incubation with scalaradial in a concentration range of 0.1 to 2 μM, corresponding to an IC50 of 210 nM. The potency of scalaradial was also dependent on the time of scalaradial incubation, as demonstrated in Figure 2D for 2 μM scalaradial, which shows an increasing inhibition of current with increasing scalaradial incubation time. This is further analyzed in Figure 2E, where inhibition of TRPM2 by 100 nM, 500 nM, or 2 μM scalaradial is binned in 10 min time baskets of drug incubation. The data indicate that at 2 μM scalaradial requires ~20 min incubation time to reach full potency, which may suggest an indirect mode of inhibition or covalent binding of TRPM2 channels by scalaradial. Recovery from inhibition was assessed by incubating TRPM2-HEK293 cells for 60 min in 2 μM scalaradial, upon which TRPM2 currents were assessed by patch-clamp with (2 μM SD) or without (recovery) scalaradial in the bath solution (Figure 2G) and compared to control (no exposure to scalaradial). Recovery of TRPM2 currents could be observed within 5 min of drug removal.
Figure 2.

Scalaradial inhibits TRPM2 in a concentration- and time-dependent manner. (A) Average normalized whole-cell current development of human TRPM2 overexpressed in HEK293 cells.3 Internal solution was standard K-glutamate-based solution with Ca2+ buffered to 300 nM and supplemented with 100 μM ADPR. Currents were elicited by voltage ramps from −100 mV to 100 mV over 50 ms at 0.5 Hz intervals. Current amplitudes were extracted at −80 and +80 mV, normalized to the control current assessed at 100 s, averaged, and plotted versus time of the experiment. Induction time for tetracycline was 6–9 h. Application of external solution supplemented with either vehicle (0.17% MeOH; open circles, control (n = 4)) or 2 μM scalaradial (closed circles (n = 7)) as indicated by the black bar. No statistical significance. Error bars are not shown. (B) Average normalized whole-cell current development of human TRPM2 overexpressed in HEK293 cells.3 Cells were incubated with the indicated concentrations of scalaradial for 30–60 min in standard external Ringer’s solution before patching (control: closed circles, n = 20; 100 nM: open circles, n = 10; 500 nM: closed squares, n = 15; 2 μM: open squares, n = 11). Solutions and data acquisition as in A. Error bars indicate SEM. (C) Current–voltage (I/V) relationship of TRPM2 currents extracted from representative cells at 100 s into the experiment and exposed to increasing concentrations of scalaradial between 30 and 60 min (control = black trace, 100 nM = green trace, 500 nM = blue trace, 2 μM = red trace). (D) Data cloud plot of time-dependent TRPM2 current inhibition as extracted from individual cells at −80 mV and 100 s and plotted against incubation time with 2 μM scalaradial. (E) Time-dependency of 0.1, 0.5, and 2 μM scalaradial on TRPM2 peak currents expressed in HEK293 cells. Scalaradial was added to TRPM2-overexpressing HEK293 cells in standard external solution. TRPM2 currents of individual cells were measured, and data from individual cells were pooled for different time periods with regard to incubation with scalaradial (0–20, 21–30, 31–40, 41–50, and 51–60 min). Induction time was 6–9 h. Internal solution as in panel A. Current amplitudes were extracted at −80 mV and 100 s and normalized to percent of controls recorded at the same day after a similar induction period (n = 4–12 per bin except n = 1 for bin 100 nM at 41–50 min; n = 20 for control). Error bars indicate SEM. (F) Concentration-response curve of TRPM2 currents to increasing concentrations of scalaradial. Normalized data were extracted at −80 mV and 100 s from panel B and plotted versus the respective scalaradial concentration. A dose-response fit to the data measured an IC50 of 210 nM with a Hill coefficient of 1.3. (G) Current size in % of TRPM2 currents without exposure to scalaradial (control), with 60 min of exposure to scalaradial and current assessment in external solution in the absence (recovery) or presence of 2 μM scalaradial (SD). Current amplitudes were assessed at −80 mV and 100 s into the experiment, evaluated as % of control, and averaged for display and statistical analysis. Error bars are SEM. Star indicates statistical significance with p < 0.001.
Scalaradial Suppresses TRPM7, but Fails to Inhibit CRAC, TRPM4, or TRPV1 Currents.
After finding that scalaradial inhibited TRPM2-mediated currents in TRPM2-overxpressing HEK293 cells in a concentration and time-dependent manner, we explored the selectivity of the observed effect against other ion channels. First, we tested effects of scalaradial on heterologously overexpressed mouse TRPM7 currents in tetracycline-inducible HEK293 cells (Figure 3A,B).24,25 This revealed that scalaradial inhibits TRPM7 in a time-dependent manner (Figure 3C), albeit more than 3-fold less potent, with an apparent IC50 of 760 nM (Figure 3D). We therefore did not pursue TRPM7 inhibition by scalaradial further. Second, we tested effects of scalaradial on calcium release-activated calcium channel (CRAC)-like currents recorded from Orai1/STIM1-overexpressing HEK293 cells (Figure 3E,F)26 and observed no significant inhibition of current at 2 μM. A second member of the TRP melastatin subfamily, endogenous TRPM4 in Jurkat T lymphocytes,27,28 was exposed to 500 nM scalaradial, without affecting the current (Figure 3G). Effects of 500 nM scalaradial on TRPV1 in INS-1 cells were also evaluated (Figure 3H),29 and results indicated scalaradial does not inhibit TRPV1 current. Although inhibition against other channels cannot be ruled out, these data demonstrate that scalaradial possesses some level of selectivity and potency in inhibiting cation conductance via TRPM2 channels and to a lesser degree TRPM7.
Figure 3.

