

Supplemental Digital Content is Available in the Text.
Baseline mechanical pain threshold does not predict analgesic response to pregabalin in painful chemotherapy-induced peripheral neuropathy caused by docetaxel, paclitaxel, or oxaliplatin.
Keywords: Chemotherapy-induced peripheral neuropathy, Pregabalin, Quantitative sensory testing, Mechanical pain threshold, Oxaliplatin, Paclitaxel, Docetaxel
Abstract
Painful chemotherapy-induced peripheral neuropathy (CIPN) is a debilitating and treatment-resistant sequela of many chemotherapeutic medications. Ligands of α2δ subunits of voltage-gated Ca2+ channels, such as pregabalin, have shown efficacy in reducing mechanical sensitivity in animal models of neuropathic pain. In addition, some data suggest that pregabalin may be more efficacious in relieving neuropathic pain in subjects with increased sensitivity to pinprick. We hypothesized that greater mechanical sensitivity, as quantified by decreased mechanical pain threshold at the feet, would be predictive of a greater reduction in average daily pain in response to pregabalin vs placebo. In a prospective, randomized, double-blinded study, 26 patients with painful CIPN from oxaliplatin, docetaxel, or paclitaxel received 28-day treatment with pregabalin (titrated to maximum dose 600 mg per day) and placebo in crossover design. Twenty-three participants were eligible for efficacy analysis. Mechanical pain threshold was not significantly correlated with reduction in average pain (P = 0.97) or worst pain (P = 0.60) in response to pregabalin. There was no significant difference between pregabalin and placebo in reducing average daily pain (22.5% vs 10.7%, P = 0.23) or worst pain (29.2% vs 16.0%, P = 0.13) from baseline. Post hoc analysis of patients with CIPN caused by oxaliplatin (n = 18) demonstrated a larger reduction in worst pain with pregabalin than with placebo (35.4% vs 14.6%, P = 0.04). In summary, baseline mechanical pain threshold tested on dorsal feet did not meaningfully predict the analgesic response to pregabalin in painful CIPN.
1. Introduction
Chemotherapy-induced peripheral neuropathy (CIPN) affects 25% to 90% of patients treated with anticancer treatments such as platinum compounds, taxanes, and vinca alkaloids. Chemotherapy-induced peripheral neuropathy is frequently a dose-limiting side effect of these medications, which can increase the rate of treatment failure.12,39 In addition, chronic CIPN, which affects anywhere from 10% to 50% of patients treated with these drugs,4,33,40 reduces quality of life even in patients whose cancers are in remission.22
Evidence of successful treatment of painful CIPN is very limited; only one large, positive, randomized controlled study on the treatment of CIPN has been published. This study demonstrated marginal effectiveness of duloxetine, a serotonin–norepinephrine reuptake inhibitor.38 Venlafaxine, another serotonin-norepinephrine reuptake inhibitor, has shown efficacy in the possible secondary prevention of oxaliplatin-induced neuropathy after the onset of acute neuropathy symptoms.11 Numerous medications have failed to show efficacy compared with placebo,5,25,36 although most studies have used composite neuropathy scores or symptom scores, rather than pain intensity, as primary outcomes.
The mechanisms of injury in CIPN, and the transition from acute to chronic neurotoxicity, are not fully understood, although many chemotherapy agents share pathological findings in CIPN, eg, common final pathway of axonal mitochondrial damage.29,35 In rats, CIPN caused by oxaliplatin and taxanes (paclitaxel and docetaxel) results in overexpression of α2δ subunits of voltage-gated Ca2+ channels in the dorsal root ganglion16 and spinal cord dorsal horn.41 This suggests that modulating neuronal Ca2+ channel function by α2δ ligands may be effective in treating CIPN. Administration of α2δ ligands gabapentin27,28,41 and pregabalin2,34 reduced mechanical and thermal hypersensitivity in rat models of CIPN caused by oxaliplatin and taxanes.
Neuronal voltage-gated Ca2+ channels as targets for treating CIPN remain largely unexplored clinically. The only published randomized clinical trial of gabapentin in CIPN was negative,36 although it did not have pain as an inclusion criterion; observational data suggest clinical effectiveness of pregabalin in oxaliplatin- and taxane-induced peripheral neuropathy.32
In neuropathic pain, the “one size fits all” pharmacotherapy approach yields unsatisfactory effect sizes.14 A few studies have prospectively demonstrated that sensory phenotyping based on quantitative sensory testing (QST) predicts response to treatment. For example, Demant et al.9 demonstrated that the irritable nociceptor phenotype predicts response to oxcarbazepine. However, most such data come from post hoc analyses of clinical trials7,37 or from uncontrolled trials.42 For example, post hoc analysis showed that treatment of neuropathic pain with botulinum toxin A was more efficacious in patients with allodynia identified by QST.3 Similarly, in an HIV sensory neuropathy study that was negative overall, patients with hyperalgesia to pinprick reported a significant analgesic response to pregabalin.37 The application of such phenotyping to CIPN remains unexplored.
Based on findings that pinprick hypersensitivity may predict responsiveness to pregabalin, together with animal data and observational clinical data supporting the usefulness of pregabalin in CIPN, we hypothesized that mechanical hypersensitivity to pinprick (lower mechanical pain threshold [MPT]) may predict better analgesic response to pregabalin treatment in patients with painful CIPN.
The objective of this prospective, randomized, double-blind, placebo-controlled crossover trial was to determine whether baseline MPT predicts the response to pregabalin in patients with painful CIPN.
2. Methods
This investigator-initiated study was approved by the Washington University Institutional Review Board (#201501067). Written informed consent was obtained from all participants, and the study was registered at ClinicalTrials.gov (NCT02394951). The IRB-approved study protocol is available at http://anest.wustl.edu/HaroutounianProtocols/Pregabalin_in_CIPN_IRB-APPROVED_PROTOCOL.pdf.
2.1. Participants
Participants were eligible for this study if they met the following criteria: (1) age >18 years; (2) distal, symmetric pain in both feet that appeared within 12 weeks of treatment with paclitaxel, docetaxel, or oxaliplatin (or any combination of these); (3) pain persistence for at least 2 months; (4) scores ≥4 on the Douleur Neuropathique 4 (DN4) questionnaire6; (5) pain of average daily intensity >3 on 0 to 10 numerical rating scale (NRS) in the past week; and, (6) if of childbearing potential, agreed to use contraception for the duration of the study. Exclusion criteria were hypersensitivity to or current treatment with pregabalin, CIPN that may be associated with previous treatment with a vinca alkaloid, current treatment with a vinca alkaloid, any prechemotherapy signs of distal symmetric polyneuropathy, creatinine clearance <30 mL/minute, ALT or AST >3 times the normal limit, planned surgeries or radiation treatment within 10 weeks after study inclusion, inability to complete pain self-report, current or planned pregnancy or lactation, seizure disorder treated with anticonvulsants, or current participation in a trial with another investigational agent. Vinca alkaloids were specifically excluded because some evidence suggests these chemotherapeutics may not lead to Ca2+channels α2δ subunit upregulation in the dorsal horn or dorsal root ganglia in CIPN in the manner than platinum or taxane medications do.16 Participants receiving gabapentin, anticonvulsants, or NSAIDs for pain were required to discontinue these medications before study initiation. Participants could continue tricyclic antidepressants and serotonin and norepinephrine reuptake inhibitors. Participants could continue standing dose of opioids at a maximum dose of 60 mg oral morphine equivalent per day, but as-needed doses of opioids were not allowed for treatment of CIPN. However, as-needed short-acting opioids (with or without acetaminophen) for pain other than CIPN were allowed up to 4 daily doses, with daily recording of analgesic consumption.
2.2. Acquisition of baseline characteristics
The sequence of events for each participant enrolled in the study is shown in Supplementary Figure 1 (available at http://links.lww.com/PAIN/A782). All participants attended an initial screening visit. At this visit, participants underwent sensory mapping of their lower extremities to sensory perception of static mechanical stimulation (#6.10 Semmes-Weinstein monofilament; North Coast Medical, Gilroy, CA), dynamic mechanical stimulation (SENSELab Brush-05; Somedic, Norra Mellby, Sweden), and 40°C warmth and 20°C cold (Rolltemp II; Somedic). These stimuli are not routinely painful to healthy people, or when applied to unaffected areas of skin in patients with neuropathy. For each sensory modality, participants were asked whether they felt that their sensation of the stimulus was decreased or increased compared with the sensation on unaffected skin and whether the sensation was painful. Participants were designated as having hypoesthesia, hyperesthesia, or allodynia if they had decreased sensation, increased sensation, or pain (respectively) at the site of their spontaneous neuropathic pain (dorsal feet). Hyperesthesia and allodynia could be present at the same site. Participants also completed the following questionnaires: Brief Pain Inventory (BPI) (from which baseline average and worst daily pain values were drawn, using a 0-10 NRS); Neuropathic Pain Symptom Inventory (NPSI); Depression, Anxiety, and Positive Outlook Scale (DAPOS); and the 9-item Sleep Problems Index (SPI II). Participants were trained in the use of tablet computers on which they documented daily pain scores, study drug use, and side effects. All participants underwent a QST battery per details below.
2.3. Quantitative sensory testing protocol
Quantitative sensory testing was performed at the first and third study visits at Washington University Clinical Trials Research Unit (CTRU). Testing was performed on the dorsal midfoot of the more painful foot. If feet were equally painful, one was chosen randomly for testing. The ipsilateral shoulder served as a control area. Shoulder was chosen as control site (in lieu of typically used volar forearm), as we suspected some subjects to have sensory changes in distal upper extremities.40
Quantitative sensory testing was performed following a previously published method.21 We tested, using the method of limits: mechanical detection threshold using Semmes-Weinstein monofilaments (North Coast Medical), MPT and wind-up ratio using weighted metal PinPrick stimulators (MRC Systems GmbH, Heidelberg, Germany), cold detection threshold, warmth detection threshold, cold pain threshold, heat pain threshold (HPT) (using Thermal Sensory Analyzer; TSA-II, Medoc, Ramat Yishai, Israel), and vibration detection threshold using a 64-Hz tuning fork. For a given parameter, the extent of hyposensitivity/hypersensitivity was measured by the value of the parameter at the affected site minus the value at the control site.
We measured conditioned pain modulation (CPM) using the method of levels via the Q-Sense Thermal Analyzer (Medoc). This machine is equipped with 2 Peltier thermodes whose temperature could be programmed independently. The testing stimulus was determined by temperature that elicits heat pain of 60 on a 0 to 100 NRS (Pain-60). At the time of protocol writing, the conditioning stimulus was planned as a hot stimulus set to a temperature that elicits a pain of 30 on a 0 to 100 NRS. After pilot testing before study initiation, the protocol was modified to deliver a cold conditioning stimulus of 16°C because it provided more consistent CPM response (unpublished data).
At first, the test stimulus was applied at the nondominant forearm twice, 30 seconds apart, and the average pain value on 0 to 100 NRS was obtained. After a 10-minute break, the conditioning stimulus was applied to the dominant forearm for 60 seconds; during the last 30 seconds of conditioning, 2 Pain-60 heat stimuli were applied to the nondominant forearm. The procedure was repeated twice. Conditioned pain modulation was measured as the difference between NRS of Pain-60 stimuli with and without the contralateral cold conditioning (average score of 2 tests). Conditioned pain modulation was measured at the participants' forearms only and not at the affected sites.
All sensory assessments were performed by the same trained research coordinator, who remained blinded to treatment sequence allocation throughout the study.
2.4. Study medication administration periods
Patients were randomized to receive pregabalin, followed by placebo, or vice versa with a 1:1 allocation ratio. The study pharmacist used an electronic random number generator to randomize participants in blocks of 8 without stratification. All study team members were blinded to the treatment allocation except the study pharmacist, who did not participate in patient enrollment or any data collection. Pregabalin (or identical looking placebo capsules, provided by Pfizer Inc) was administered for 4 weeks in each study period. The dose was increased as follows: 75 mg twice a day (BID) for 3 days, 150 mg BID for 3 days, then 300 mg BID until and including day 28.