Scalaradial inhibits TRPM7 but not CRAC currents. (A) Average whole-cell current development of mouse TRPM7 overexpressed in HEK293. Cells were incubated with 100 nM (open circles; n = 9), 500 nM (filled triangle; n = 10), or 2 μM scalaradial (open squares, n = 11) for 15 min in cell culture medium before whole-cell patch clamp or not (control, solid circles, n = 10). Maximum exposure time to scalaradial was 60 min. Tetracycline induction time was 19–22 h. Internal solution was standard K-glutamate solution supplemented with 10 mM K-BAPTA. Data were acquired and analyzed as in Figure 2A. Error bars are SEM. (B) Averaged current–voltage (I/V) relationship of TRPM7 currents extracted at 200 s into the experiment from data in panel A and exposed to control solution or solution supplemented with increasing scalaradial concentrations. (C) Analysis of time-dependent inhibition of TRPM7 by 100 nM, 500 nM, and 2 μM scalaradial compared to control. Same data as averaged in panel A (n = 2–3 per bin, except 100 nM at 51–60 min (n = 1)). (D) Concentration–response curve of TRPM7 currents to increasing concentrations of scalaradial. Normalized data were extracted at +80 mV at 200 s from panel A and plotted versus the respective scalaradial concentration. A concentration–response fit to the data measured an IC50 of 760 nM with a Hill coefficient of 2. (E) Average whole-cell CRAC currents measured in HEK293 cells transiently overexpressing Orai1 and stably expressing STIM1.26 Exposure to 2 μM scalaradial between 30 and 60 min (open circles, n = 9) did not significantly affect CRAC currents compared to control (closed circles, n = 7). Extracellular solution contained 10 mM CaCl2 and was NaCl-based (see Experimental Section). Intracellular solution was Cs glutamate-based and supplemented with 20 mM Cs- BAPTA and 20 μM IP3. Currents were elicited using a voltage ramp from −150 mV to +150 mV over 50 ms at 0.5 Hz. Current amplitudes were assessed at −80 mV, averaged, and plotted versus time of the experiment. Error bars indicate SEM. (F) Current–voltage (I/V) relationship of CRAC currents extracted from representative cells at 100 s into the experiment and exposed to control solution (black trace) or solution supplemented with 2 μM scalaradial (red trace). (G) Average whole-cell current development of endogenous TRPM4-like currents in Jurkat T lymphocytes. Cells were incubated with 500 nM scalaradial (open circles, n = 5) or no scalaradial (control, solid circles, n = 6) in standard external solution before whole-cell patch clamp. Internal solution was standard Cs-glutamate solution with intracellular Ca2+ clamped to 2 μM. Recordings were done within the first 60 min of incubation. Data were acquired and analyzed as in Figure 2A. (H) Average whole-cell current development of endogenous TRPV1-like currents in rat beta pancreatic INS-1 cells. Cells were incubated with 500 nM scalaradial (open circles, n = 8) in standard external solution before whole-cell patch clamp or no scalaradial (control, solid circles, n = 7). Recordings were performed with the first 60 min of incubation. Internal solution was standard Cs-glutamate solution with Ca2+ left unbuffered. Capsaicin (10 μM) in external solution was applied as indicated by the black bar. Data were acquired and analyzed as in Figure 2A.
Scalaradial’s Effect on TRPM2 Is Not via Disruption of ADPR Metabolism.
ADPR is the principal physiological ligand gating TRPM2 channels, and its cellular concentration is regulated in part by PARP-1 and PARG activity.30 For our Ca2+ imaging assays, TRPM2-mediated Ca2+ flux is elicited by H2O2, which appears to increase cellular ADPR concentrations.6 In this assay the PARP-1 inhibitors N-(6-oxo-5,6-dihydrophenan-thridin-2-yl)-(N,N-dimethylamino)acetamide hydrochloride hy drate (PJ34) and 3,4-dihydro-5-[4-(1-piperidinyl)butoxyl]-1(2H)-isoquinolinone (DPQ)31 effectively blocked the TRPM2-mediated Ca2+ flux with IC50 values of 25 and 600 nM, respectively (Figure 4A), values which were comparable to previously reported data.32 In contrast to the cell-based assay, ADPR is applied directly in the internal solution during our patch-clamp experiments. Consistent with the idea that inhibition of PARP-1 is irrelevant when providing ADPR within the patch pipet, the PARP-1 inhibitors PJ34 and DPQ did not affect TRPM2-mediated currents (Figure 4B).6 However, the fact that scalaradial does inhibit TRPM2-mediated currents in our patch-clamp experiments, despite the exogenously applied ADPR through the patching pipet, suggests that scalaradial works through a mechanism independent of endogenous ADPR generation/metabolism.
Figure 4.

Effect of PARP-1, sPLA2, and PI3K/Akt pathway inhibitors on TRPM2. (A) In a calcium imaging assay, TRPM2-HEK cells were plated and induced for 20 h. Following fura-2 AM loading, the PARP-1 inhibitors PJ34 and DPQ were added. Cells were stimulated with 1.0 mM H2O2 30 s after addition of the PARP-1 inhibitors, and the changes in intracellular [Ca2+] were followed for 120 s. Change of the Ca2+ signal is expressed as the maximum ratio change of fluorescence (at 510 nm, after excitation at 340/380 nm) normalized to vehicle as control (=100%). Each data point is the mean (±SEM) of quadruplicate determinations. Data are from one representative experiment. (B) In patch clamp experiments, TRPM2-HEK293 cells were incubated with the indicated concentrations of PARP-1 inhibitors in standard external Ringer’s solution, and whole-cell currents were recorded during the first 60 min. Internal solution was standard K-glutamate solution with 100 μM ADPR. Current amplitudes were extracted at −80 mV and normalized to percent of controls recorded on the same day. Bar graphs show means of n = 3–17. Error bars are SEM. (C) In patch clamp experiments, TRPM2-HEK293 cells were incubated with the indicated concentrations of sPLA2 inhibitor in standard external Ringer’s solution, and whole-cell currents were recorded during the first 60 min. Internal solution was standard K-glutamate solution with 100 μM ADPR. Current amplitudes were extracted at −80 mV and normalized to percent of controls recorded on the same day. Bar graphs show means of n = 3–17. Error bars are SEM. (D) In patch clamp experiments, TRPM2-HEK293 cells were preincubated for 60 min with the indicated concentrations of Akt and PI3K inhibitor in serum-containing medium, before being transferred into standard external Ringer’s solution. Currents were recorded in the continued presence of the inhibitors. Internal solution was standard K-glutamate solution supplemented with 500 μM ADPR. Current amplitudes were extracted at −80 mV and normalized to percent of controls recorded on the same day. Bar graphs show means of n = 10–19. Error bars are SEM.
Scalaradial’s Effect on TRPM2 Channels Is Independent of Its Ability to Inhibit sPLA2.