In the event of any grade 2 adverse events (Common Terminology Criteria for Adverse Events—CTCAE version 4.0), the dose was reduced to the last dose before dose increase. If the adverse effect diminished to ≤grade 1, one attempt to dose escalate was allowed after 3 additional days. If the dose escalation was not tolerated (ie, recurrent grade 2 adverse event), the treatment was continued at the previous (lower) dose until day 28. Any CTCAE ≥grade 3 triggered participant withdrawal from the study. During the first treatment sequence, the participants were asked to guess whether they were receiving placebo or pregabalin.
At the end of treatment sequence, the dose was tapered over 3 days (eg, 150 mg BID for 2 days, then 75 mg BID for 1 additional day) followed by 10 days of study treatment-free washout (end of week 6). In participants with creatinine clearance 30 to 60 mL/minute, the target (maximum) dose was 300 mg/day. In these participants, the dose was tapered over 3 days of 75 mg BID dosing. If participant's highest tolerable maintenance dose was 75 mg BID, the taper schedule was 75 mg once a day over 3 days.
Acetaminophen (paracetamol) 500 mg tablets were provided as rescue medication, as the only allowed analgesic to use for breakthrough pain, in doses up to 4000 mg/day.
We used an electronic diary system on a tablet device provided to each participant for daily assessment of pain ratings, adherence to medications, and side-effect reporting. On each day beginning with the first day of study medication administration, participants recorded via the tablet device the amount (in pills) of study medication used, their average/worst pain severity, any adverse effects, and whether they believed such effects were due to the study medication. Medication consumption was not directly observed by members of the study team, but pill count (both for study drug and acetaminophen) was performed at the end of each treatment sequence. The data entered to the tablet device were stored on an encrypted and password-protected cloud server; these data were accessible in real time to study team members for adverse effect monitoring.
The participants were scheduled to return on day 28 ± 2 days for visit 2 to complete the questionnaires. After the washout period (end of week 6), the participants returned for visit 3, before the second treatment sequence. The participants underwent QST and CPM procedures and completed appropriate questionnaires (BPI, NPSI, DAPOS, and SPI II). Thereafter, the second 4-week treatment sequence was scheduled for weeks 7 to 10, according to the same titration schedule as sequence 1. Three-day dose tapering was practiced at the end of the second treatment sequence in the same manner as was performed for the first treatment. The participants returned for the final assessment visit (visit 4) approximately 3 days after end of week 10. Safety and efficacy data, blood draw for determining plasma pregabalin concentration, and an optional skin biopsy in participants who agreed were obtained at visit 4. Pregabalin presence in plasma was determined to confirm adherence to treatment and correct pregabalin vs placebo allocation.
2.5. Pregabalin concentration assay
The analytical method for determining the concentration of pregabalin in plasma was adapted from Martinc et al.30 Specifically, Agilent 1260 HPLC system equipped with fluorescent detector was used. Separation was achieved with Agilent InfinityLab Poroshell 120 EC-C18 column (100 × 4.6 mm, 2.7 µm) and a gradient flow of methanol: acetonitrile: ammonium acetate buffer pH 5.0. Gabapentin was used as an internal standard. Pregabalin retention time was 11.2 minutes, and limit of quantification (LOQ) was 100 ng/mL.
2.6. Skin biopsy
In patients who provided consent, a 3-mm skin punch biopsy in the lower leg (10 cm above the lateral malleolus) was performed at the final study visit. The biopsy specimen was placed in Zamboni's fixative overnight, then transferred to a glycerol-based cryoprotection fluid, and subsequently frozen and stored at −80°C. Frozen tissues were cut into 50-µm sections before immunostaining with PGP 9.5 antibody for the determination of intraepidermal nerve fiber density (IENFD) per published guidelines.23,26 A researcher blinded to patient allocation/results analyzed the skin biopsies.
2.7. Statistical methods
The primary outcome of the study was the comparison of the slopes of 2 correlations: baseline MPT vs percent pain reduction with pregabalin and baseline MPT vs percent pain reduction with placebo.
Pain reduction (both for average and worst pain) was determined using the difference in pain intensity from baseline to the average of daily pain on days 24 to 28 (ie, last 5 days) of each treatment phase, obtained from the patient diaries. For patients whose data for the last week was missing, we used the last observation carried forward method of imputation.
The sample size calculation was performed for detecting a significant difference between correlations on percent pain reduction vs MPT slopes of pregabalin and placebo in linear bivariate regression. To detect 0.3 (SD = 0.5) difference in correlation coefficients between the 2 slopes, with 85% power and α = 0.05, 27 participants were required in this crossover design. We aimed to recruit 35 patients to account for possible 20% dropout rate.
As secondary analyses, we compared pain reduction with pregabalin (day 0-28) vs placebo for both average and worst daily pain, using paired t-tests. In addition, we performed the Fisher exact test to compare the proportion of patients with ≥30% and ≥50% reduction in average and worst pain intensity from baseline with pregabalin vs placebo. Number needed to treat (NNT) was calculated from this proportion of responders to pregabalin and placebo.13 We compared the rates of adverse events and the proportions of participants experiencing any adverse event using the Fisher exact test.
In prespecified exploratory analyses, we examined the associations between other QST variables (mechanical detection threshold, warmth detection threshold, cold detection threshold, heat pain threshold, cold pain threshold, vibration detection threshold, wind-up ratio, and conditioned pain modulation) and response to pregabalin and placebo. In addition, we examined the association between hyperesthesia or allodynia to cold, warmth, static mechanical stimulation, and dynamic mechanical stimulation, and the response to pregabalin and placebo. We also examined the value of clinically significant scores in each NPSI domain (defined as a score ≥3) in predicting response to pregabalin vs placebo. Because the majority of patients' CIPN was associated with oxaliplatin treatment, we performed a post hoc subgroup analysis examining the efficacy outcomes in patients with only oxaliplatin-induced peripheral neuropathy.
3. Results
Study recruitment began in April 2015. Due to slower-than-anticipated recruitment, the study sponsor requested the study to be terminated in March 2018, before meeting the recruitment goal. Overall, 26 patients were enrolled in the study (Fig. 1). Five participants (19%) voluntarily withdrew during the pregabalin treatment phase, reporting hypoglycemia (n = 1), urticaria (n = 1), stomach pain and constipation (n = 1), dizziness, blurred vision, somnolence, and urticaria (n = 1), and small bowel obstruction (n = 1). One participant (4%) voluntarily withdrew during the placebo treatment phase, reporting uncontrolled pain after discontinuing prestudy analgesics. Of these, 2 participants (8%) did not begin the second arm of the study (pregabalin for one, placebo for the other); these patients were censored from any outcomes examining changes in pain. One participant (4%) initiated both arms but refused the visit 3 QST; this participant was included in analysis of changes in pain but not in analysis of QST as a predictor of response to treatment.
Figure 1.
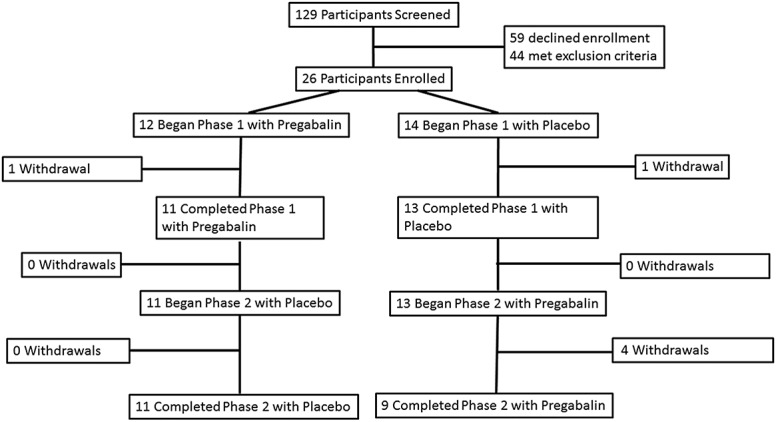
Enrollment flowchart. Flow of participants through the study.
Table 1 shows participant demographics (Supplementary Table 1 shows baseline QST parameters; available at http://links.lww.com/PAIN/A782). Notably, most patients (88%) initially developed neuropathy symptoms while actively receiving chemotherapy, and oxaliplatin was the causative chemotherapeutic in 81% of cases. Average and worst daily pain severity in our cohort before therapy were 5.3 and 5.6 on a 0 to 10 NRS, respectively.
Table 1.
Participant demographic information divided by study medication administration order.

Figure 2 and Table 2 show changes in average and worst pain over time. There were no significant differences in baseline average pain (4.4 ± 2.1 vs 4.3 ± SD 1.9, P = 0.41) or worst pain (5.1 ± 2.2 vs 5.3 ± 2.3, P = 0.41) before initiating pregabalin or placebo treatment. Overall, the absolute reduction in average daily pain on 0 to 10 NRS was 0.8 ± 1.1 with pregabalin, and 0.5 ± 1.1 with placebo (P = 0.08); this represented a 22.5% and 10.7% reduction in average daily pain, respectively (P = 0.23). There was no association between tolerable daily dose and percent reduction in average daily pain (R2 < 0.001) or worst daily pain (R2 = 0.002). Numbers of patients needed to treat (NNT) with pregabalin for achieving ≥30% and ≥50% reductions in average pain were 6.2 and 12.5, respectively. The absolute reduction in worst daily pain on 0 to 10 NRS was 1.4 ± 1.3 with pregabalin use and 0.9 ± 1.7 with placebo (P = 0.25); this represented a 29.2% and 14.0% reduction in worst daily pain, respectively (P = 0.13). The NNT for achieving ≥30% and ≥50% reductions in worst pain with pregabalin were 4.2 and 8.3, respectively. Average daily acetaminophen intake was not significantly different between pregabalin vs placebo treatment phases; 213 mg ± 364 mg vs 374 mg ± 712 mg, respectively (P = 0.19).
Figure 2.
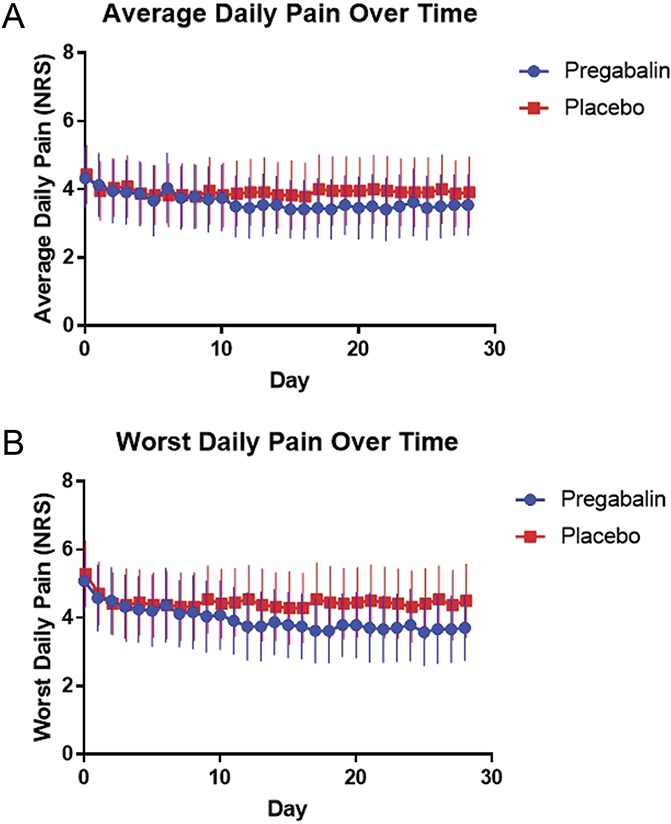
Changes in pain over time (by medication). (A) Changes in average pain over time. (B) Changes in worst pain over time. Blue/circles—pregabalin, red/squares—placebo. Points show mean daily pain, and lines represent the bounds of the 95% confidence interval. NRS, Numerical Rating Scale.