Scalaradial’s best-known physiological targets are members of the subfamily of small secreted phospholipase A2 (sPLA2) enzymes such as bee venom PLA2 and human-type II sPLA2, against which scalaradial has reported inhibitory potencies (IC50) of 0.07 and 5.4 μM, respectively.33,34 In order to explore whether TRPM2 inhibition by scalaradial is an event downstream of its inhibitory action on sPLA2s, we tested two unrelated sPLA2 inhibitors for their effect on TRPM2-mediated currents. Figure 4C shows the lack of effect of 1 μM MJ33 (1-hexadecyl-3-(trifluoroethyl)-sn-glycero-2-phosphomethanol, lithium) and 5 μM “sPLA2 inhibitor” (5-(4-benzyloxyphenyl)-4S-(7-phenylheptanoylamino)pentanoic acid) when applied under the same conditions as scalaradial in patch-clamp experiments with our TRPM2-overexpressing HEK293 cells. The respective concentrations for MJ33 and “sPLA2 inhibitor” were based on literature values 35–38 and titrated in order to avoid significant cell death during a 1 h recording session, as observed at 5–10-fold higher concentrations (data not shown). From these experiments we concluded that scalaradial is exerting its effect on TRPM2 channels independent of its sPLA2 inhibitory action.
Scalaradial’s Effect on TRPM2 Is Not Mimicked by Known Inhibitors of the PI3K/Akt Pathways.
Xie and co-workers39 have shown that scalaradial interacts with the extracellular signal-regulated kinases (ERK) and Akt pathways in a biphasic manner. Scalaradial’s inhibitory action on sPLA2 was shown to cause an early (but transient) inhibition of ERK1/2-phosphorylation. In addition, scalaradial also inhibited the EGF-stimulated phosphorylation of Akt, which in turn prevented Raf-1 phosphorylation (by Akt), and therefore caused a disinhibition (late stimulation) of ERK phosphorylation. In order to explore whether scalaradial inhibition of TRPM2 currents is a downstream event of scalaradial inhibiting Akt phosphorylation, we reasoned that Akt inhibitors should have a similar effect on TRPM2 currents to scalaradial. We used “Akt inhibitor VIII” (1,3-dihydro-1-(1-((4-(6-phenyl-1H-imidazo[4,5-g]quinoxalin-7-yl)phenyl)methyl)-4-piperidinyl)-2H-benzimidazol-2-one), a potent Akt1/Akt2- (over Akt3) preferring inhibitor,40 and wortmannin, a potent and irreversible inhibitor of phosphatidylinositol 3′-kinases (PI3K),41,42 thus, a general inhibitor of PI3K-Akt pathways.43 TRPM2-expressing HEK293 cells were incubated for 60 min with either one of the two tool compounds before TRPM2-mediated currents were measured. As shown in Figure 4D, 200 nM of the “Akt inhibitor VIII” or 20 nM of wortmannin both failed to affect TRPM2-mediated currents, suggesting that this pathway is not upstream of scalaradial’s effect on TRPM2 channels. Fivefold higher concentrations of either compound still showed current amplitudes comparable to control cells, but affected cell viability (causing cell rounding, granule formation, and blebbing; data not shown).
Scalaradial Inhibits the Endogenous TRPM2 Channel in a Rat Insulinoma Cell Line.
In order to explore whether the pathway by which scalaradial inhibits TRPM2-mediated currents is of physiological relevance, a rat insulinoma cell line (INS-1 cells), which endogenously expresses functional TRPM2 channels,7 was patched under similar conditions. When applying 0.5–2.0 μM scalaradial to these cells in whole-cell patch-clamp analysis, the ADPR-induced currents were largely abolished (Figure 5A,B). A concentration–response was observed between 10 nM and 2 μM, with an IC50 value of 330 nM (Figure 5C), comparable with the inhibition of TRPM2 in the overexpression system. At 2 μM scalaradial, a concentration suitable for the TRPM2-HEK293 cells, INS-1 cells rapidly deteriorated (rounding, granule formation, and blebbing), which complicated patching at the highest concentration. However, cells appeared healthy for at least 1 h with continual exposure to 500 nM scalaradial and lower concentrations.
Figure 5.

Scalaradial inhibits native TRPM2 currents in rat INS-1 beta pancreatic cells. (A) Time-course of TRPM2 current development in rat insulinoma beta pancreatic cells (INS-1) in whole-cell patch-clamp experiments. Cells were incubated with the indicated concentrations of scalaradial in standard external solution, and currents were recorded during the first 60 min of scalaradial incubation (control: closed circles, n = 27; 100 nM scalaradial: open circles, n = 5; 500 nM scalaradial: closed squares, n = 15; 2 μM scalaradial: open squares, n = 5). Data acquisition as in Figure 2A. Current amplitudes were extracted at −80 and +80 mV, averaged, and plotted versus time of the experiment. Internal solution was Cs glutamate-based and supplemented with 500 μM ADPR. Error bars are SEM. (B) Examples shown of current–voltage relationship of TRPM2 currents measured in INS-1 cells exposed to 100 nM (blue trace), 500 nM (red trace), or no scalaradial and extracted at 100 s experimental time. (C) Inhibition of TRPM2 currents exposed to increasing concentrations of scalaradial. Current amplitudes were extracted at −80 mV and 50 s experimental time, normalized as percent inhibition to control, and plotted versus the respective scalaradial concentration. Error bars are SEM. A dose–response fit calculated an IC50 of 330 nM with a Hill coefficient of 4. Same data set as in panel A.
DISCUSSION
The TRPM2 channel is a voltage-insensitive, nonselective cation channel. Its widespread expression pattern and role in Ca2+ mobilization, both from extracellular and intracellular compartments, makes TRPM2 a potential key regulator of a range of physiological and pathophysiological conditions.1 Due to TRPM2 activation by H2O2 and subsequent entry of Ca2+ into the cytosol, it is believed that TRPM2 plays a role in sensing oxidative stress and is involved in pathophysiological states characterized by an increase in production of ROS, such as cardiac ischemia, stroke, neurodegenerative diseases, and even diabetes.1 Therefore, TRPM2 must be considered an attractive target for therapeutic intervention. We here identify scalaradial as a potent inhibitor of TRPM2, with lower potency on TRPM7 channels, but no significant effect on CRAC channels, TRPM4, and TRPV1.
The known TRPM2 inhibitors suffer from low potency and/or specificity, which makes it difficult to use them as tools to elucidate the details of TRPM2 physiology. Flufenamic acid (FFA) and imidazole derivatives such as clotrimazole and econazole were the first described TRPM2 inhibitors,19,20 yet suffered from low solubility and poor selectivity. N-(p-Amylcinnamoyl)anthranilic acid (ACA) has an IC50 of 4.5 μM on TRPM2-transfected HEK cells, yet blocks several TRP channels such as TRPC6, TRPM8, TRPC3, and TRPV1.22 2-APB was shown to have an IC50 of 1 μM on TRPM2-transfected HEK cells,21 yet also suffers from low specificity and is known to inhibit IP3 receptors44 and multiple TRPC channels,45 activate a several TRPV channels,46 and enhance or inhibit other TRPM47 and store-operated calcium entry26 channels. Finally, the currently most potent inhibitor is curcumin, which blocks TRPM2 currents with preincubation in the low nanomolar range,23 although again suffers from relative lack of specificity.48
In the search for novel bioactive metabolites, we have focused on screening marine-derived extracts including those from soft-bodied marine animals, which are known to produce an abundance of secondary metabolites exhibiting a wide variety of biological activity. Such compounds are ostensibly meant to serve an ecological purpose, such as chemical defenses against predators. Among our active extracts was that of a sponge of the genus Cacospongia. Subsequent activity-guided fractionation of the extract led us to isolate scalaradial and 12-deacetylscalaradial as potent inhibitors of TRPM2.