Table 2.
Effects of pregabalin and placebo on daily pain.

Figure 3 shows the correlation between MPT and percent reduction in average (Fig. 3A) and worst (Fig. 3B) daily pain with pregabalin (blue) and placebo (red). There was no significant association between MPT and reduction in average pain (R2 = 0.08, P = 0.20) and worst pain (R2 = 0.08, P = 0.19) with pregabalin. These associations were not significantly different from those seen between MPT and reduction in average pain (R2 = 0.05, P = 0.31) and worst pain (R2 = 0.14, P = 0.07) with placebo. There was no significant difference in the slopes of MPT vs pain reduction between the 2 arms for either average pain (P = 0.92) or worst pain (P = 0.60). The associations between baseline MPT and changes in additional outcomes (BPI interference, BPI severity, NPSI total, and SPI II scores) were also uniformly low (ranging from R2 = 0.002 to R2 = 0.017) and nonsignificant. Of the 8 other QST parameters examined, none of the correlations between the QST parameter at baseline and the reduction in average daily pain with pregabalin was significant.
Figure 3.
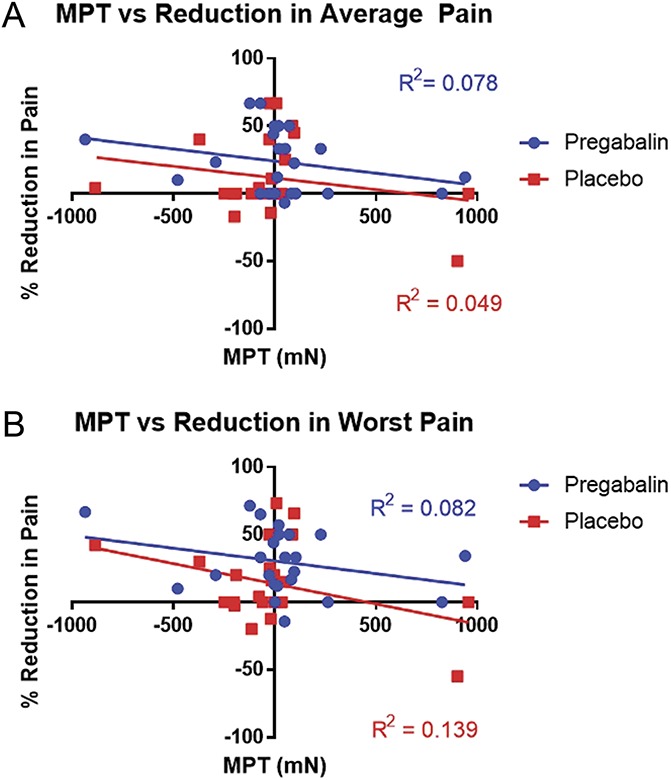
Correlations between MPT and reduction in pain with pregabalin and placebo. (A) Correlations between MPT and percent reduction in average pain. (B) Correlations between MPT and percent reduction in worst pain. Blue/circles—pregabalin, red/squares—placebo. MPT, mechanical pain threshold.
The aforementioned primary outcome measures were based on data collected on days 23 to 28 of treatment. Secondary outcome measures (including NPSI, SPI II, BPI, and DAPOS) were collected at a single time point occurring 1.5 ± 1.7 days, on average, after the last day of full dosing of the study drug; participants received a tapered dose of the study drug for 3 days after the last day of full dosing. Table 3 shows changes in NPSI, SPI II, BPI, and DAPOS with pregabalin and placebo. Neuropathic Pain Symptom Inventory subscales and the NPSI total score decreased between 23% and 32% with pregabalin vs 3% to 27% with placebo. The change in scores for pressing pain (−31.5% vs +26.3%, P = 0.02), evoked pain (−30.3% vs +12.2%, P = 0.02), paresthesias/dysesthesias (−28.3% vs −6.1%, P = 0.05), and total NPSI score (−29.5% vs +6.0%, P = 0.01) were more significant with pregabalin than with placebo. Baseline MPT was not correlated with the changes in any of these scales. As a post hoc analysis, we tested whether any other QST parameters correlated with decrease in total NPSI score; only HPT trended with a change in NPSI (P = 0.049). There were no significant differences between pregabalin and placebo in changes on SPI II or any of the subscales of BPI or DAPOS.
Table 3.
Changes in pain and quality-of-life questionnaires after treatment.

After identifying that pregabalin induced significant decrements in certain domains of the NPSI, we performed a post hoc analysis testing whether the preintervention presence of clinically meaningful intensity on any dimension of the NPSI (defined as a score ≥3) was predictive of change in pain with pregabalin vs placebo. There was no significant predictive value of any dimension with regard to changes in average or worst pain (P = 0.42-0.96; see Supplementary Table 2, available at http://links.lww.com/PAIN/A782).
Post hoc subgroup analysis was performed examining participants with oxaliplatin-induced CIPN (n = 19 starting both arms, n = 18 completing QST). With regard to reduction in average daily pain, the magnitude of pain reduction seen with pregabalin vs placebo either in absolute change (1.05 ± 1.2 vs 0.38 ± 1.2 on 0-10 NRS, P = 0.12) or percent pain reduction (24.4% vs 11.9%, P = 0.16) was not significant. The magnitude of reduction in worst pain was 1.72 ± 1.2 vs 0.84 ± 1.7 (P = 0.14) for absolute pain reduction, and 35.4% vs 14.6% (P = 0.04) for percent pain reduction in oxaliplatin-induced peripheral neuropathy. As with the overall sample, correlations between MPT and percent pain reduction were low (R2 = 0.08 for average pain; R2 = 0.13 for worst pain) and were not significantly greater than the correlations for placebo for average pain (P = 0.56) or worst pain (P = 0.60).
Proportions of participants experiencing adverse events with each study medication are shown in Table 4. The majority of participants experienced at least one adverse effect from both pregabalin (92%) and placebo (84%), although the proportion of patients experiencing any adverse effect did not differ between the 2 arms (P = 0.67, number needed to harm [NNH] = 12.5). Two types of adverse effects were experienced with a higher frequency with pregabalin than with placebo: dizziness (17 [68%] vs 4 [16%], P < 0.001, NNH = 1.9) and somnolence (22 [88%] vs 11 [44%], P = 0.002, NNH = 2.3). No adverse events were experienced more frequently with placebo.
Table 4.
Adverse events.

Twelve participants consented to punch biopsy of the distal leg, all of whom completed both arms of the study and both QST sessions. There was no significant correlation between IENFD and percent reduction in average pain (R2 < 0.001) or worst pain (R2 = 0.10) with pregabalin in these participants.
Results of static mechanical, dynamic mechanical, cold, and warm stimulation to the distal leg are shown in Tables 5 and 6. In exploratory analyses, we examined whether hyperesthesia or allodynia to these sensory modalities predicted either greater reduction in average or worst daily pain with pregabalin vs placebo (Table 5), or predicted a greater likelihood to achieve a clinically meaningful improvement of ≥30% in average or worst daily pain (Table 6). The magnitude of difference between pregabalin and placebo for reduction in average daily pain and worst daily pain was greatest for participants with static mechanical allodynia (33.5% ± 36.4% vs 6.1% ± 37.1% in average pain and 45.1% ± 33.6% vs 7.3% ± 36.9% in worst pain) as compared to cold, warmth, or dynamic mechanical allodynia. However, none of these tests had statistically significant predictive value (P = 0.12-0.95). In addition, although participants with baseline cold hyperesthesia were more likely to show a significant analgesic response to pregabalin (67% of participants with hyperesthesia vs 23% participants without hyperesthesia achieved ≥ 30% reduction in average pain, P = 0.05; not corrected for multiple comparisons), none of the other evoked hyperesthesia/allodynia phenotypes were predictive of response to pregabalin or placebo.
Table 5.
Analgesic responses to pregabalin vs placebo for participants with and without allodynia to static mechanical, dynamic mechanical, cold, and warm stimulation.

Table 6.
Baseline hyperesthesia and allodynia to static mechanical, dynamic mechanical, cold, and warm stimulation as predictors of ≥30% decrease in average daily pain.

When asked to guess which treatment they were receiving, 19 (73%) participants correctly identified their treatment allocation. The result was significantly different from randomly guessing the treatment allocation (P = 0.02).
Plasma pregabalin concentrations were analyzed at the end of the pregabalin arm for 20 participants and at the end of the placebo arm for 23 participants. No participants had a detectable pregabalin level at the end of the placebo arm. Of participants from whom pregabalin levels were collected at the end of the pregabalin arm of the study, the mean pregabalin dosing was 375 mg ± 165 mg/day. Mean time from last dose to sample collection was 7.8 ± 12.9 hours (5.0 ± 4.1 hour when excluding one outlier with time between last dose and sample collection of 60 hours). Mean plasma concentration of pregabalin was 4979 ng/mL ± 2779 ng/mL. Notably, 2 of 23 participants at the end of pregabalin treatment had plasma pregabalin concentration below LOQ. Both of these participants were receiving the lowest daily drug dose (75 mg) at the end of dose taper off, and both had the longest intervals between last pregabalin dose and collection of sample (60 and 13 hours).
4. Discussion
Chemotherapy-induced peripheral neuropathy is an iatrogenic complication of several chemotherapeutic agents and is a neuropathic pain condition where little evidence exists to support efficacious treatments. Several studies have suggested that somatosensory phenotyping in neuropathic pain can help identifying subgroups of patients who are more likely to respond to certain pharmacological interventions9,37,42 with the goal of developing mechanism-based treatment approaches.20 Because animal models suggest that pregabalin is effective in alleviating mechanical allodynia/hyperalgesia in neuropathic pain,2,18,31 we hypothesized that patients with neuropathic pain due to CIPN with greater mechanical sensitivity may be more likely to respond to pregabalin.
Our results suggest that baseline MPT, a quantitative measure of mechanosensitivity, is not a useful predictor of analgesic response to pregabalin in patients with painful CIPN. It also did not predict the degree of response to placebo. Both in our entire cohort and in the subgroup of participants with oxaliplatin-induced peripheral neuropathy, the associations between MPT and reduction in average or worst daily pain with the study drug were not significant. Similarly, when increased mechanosensitivity was operationalized dichotomously based on allodynia to static mechanical stimulation, there was no significant difference in response to pregabalin vs placebo in participants with and without allodynia. No other baseline QST parameter was found to be a significant predictor of analgesic response to pregabalin in this setting.
The overall reduction in average daily pain or worst daily pain over 4 weeks of treatment with a target dose of 600 mg/day pregabalin was not different from placebo. The NNTs for achieving ≥50% reduction in average or worst daily pain with pregabalin were 12.5 and 8.3 respectively, which is somewhat larger than the recently reported NNTs of pregabalin in neuropathic pain in general.14 Other outcome measures such as BPI pain severity or interference, sleep, and DAPOS scores were not different between the arms. However, several of the neuropathic pain-specific measures on NPSI questionnaire (including evoked pain, paresthesia/dysesthesia, and total NPSI score) showed larger improvement with pregabalin than with placebo. Post hoc analysis suggested that the improvement in worst pain was better with pregabalin than with placebo in the subgroup of patients with oxaliplatin-induced neuropathy, although this was not a prespecified outcome measure.