Our screening assay, which utilizes the Ca2+-sensitive fluorescent dye fura-2 to measure cytosolic increase in Ca2+ due to TRPM2 activation, is a validated approach for identifying TRPM2 activation and inhibition.49 Direct activation of TRPM2 by H2O2 does not appear to be a dominant pathway in our TRPM2-overexpression assay; rather, TRPM2 is activated by ADPR produced by the synchronized activity of PARP and PARG, a pathway activated in response to the DNA damage caused by H2O2.6 This is corroborated by our observation that the PARP-1 inhibitors PJ34 and DPQ attenuated H2O2-mediated Ca2+ influx in our Ca2+ imaging assays, while use of the inhibitors in patch-clamp experiments with internally applied ADPR showed no effect on the current (Figure 4A,B).
Scalaradial inhibition of TRPM2-mediated Ca2+ influx was confirmed in whole-cell patch-clamp using TRPM2-transfected HEK cells as well as rat insulinoma (INS-1) cell lines exposed to intracellular ADPR, with IC50 values of 210 and 330 nM, respectively. The potency of scalaradial on TRPM2-transfected HEK cells was dependent on the preincubation period with scalaradial prior to the whole-cell current measurement, necessitating approximately 20 min to reach full potency. When scalaradial was added to the cell internally (data not shown), the time dependency was not affected. These data may suggest a metabolic activation of scalaradial, an indirect effect on TRPM2 by scalaradial interaction on a regulatory pathway upstream of TRPM2, effects on the subcellular relocalization of TRPM2 by scalaradial, or a functional modification of TRPM2 through covalent binding of scalaradial.
Several studies have shown that scalaradial or its epimer inhibits the activity of several proteins involved in a number of interconnecting signal transduction cascades. The first and perhaps most well studied targets of scalaradial are the members of the Ca2+-dependent50 secreted subfamily of phospholipase A2 (sPLA2). Scalaradial is used to study various physiological and pathophysiological pathways due to its ability to distinguish between sPLA2s from cytosolic PLA2s at low concentrations.34 sPLA2’s catalytic activity releases signaling molecules which are involved in many inflammation and pain-related pathways, one of which, arachidonic acid, is known to be a positive regulator of TRPM2.51 Although sPLA2-mediated arachidonic acid release can be triggered by oxidative stress,52 no change in scalaradial inhibition of TRPM2 was found in TRPM2-transfected HEK cells when treated with specific sPLA2 inhibitors. This suggests that TRPM2 activity is not dependent on sPLA2 inhibition. Similarly, in many previous studies the use of ACA was initially used as an inhibitor of PLA2s, but was also found to inhibit TRPM2 independent of PLA2 inhibition.22 Previous studies using scalaradial to inhibit sPLA2 should be reinterpreted in light of the discovery that scalaradial has an independent inhibitory effect on TRPM2. Interestingly, numerous studies that use scalaradial to identify sPLA2 activity are conducted in models that also highly express TRPM2, such as various CNS tissues,53 macrophages,54 and lymphocytes.55 While it is not clear what effect sPLA2 has on TRPM2, sPLA2’s involvement in inflammatory and apoptotic55 signaling cascades and dependence on Ca2+48 point to a possible downstream relationship to TRPM2 that should not be overlooked. True to this, while studies have supported that the use of specific sPLA2 inhibitors indoxam56 and PX-1857 ameliorated ischemic injury, use of scalaradial showed the opposite effect and increased ischemic injury.58
Scalaradial also inhibits epidermal growth factor receptor-stimulated Akt phosphorylation independent of scalaradial’s inhibitory effects on sPLA2 and was also shown to inhibit PDK-1 translocation and PI3K activation, both upstream regulators of Akt.59 Later, Xie and co-workers demonstrated scalaradial’s biphasic effect on phosphorylation of ERK1/2, which was dependent on both sPLA2 and Akt inhibition, clearly demonstrating intersections between the PI3K/Akt and the Raf/MEK/ERK pathways.37 Because the use of either Akt or general PI3K pathway inhibitor failed to reproduce scalaradial’s effect on TRPM2, we have concluded that scalaradial inhibition of TRPM2 is not downstream of PI3K, Akt, or ERK1/2 pathway inhibition. Several studies have demonstrated that activation of Akt is dependent on cytosolic Ca2+ levels 60–62 and may point to TRPM2’s putative role in playing upstream from Pi3K/Akt.
Scalaradial shows some selectivity, as it fails to inhibit CRAC, TRPM4, and TRPV1 currents in patch-clamp experiments at comparable concentrations, demonstrating that scalaradial is not a general ion channel inhibitor and is able to distinguish within the TRPM subfamily. However, scalaradial was found to inhibit heterologous TRPM7 about 3-fold less efficiently but with a similar time course in tetracycline-induced TRPM7-HEK cells. Aarts and co-workers63 reported that suppression of TRPM7 with siRNA simultaneously reduced TRPM2 ex-pression independent of sequence homology, suggesting expression of both channel subunits are interdependent and may form heterotetramers. However, later work from this group using viral vectors bearing shRNA against TRPM7 to prevent ischemia-induced neuronal damage could not find concomitant TRPM2 suppression.64 This leaves the link between TRPM2 and TRPM7 inhibition by scalaradial as largely unanswered, but a bifunctional TRPM2/TRPM7 inhibitor could potentially mediate neuroprotective effects through multiple mechanisms.
We have demonstrated that scalaradial is a potent TRPM2 inhibitor and confirmed its specificity against several other ion channels. While we cannot rule out that scalaradial may have effects on other ion channels, the nanomolar concentration needed to inhibit TRPM2 makes scalaradial to date among the most potent inhibitors of TRPM2. Scalaradial also exhibits a novel chemical scaffold compared to other TRPM2 inhibitors, which may guide research into structure–activity relationships and the cellular pathways of TRPM2 functional regulation.