There are multiple explanations for the lack of correlation between MPT and response to pregabalin, which contrasts with findings from rodent models. First, animal models of painful CIPN may incompletely recapitulate the physiology of humans CIPN. For example, rodent CIPN models consistently show mechanical hypersensitivity of the paw.10,15,19 By contrast, only 5 of the 24 participants we tested (21%) reported static mechanical allodynia. This decreased mechanosensitivity is concordant with prior research in humans with CIPN.40 In addition, voltage-gated Ca2+ channels in the spinal cord dorsal horn are important mediators of mechanical hypersensitivity in rodent models but may play a lesser role in humans. Alternatively, a single QST modality such as MPT may be an insufficiently sensitive test for assessing the degree of α2δ overexpression-associated neuronal hypersensitivity. As a psychophysical test, MPT represents parameters beyond the degree of neuronal hypersensitivity in the spinal cord dorsal horn.17 Recent evidence suggests that tests involving less subjective input from the patient better reflect the structural and functional changes in peripheral neuropathic pain.24 Another important consideration is that rodent pain models use mechanical hypersensitivity as an outcome measure. The favorable response to pregabalin in the evoked pain component of NPSI suggests that outcome measures of evoked hypersensitivity better reflect the tactile hypersensitivity in animal models. Spontaneous pain, the key outcome measure in our study, does not have parallel accurate representation in rodents; additional research is required to align the phenotypic components of pain vs outcome measures in animal to human translation. Furthermore, animal studies are beset by translational challenges such as the intrinsic differences between species' physiology, which may affect both the response to neurotoxic chemotherapy (single-agent in rodents vs combination multidrug protocols in humans) and to therapeutic interventions, as well as differences in immune and psychological response to cancer and cancer therapy.1 Finally, the human pain experience is a complex interplay of signal transduction, transmission, perception, and modulation; therefore, even if the calcium channel α2δ subunit is an appropriate therapeutic target, and MPT is an accurate assay of its spinal overexpression or hyperactivity, our approach might have been too simplistic to capture the psychosocial factors that contribute to the experience of human pain in the setting of CIPN.
Adverse effects were common for both pregabalin and placebo. However, most were low grade. Of the adverse events leading to withdrawals, 2 were possibly or likely unrelated to the study treatment. One participant withdrew from the pregabalin arm after a hypoglycemic episode, but this was in the setting of introduction of an antihyperglycemic medication outside the study. Another withdrew from the pregabalin arm because of a small bowel obstruction in the setting of a new oncologic diagnosis. Two of the remaining 23 participants found their symptoms while taking pregabalin bothersome enough to withdraw from the study, which exemplifies the need to identify which patients are most likely to respond to which analgesics because trialing multiple medications is not necessarily a benign process.
This study has notable limitations. Most importantly, the study was terminated early for slow recruitment. Overall, 23 participants were included in the analysis of primary and secondary outcome measures, when our initial goal was to enroll 27 participants. This may have limited our power to detect significant changes, but the primary outcome was overwhelmingly negative (P = 0.97), and it is unlikely that it would have been affected by recruiting 4 additional participants. Our primary outcome assessed for a linear correlation between MPT and analgesic response. However, as neither the primary outcome nor the secondary analysis, using dichotomous operationalization of allodynia to static mechanical stimulation, significantly predicted response to pregabalin, it is unlikely that a strong relationship exists between increased mechanosensitivity to pinprick in CIPN and preferential analgesic response to pregabalin. On the other hand, the reduced number of participants may have influenced the effect sizes of some of the secondary outcomes, particularly considering that most participants completed the posttreatment NPSI, BPI, DAPOS, and SPI II questionnaires while tapering off the study drug.
Our study was not powered to allow for controlling for multiple variables in the efficacy analysis. Furthermore, 6 participants had painful CIPN lasting less than 6 months. Although reduction of pain intensity with pregabalin and placebo was modest at best, it is possible that some of these participants experienced spontaneous improvement in their pain during the study, as painful CIPN improves in some patients in the first 12 months after onset. Finally, we cannot extrapolate these results to patients with CIPN from chemotherapy agents beyond those included in this study, such as vinca alkaloids, bortezomib, and others.16
Skin biopsy results showed no correlation between IENFD and changes in average or worst pain with pregabalin. Notably, no participants who consented to biopsy had spontaneous pain extending proximally to the biopsy site (10 cm proximal to the lateral malleolus). This site was chosen based on the guidelines of the European Federation of Neurological Societies (EFNS).26 Recent evidence suggests that the correlation between biopsy and functional measures in length-dependent neuropathies may vary depending on the site24; therefore, results may have differed in patients with spontaneous pain extending proximally to the ankle.
The study has notable strengths. It is one of the first studies aimed primarily at assessing phenotypic predictors of response to treatment in CIPN. We applied rigorous inclusion criteria and performed detailed somatosensory phenotyping both with comprehensive and bedside QST approaches. We used electronic diaries for documentation of pain and adverse effects as well as for adherence improvement and monitoring. Given data on the superiority of electronic data capture methods (including improved protocol compliance, lower rates of data falsification, and fewer data entry errors8), this method is likely to result in more reliable patient-reported outcomes. Finally, our pregabalin plasma concentration assays suggest that participants reliably adhered to their allocated treatment arms. No participants in the placebo arm demonstrated detectable pregabalin levels, and 91% of participants in the pregabalin arm demonstrated detectable pregabalin levels. Two subjects in the pregabalin arm had concentrations below LOQ. It is possible that these subjects did not adhere to study protocol; however, both took the lowest presampling pregabalin dose at the end of drug taper off, and the period between last pregabalin dose and sample collection for both was more than 2 SDs longer than the average period for the remaining subjects.
In conclusion, MPT does not meaningfully predict response to pregabalin for painful CIPN. Over a 4-week treatment period, average or worst spontaneous pain reduction in CIPN was not significantly larger with pregabalin compared to placebo. Several neuropathic pain–specific measures on NPSI questionnaire demonstrated better improvement with pregabalin than with placebo. Dizziness and somnolence were significantly more frequent in the pregabalin group. Additional research is needed to identify critically important phenotypic and outcome measure parallels between animal pain models and clinical conditions to improve the translatability of pain research for achieving better, personalized treatment of neuropathic pain.
Conflict of interest statement
The authors have no conflict of interest to declare.
Supplementary Material
{kind=link}
Acknowledgements
The research was supported by ASPIRE neuropathic pain program grant from Pfizer, Inc., the manufacturer of Lyrica. Pregabalin and identical placebo pills were provided by Pfizer Inc. No direct payments from Pfizer were made to any investigators. No representative from Pfizer Inc has contributed to data collection, interpretation, or analysis. Research reported in this publication was supported by the Washington University Institute of Clinical and Translational Sciences grant UL1TR002345 from the National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health (NIH). The content is solely the responsibility of the authors and does not necessarily represent the official view of the NIH.