EXPERIMENTAL SECTION
Collection and Identification of Sponge Biomass.
The sponge sample was collected from caves and under overhangs near Pupukea Beach Park, Oahu, Hawaii, on July 22, 1997, from a depth of 8 m. In life, the sponge formed a thick encrustation with rounded projections, the texture was firm and brittle when torn, the surface was covered in tiny, regularly spaced, conules, and the external color was dark brownish-black with a cream interior. The skeleton is composed of laminated, pithed, and cored primary fibers, which are regularly distributed throughout the sponge and joined by smaller, pithed, secondary fibers, clear of coring material. Based on its morphology, coloration, and skeletal architecture, the sponge is identified as an undescribed species of Cacospongia (order Dictyoceratida, family Thorectidae). The freshly collected sample was freeze-dried and stored vacuum-packed at −20 °C until use. A voucher was deposited in the Natural History Museum, London (BMNH 1998.8.12.2, BMNH 1998.8.12.3, and BMNH 1998.8.12.4).
Screening of the Sponge Organic Extract for TRPM2 Inhibition.
The freeze-dried biomass of Cacospongia sp. was extracted by repeatedly macerating in 2-propanol/dichloromethane (1:1), and the combined extracts were dried under vacuum to give a light brown crystalline residue that was stored at −20 °C until use. Prior to bioassay, a small aliquot of the extract residue was dissolved in methanol/ethyl acetate/tert-butyl methyl ether (60:30:10) and diluted in Krebs–Ringer–HEPES (KRH) buffer (composition in mM: 135 NaCl, 5 KCl, 1.5 MgCl2, 1.5 CaCl2, 20 HEPES, and 0.1% glucose). The sample was tested for effects on TRPM2-mediated Ca2+ flux in a Ca2+ imaging assay (described below) in duplicate on separate days at a final concentration of 30 μg/mL (0.5% organic solvent).
Bioassay-Linked Fractionation of Cacospongia sp. Organic Extract.
The TRPM2-active extract (2.5–3.5 mg) was fractionated by reversed-phase HPLC (Agilent 1100; Phenomenex Luna C18(2) column, 5 μm, 4.6 × 250 mm; mobile phase: acetonitrile/10 mM aqueous NH4OAc, 86:14, from 0 to 2 min, linear gradient from 86:14 to 95:5 from 2 to 7 min, 95:5 from 7 to 30 min; flow rate: 1.0 mL/min; UV detection, 222–238 nm), and the eluent (0.5 or 1.0 min fractions) was collected into 96-well polypropylene deep-well plates. The eluent wells were aliquoted in equal volumes into test plates, dried under vacuum, and stored at −20 °C until use. Prior to assay, plate well fractions were reconstituted in either methanol or methanol/ethyl acetate/tert-butyl methyl ether (60:30:10) and diluted in KRH buffer. Selected wells inhibiting TRPM2-mediated Ca2+ conductance in the Ca2+ imaging assay (at final average test concentrations of 6–50 μg/well) were tested in TRPM2 patch-clamp experiments described below. The TRPM2 activity was focused in the fractions eluting between 12.5 and 13.5 min, and LCMS analysis (Thermo LCQ Deca XP Max APCI-ion trap MS, 400 °C source temperature, 10 μA discharge current) indicated molecular weights of 386 (MH+ m/z 387.1) and 428 (MH+ m/z 429.1) of the eluting components. A search of potential molecular structures in the Dictionary of Natural Products (CRC Press, v17.1) for Cacospongia spp. as the biological source and limited by exact molecular masses of 428.0–428.5 and 386.0–386.5 mmu returned seven scalarane-type sesterterpenes, neither of which possessed reported cation channel inhibitory activity. As a prioritized sample, subsequent scaled-up isolation of the suspected active compounds was carried out.
Isolation of Scalaradial and 12-Deacetylscalaradial.
Freeze-dried sponge material was extracted with methanol, and the residue was repeatedly chromatographed to >95% purity by reversed-phase HPLC (Agilent 1100; Phenomenex Luna C18(2) column, 10 μm, 10 × 250 mm; mobile phase: acetonitrile/10 mM aqueous NH4OAc, 89:11; flow rate: 3.8 mL/min; UV detection, 222–238 nm). Scalaradial and 12-deacetylscalaradial were identified by comparison of 1H and 13C NMR, MS, and polarimetry data with literature values.65,66 Prior to bioassay, the purity of scalaradial and 12-deacetylscalaradial were determined by analytical HPLC by UV detection [Agilent 1100; Phenomenex Luna C18(2) column, 5 μm, 2.0 × 250 mm; mobile phase: acetonitrile/10 mM aqueous NH4OAc,50:50 for 2 min, then linear gradient from 50:50 to 100:0 over 30 min; flow rate: 0.20 mL/min; UV detection, 222–238 nm].
Cell Culture.
Wild-type (nontransfected) human embryonic kidney (HEK293) cells and tetracycline-inducible HEK293 cells stably transfected with either a FLAG-human TRPM2/pcDNA4/TO construct3 or a FLAG-murine TRPM7/pCDNA4/TO construct 24 were cultured at 37 °C with 5% CO2 in DMEM supplemented with 10% fetal bovine serum. For the TRPM2- and TRPM7-transfected cells, the medium was supplemented with blasticidin (5 μg/mL; Invitrogen) and zeocin (0.4 mg/mL; Invitrogen). TRPM2 and TRPM7 overexpression was induced by adding 1 μg/mL tetracycline to the media 5–22 h before experiments.
For the electrophysiological analysis of CRAC-like currents, we used HEK293 cells stably transfected with an STIM1-pIRESneo vector construct (cell line kindly provided by D. L. Gill’s lab),67 which, in addition, were transiently transfected with a CRACM1-pIRES2-EGFP vector construct.68,69 Cells were cultured at 37 °C with 5% CO2 in DMEM supplemented with 10% fetal bovine serum. CRAC recordings were performed 24–48 h post-transfection, with cotransfected cells being identified by the fluorescence of EGFP.
The rat insulinoma cell line (INS-1) was kept at 37 °C with 5% CO2 in RPMI containing 10% fetal bovine serum.
Calcium Imaging Assay for TRPM2 Inhibitors.
Changes in cytosolic [Ca2+] were monitored as changes in fluorescence using the Ca2+-indicator dye fura-270 and a scanning plate reader (FlexStation, Molecular Devices). Data were collected and processed using SoftMax Pro software (v5.2, Molecular Devices). TRPM2-HEK293 cells were plated in poly-l-lysine-coated 96-well plates, and TRPM2 expression was induced 2–3 h after plating by addition of 2–10 μg/mL tetracycline. The culture medium was completely removed at 16–20 h post induction and replaced with the fura-2 loading buffer: KRH buffer supplemented with 2 μM fura-2 acetoxymethyl ester (fura-2 AM; Calbiochem) and 0.1% Pluronic F-127. Following incubation (45 min at 37 °C) the loading buffer was removed, and the cells were washed once with KRH before the addition of assay buffer (KRH). The plates were then transferred to the prewarmed FlexStation (37 °C), which also contained a compound plate with separate wells containing the appropriate test substance solutions (i.e., vehicle, extracts/fractions of marine organisms, and compounds) or H2O2 dissolved in KRH. Cells were initially incubated with the appropriate drugs or vehicle for ~20 min or as indicated prior to addition of a final 250–1000 μM H2O2. In all assays performed, vehicle-receiving, induced cells served as positive control for the activation of a TRPM2-mediated Ca2+ influx, while wells containing noninduced cells (receiving vehicle) periodically served as negative controls to define the baseline for the changes of cytosolic [Ca2+]. Intracellular [Ca2+] was monitored for up to ~2 min following compound/vehicle addition (or the entire compound/vehicle incubation time, if shorter) and for 2~3 min following H2O2 addition as the fluorescence intensity was measured at 510 nm after excitation at 340 nm (Ca2+-bound fura-2). The change in relative fluorescence was used to monitor changes in intracellular [Ca2+].
Electrophysiology.
Patch-clamp experiments were performed in the whole-cell configuration at 21–25 °C. All data were acquired with PatchMaster (HEKA) software controlling an EPC-9 amplifier (HEKA, Lambrecht, Germany) and analyzed using FitMaster (HEKA) and Igor Pro (Wavemetrics). Voltage ramps of 50 ms spanning from −100 to +100 mV were delivered from a holding potential of 0 mV at a rate of 0.5 Hz. Voltages were corrected for liquid junction potentials (10 mV). Currents were filtered at 2.9 kHz and digitized at 100 μs intervals. Capacitive currents were determined and corrected before each voltage ramp. For analysis, current amplitudes were extracted at −80 mV, but for TRPM2 and TRPM7 currents were not corrected for the cell capacitance, and TRPM7 was assessed at +80 mV. Usual cell capacitance was 12–20 pF for HEK293 cells and 7–12 pF for INS cells. CRACM1/STIM1 currents were measured using a voltage ramp ranging from −150 to +150 mV within 50 ms.
Solutions and Chemicals.
For patch-clamp experiments cells were kept in standard external Ringer’s solution (in mM): 140 NaCl, 2.8 KCl, 1.0 CaCl2, 2.0 MgCl2, 10 glucose, and 10 HEPES-NaOH (pH 7.2 adjusted with NaOH), 310 mOsm. Standard internal pipet-filling solutions contained (in mM) 140 Cs-glutamate for (INS-1 and Jurkat T cells), 140 K-glutamate (for HEK293 Flag-TRPM2 and Flag-TRPM7 expressing cells), 8.0 NaCl, 1.0 MgCl2, and 10 HEPES (pH 7.2 adjusted with CsOH/KOH). For recordings of TRPM2- and TRPV1-mediated currents, Ca2+ was left unbuffered by leaving out Ca2+ chelators. For TRPM2, ADPR was added to its final concentrations of 100 or 500 μM. For recordings of TRPM7-mediated currents, intracellular Ca2+ was buffered with 10 mM BAPTA. Solutions for the recordings of CRAC currents were the following: standard external solution (in mM): 120 NaCl, 10 CsCl, 2.8 KCl, 2.0 MgCl2, 10 CaCl2, 10 TEA-Cl, 10 HEPES, and 10 glucose, pH was adjusted to 7.2 with NaOH, 300 mOsm. Internal solution was as follows (in mM): 120 Cs-glutamate, 20 Cs-BAPTA, 3.0 MgCl2, 10 HEPES, and 0.020 inositol 1,4,5-trisphosphate (IP3), pH was adjusted to 7.2 with CsOH, 300 mOsm. All reagents and drugs were purchased from Sigma-Aldrich except for Akt inhibitor VIII (Calbiochem, 1,3-dihydro-1-(1-((4-(6-phenyl-1H-imidazo [4,5-g]quinoxalin-7-yl)-phenyl)methyl)-4-piperidinyl)-2H-benzimidazol-2-one or synonym: Akti-1/2) and fura-2 AM. Scalaradial was isolated and purified as described above, reconstituted in methanol (2.0 mM), and diluted with KRH to a working solution of 200 μM.
Statistical Analysis.
Patch-clamp data were acquired with PatchMaster software and exported to IGOR Pro (Wavemetrics). Current amplitudes were extracted from IGOR Pro and transferred to Excel (MS Office 2007), where all values for mean, standard deviation, and standard error of the mean were calculated. In some cases p-values based on unequal variances t test (unpaired, 2-tail) were calculated (Excel, TTEST function). Comparison of time course data was performed using one-way ANOVA with Tukey’s multiple comparison test. Significance level was set at 0.05.
Calcium imaging data were processed in SoftMax Pro. Maximum rate of Ca2+ influx was determined for each test well by line fitting 5 points (spanning ~16 s) that give the largest slope (5-point “Vmax” reduction applied). Resulting slopes were normalized to vehicle control. Fifty-percent inhibitory concentration curves for patch-clamp and Ca2+ imaging were fitted in IGOR Pro.
ACKNOWLEDGMENTS
This work was supported in part by grants from the Hawaii Community Foundation (Grant No. 20061480) (J.S.), NIH R01GM070634-04 (A.F.), NIH R01GM063954 (R.P.), and NIH P20GM103466 (F.D.H.). We thank B. Castillo, C. Kuo, J. Reinicke, J. Sahara, Tifanie Vansach, and S. Johne for excellent technical support. We thank I. Lange for critical feedback.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Faouzi M; Penner R Handb. Exp. Pharmacol 2014, 222, 403–426. [DOI] [PubMed] [Google Scholar]
- (2).Fleig A; Chubanov V Handb. Exp. Pharmacol 2014, 222, 521–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Perraud AL; Fleig A; Dunn CA; Bagley LA; Launay P; Schmitz C; Stokes AJ; Zhu Q; Bessman MJ; Penner R; Kinet JP; Scharenberg AM Nature 2001, 411, 595–599. [DOI] [PubMed] [Google Scholar]
- (4).Starkus J; Beck A; Fleig A; Penner R J. Gen. Physiol 2007, 130, 427–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Miller BA Br. J. Pharmacol 2004, 143, 515–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Fonfria E; Marshall IC; Benham CD; Boyfield I; Brown JD; Hill K; Hughes JP; Skaper SD; McNulty S Br. J. Pharmacol 2004, 143, 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lange I; Yamamoto S; Partida-Sanchez S; Mori Y; Fleig A; Penner R Sci Signaling 2009, 2, ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Fonfria E; Mattei C; Hill K; Brown JT; Randall A; Benham CD; Skaper SD; Campbell CA; Crook B; Murdock PR ; Wilson JM; Maurio FP; Owen DE; Tilling PL; McNulty S J. Recept. Signal Transduction Res 2006, 26, 179–198. [DOI] [PubMed] [Google Scholar]
- (9).Chung KK; Freestone PS; Lipski J J. Neurophysiol 2011, 106, 2865–2875. [DOI] [PubMed] [Google Scholar]
- (10).Park L; Wang G; Moore J; Girouard H; Zhou P; Anrather J; Iadecola C Nat. Commun 2014, 5, 5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Takahashi K; Sakamoto K; Kimura J J. Pharmacol. Sci 2012, 118, 186–197. [DOI] [PubMed] [Google Scholar]
- (12).Cook NL; Vink R; Helps SC; Manavis J; van den Heuvel C J. Mol. Neurosci 2010, 42, 192–199. [DOI] [PubMed] [Google Scholar]
- (13).Togashi K; Hara Y; Tominaga T; Higashi T; Konishi Y; Mori Y; Tominaga M EMBO J. 2006, 25, 1804–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yamamoto S; Shimizu S; Kiyonaka S; Takahashi N; Wajima T ; Hara Y; Negoro T; Hiroi T; Kiuchi Y; Okada T; Kaneko S; Lange I; Fleig A; Penner R; Nishi M; Takeshima H; Mori Y Nat. Med 2008, 14, 738–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zeng X; Sikka SC; Huang L; Sun C; Xu C; Jia D; Abdel-Mageed AB; Pottle JE; Taylor JT; Li M Prostate Cancer Prostatic Dis. 2010, 13, 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Koh DW; Powell DP; Blake SD; Hoffman JL; Hopkins MM; Feng X Oncol. Rep 2015, 34, 1589–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yonezawa R; Yamamoto S; Takenaka M; Kage Y; Negoro T; Toda T; Ohbayashi M; Numata T; Nakano Y; Yamamoto T; Mori Y; Ishii M; Shimizu S Free Radical Biol. Med 2016, 90, 101–113. [DOI] [PubMed] [Google Scholar]
- (18).Ostapchenko VG; Chen M; Guzman MS; Xie YF; Lavine N; Fan J; Beraldo FH; Martyn AC; Belrose JC; Mori Y; MacDonald JF; Prado VF; Prado MA; Jackson MF J. Neurosci 2015, 35, 15157–15169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hill K; Benham CD; McNulty S; Randall AD Neuropharmacology 2004, 47, 450–460. [DOI] [PubMed] [Google Scholar]
- (20).Hill K; McNulty S; Randall AD Naunyn-Schmiedeberg’s Arch. Pharmacol 2004, 370, 227–237. [DOI] [PubMed] [Google Scholar]
- (21).Togashi K; Inada H; Tominaga M Br. J. Pharmacol 2008, 153, 1324–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kraft R; Grimm C; Frenzel H; Harteneck C Br. J. Pharmacol 2006, 148, 264–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kheradpezhouh E; Barritt GJ; Rychkov GY Redox Biol. 2016, 7, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Nadler MJS; Hermosura MC; Inabe K; Perraud A; Zhu Q; Stokes AJ; Kurosaki T; Kinet J; Penner P; Scharenberg AM; Fleig A Nature 2001, 411, 590–595. [DOI] [PubMed] [Google Scholar]
- (25).Schmitz C; Perraud AL; Johnson CO; Inabe K; Smith MK; Penner R; Kurosaki T; Fleig A; Scharenberg AM Cell 2003, 114, 191–200. [DOI] [PubMed] [Google Scholar]
- (26).Peinelt C; Vig M; Koomoa DL; Beck A; Nadler MJ; Koblan-Huberson M; Lis A; Fleig A; Penner R; Kinet JP Nat. Cell Biol 2006, 8, 771–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Launay P; Cheng H; Srivatsan S; Penner R; Fleig A; Kinet JP Science 2004, 306, 1374–1377. [DOI] [PubMed] [Google Scholar]
- (28).Launay P; Fleig A; Perraud AL; Scharenberg AM; Penner R; Kinet JP Cell 2002, 109, 397–407. [DOI] [PubMed] [Google Scholar]
- (29).Waluk DP; Vielfort K; Derakhshan S; Aro H; Hunt MC Biochem. Biophys. Res. Commun 2013, 430, 54–59. [DOI] [PubMed] [Google Scholar]
- (30).Davidovic L; Vodenicharov M; Affar EB; Poirier GG Exp. Cell Res 2001, 268, 7–13. [DOI] [PubMed] [Google Scholar]
- (31).Suto MJ; Turner WR; Arundel-Suto CM; Werbel LM; Sebolt-Leopold JS Anti-Cancer Drug Des. 1991, 6, 107–117. [PubMed] [Google Scholar]
- (32).Alano CC; Kauppinen TM; Valls AV; Swanson RA Proc. Natl. Acad. Sci U. S. A. 2006, 103, 9685–9690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Carvalho LH; Brandao MGL; Santos-Filho D; Lopes JLC; Krettli AU Brazilian J. Med. Biol. Res 1991, 24, 1113–1123. [PubMed] [Google Scholar]
- (34).Hope WC; Chen T; Morgan DW Agents Actions 1993, 39, C39–42. [DOI] [PubMed] [Google Scholar]
- (35).Arumugam TV; Arnold N; Proctor LM; Newman M; Reid RC; Hansford KA; Fairlie DP; Shiels IA; Taylor SM Br. J. Pharmacol 2003, 140, 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Hansford KA; Reid RC; Clark CI; Tyndall JD; Whitehouse MW; Guthrie T; McGeary RP; Schafer K; Martin JL; Fairlie DP ChemBioChem 2003, 4, 181–185. [DOI] [PubMed] [Google Scholar]
- (37).Singer AG; Ghomashchi F; Le Calvez C; Bollinger J; Bezzine S; Rouault M; Sadilek M; Nguyen E; Lazdunski M; Lambeau G; Gelb MH J. Biol. Chem 2002, 277, 48535–48549. [DOI] [PubMed] [Google Scholar]
- (38).Tithof PK; Elgayyar M; Cho Y; Guan W; Fisher AB; Peters-Golden M FASEB J. 2002, 16, 1463–1464. [DOI] [PubMed] [Google Scholar]
- (39).Xie Y; Yu Z; Liu L; Guo Y; Lou L Cancer Biol. Ther 2006, 5, 988–992. [DOI] [PubMed] [Google Scholar]
- (40).DeFeo-Jones D; Barnett SF; Fu S; Hancock PJ; Haskell KM; Leander KR; McAvoy E; Robinson RG; Duggan ME; Lindsley CW; Zhao Z; Huber HE; Jones RE Mol. Cancer Ther 2005, 4, 271–279. [PubMed] [Google Scholar]
- (41).Davies SP; Reddy H; Caivano M; Cohen P Biochem. J 2000, 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Powis G; Bonjouklian R; Berggren MM; Gallegos A; Abraham R; Ashendel C; Zalkow L; Matter WF; Dodge J; Grindey G; Vlahos CJ Cancer Res. 1994, 54, 2419–2423. [PubMed] [Google Scholar]
- (43).Lindsley CW; Barnett SF; Layton ME; Bilodeau MT Curr. Cancer Drug Targets 2008, 8, 7–18. [DOI] [PubMed] [Google Scholar]
- (44).Ma HT; Patterson RL; van Rossum DB; Birnbaumer L; Mikoshiba K; Gill DL Science 2000, 287, 1647–1651. [DOI] [PubMed] [Google Scholar]
- (45).Xu SZ; Zeng F; Boulay G; Grimm C; Harteneck C; Beech DJ Br. J. Pharmacol 2005, 145, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hu HZ; Gu Q; Wang C; Colton CK; Tang J; Kinoshita-Kawada M; Lee LY; Wood JD; Zhu MX J. Biol. Chem 2004, 279, 35741–35748. [DOI] [PubMed] [Google Scholar]
- (47).Li M; Jiang J; Yue L J. Gen. Physiol 2006, 127, 525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Zhang X; Chen Q; Wang Y; Peng W; Cai H Front. Physiol 2014, 5, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Song Y; Buelow B; Perraud AL; Scharenberg AM J. Biomol. Screening 2008, 13, 54–61. [DOI] [PubMed] [Google Scholar]
- (50).Dennis EA J. Biol. Chem 1994, 269, 13057–13060. [PubMed] [Google Scholar]
- (51).Hara Y; Wakamori M; Ishii M; Maeno E; Nishida M; Yoshida T; Yamada H; Shimizu S; Mori E; Kudoh J; Shimizu N; Kurose H; Okada Y; Imoto K; Mori Y Mol. Cell 2002, 9, 163–173. [DOI] [PubMed] [Google Scholar]
- (52).Boyer CS; Bannenberg GL; Neve EP; Ryrfeldt A; Moldeus P Biochem. Pharmacol 1995, 50, 753–761. [DOI] [PubMed] [Google Scholar]
- (53).Thwin MM; Ong WY; Fong CW; Sato K; Kodama K; Farooqui AA; Gopalakrishnakone P Exp. Brain Res 2003, 150, 427–433. [DOI] [PubMed] [Google Scholar]
- (54).Baek SH; Kwon TK; Lim JH; Lee YJ; Chang HW; Lee SJ; Kim JH; Kwun KB J. Immunol 2000, 164, 6359–6365. [DOI] [PubMed] [Google Scholar]
- (55).Costa-Junior HM; Hamaty FC; da Silva Farias R; Einicker-Lamas M; da Silva MH; Persechini PM Cell Tissue Res. 2006, 324, 255–266. [DOI] [PubMed] [Google Scholar]
- (56).Yagami T; Ueda K; Asakura K; Hata S; Kuroda T; Sakaeda T; Takasu N; Tanaka K; Gemba T; Hori Y Mol. Pharmacol 2002, 61, 114–126. [DOI] [PubMed] [Google Scholar]
- (57).Wang Q; Sun AY; Pardeike J; Muller RH; Simonyi A; Sun GY Brain Res. 2009, 1285, 188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Torregrosa G; Perez-Asensio FJ; Burguete MC; Castello-Ruiz M; Salom JB; Alborch E Exp. Brain Res 2007, 176, 248–259. [DOI] [PubMed] [Google Scholar]
- (59).Xie Y; Liu L; Huang X; Guo Y; Lou L J. Pharmacol. Exp. Ther 2005, 314, 1210–1217. [DOI] [PubMed] [Google Scholar]
- (60).Danciu TE; Adam RM; Naruse K; Freeman MR; Hauschka PV FEBS Lett. 2003, 536, 193–197. [DOI] [PubMed] [Google Scholar]
- (61).Sandoval AJ; Riquelme JP; Carretta MD; Hancke JL; Hidalgo MA; Burgos RA J. Leukocyte Biol 2007, 82, 1266–1277. [DOI] [PubMed] [Google Scholar]
- (62).Yano S; Tokumitsu H; Soderling TR Nature 1998, 396, 584–587. [DOI] [PubMed] [Google Scholar]
- (63).Aarts M; Iihara K; Wei WL; Xiong ZG; Arundine M; Cerwinski W; MacDonald JF; Tymianski M Cell 2003, 115, 863–877. [DOI] [PubMed] [Google Scholar]
- (64).Sun HS; Jackson MF; Martin LJ; Jansen K; Teves L; Cui H; Kiyonaka S; Mori Y; Jones M; Forder JP; Golde TE; Orser BA; Macdonald JF; Tymianski M Nat. Neurosci 2009, 12, 1300–1307. [DOI] [PubMed] [Google Scholar]
- (65).Cimino G; Stefano SD; Luccia AD Experientia 1979, 35, 1277–1278. [Google Scholar]
- (66).Yasuda F; Tada H Experientia 1981, 37, 110–111. [DOI] [PubMed] [Google Scholar]
- (67).Soboloff J; Spassova MA; He LP; Xu W; Dziadek MA; Gill DL Proc. Natl. Acad. Sci U. S. A. 2006, 103, 4040–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Lis A; Peinelt C; Beck A; Parvez S; Monteilh-Zoller M; Fleig A; Penner R Curr. Biol 2007, 17, 794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Vig M; Peinelt C; Beck A; Koomoa DL; Rabah D; Koblan-Huberson M; Kraft S; Turner H; Fleig A; Penner R; Kinet JP Science 2006, 312, 1220–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Grynkiewicz G; Poenie M; Tsien RY J. Biol. Chem 1985, 260, 3440–3450. [PubMed] [Google Scholar]