N. Wagner-Johnston has served on an advisory board for Janssen Pharmaceuticals. P. Karlsson is part of the International Diabetic Neuropathy Consortium (IDNC) research program, which is supported by a Novo Nordisk Foundation Challenge Programme grant (Grant number NNF14OC0011633). R. Shah's spouse reports receiving research funding from Celgene, Bristol-Myers Squibb, Genentech-Roche, AstraZeneca, and Verastem, and serving as a consultant for Kyowa-Hakka-Kirin. Support for L. Kagan, whose laboratory performed all pregabalin plasma concentration assays, came in part from National Institutes of Health via National Institute of General Medical Sciences grant 1R01GM124046-01A1. K. Kosicka is supported in part by the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 778051 and the Ministry of Science and Higher Education of Poland fund for supporting internationally cofinanced projects in 2018 to 2022 (agreement No 3899/H2020/2018/2). This article reflects the authors' view only. Neither the Research Executive Agency nor the Polish Ministry of Science and Higher Education may be held responsible for the use which may be made of the information contained therein.
Appendix A. Supplemental digital content
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/A782.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
References
- [1].Akhtar A. The flaws and human harms of animal experimentation. Camb Q Healthc Ethics 2015;24:407–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Aoki M, Kurauchi Y, Mori A, Nakahara T, Sakamoto K, Ishii K. Comparison of the effects of single doses of elcatonin and pregabalin on oxaliplatin-induced cold and mechanical allodynia in rats. Biol Pharm Bull 2014;37:322–6. [DOI] [PubMed] [Google Scholar]
- [3].Attal N, de Andrade DC, Adam F, Ranoux D, Teixeira MJ, Galhardoni R, Raicher I, Üçeyler N, Sommer C, Bouhassira D. Safety and efficacy of repeated injections of botulinum toxin A in peripheral neuropathic pain (BOTNEP): a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2016;15:555–65. [DOI] [PubMed] [Google Scholar]
- [4].Bao T, Basal C, Seluzicki C, Li SQ, Seidman AD, Mao JJ. Long-term chemotherapy-induced peripheral neuropathy among breast cancer survivors: prevalence, risk factors, and fall risk. Breast Cancer Res Treat 2016;159:327–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Barton DL, Wos EJ, Qin R, Mattar BI, Green NB, Lanier KS, Bearden JD, Kugler JW, Hoff KL, Reddy PS, Rowland KM, Riepl M, Christensen B, Loprinzi CL. A double-blind, placebo-controlled trial of a topical treatment for chemotherapy-induced peripheral neuropathy: NCCTG trial N06CA. Support Care Cancer 2011;19:833–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bouhassira D, Attal N, Alchaar H, Boureau F, Brochet B, Bruxelle J, Cunin G, Fermanian J, Ginies P, Grun-Overdyking A, Jafari-Schluep H, Lantéri-Minet M, Laurent B, Mick G, Serrie A, Valade D, Vicaut E. Comparison of pain syndromes associated with nervous or somatic lesions and development of a new neuropathic pain diagnostic questionnaire (DN4). PAIN 2005;114:29–36. [DOI] [PubMed] [Google Scholar]
- [7].Campbell CM, Kipnes MS, Stouch BC, Brady KL, Kelly M, Schmidt WK, Petersen KL, Rowbotham MC, Campbell JN. Randomized control trial of topical clonidine for treatment of painful diabetic neuropathy. PAIN 2012;153:1815–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dale O, Hagen KB. Despite technical problems personal digital assistants outperform pen and paper when collecting patient diary data. J Clin Epidemiol 2007;60:8–17. [DOI] [PubMed] [Google Scholar]
- [9].Demant DT, Lund K, Vollert J, Maier C, Segerdahl M, Finnerup NB, Jensen TS, Sindrup SH. The effect of oxcarbazepine in peripheral neuropathic pain depends on pain phenotype: a randomised, double-blind, placebo-controlled phenotype-stratified study. PAIN 2014;155:2263–73. [DOI] [PubMed] [Google Scholar]
- [10].Di Cesare Mannelli L, Pacini A, Bonaccini L, Zanardelli M, Mello T, Ghelardini C. Morphologic features and glial activation in rat oxaliplatin-dependent neuropathic pain. J Pain 2013;14:1585–600. [DOI] [PubMed] [Google Scholar]
- [11].Durand JP, Deplanque G, Montheil V, Gornet JM, Scotte F, Mir O, Cessot A, Coriat R, Raymond E, Mitry E, Herait P, Yataghene Y, Goldwasser F. Efficacy of venlafaxine for the prevention and relief of oxaliplatin-induced acute neurotoxicity: results of EFFOX, a randomized, double-blind, placebo-controlled phase III trial. Ann Oncol 2012;23:200–5. [DOI] [PubMed] [Google Scholar]
- [12].Eckhoff L, Knoop AS, Jensen MB, Ejlertsen B, Ewertz M. Risk of docetaxel-induced peripheral neuropathy among 1,725 Danish patients with early stage breast cancer. Breast Cancer Res Treat 2013;142:109–18. [DOI] [PubMed] [Google Scholar]
- [13].Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, Gilron I, Haanpää M, Hansson P, Jensen TS, Kamerman PR, Lund K, Moore A, Raja SN, Rice AS, Rowbotham M, Sena E, Siddall P, Smith BH, Wallace M. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol 2015;14:162–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Finnerup NB, Haroutounian S, Baron R, Dworkin RH, Gilron I, Haanpaa M, Jensen TS, Kamerman PR, McNicol E, Moore A, Raja SN, Andersen NT, Sena ES, Smith BH, Rice ASC, Attal N. Neuropathic pain clinical trials: factors associated with decreases in estimated drug efficacy. PAIN 2018;159:2339–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Flatters SJ, Xiao WH, Bennett GJ. Acetyl-L-carnitine prevents and reduces paclitaxel-induced painful peripheral neuropathy. Neurosci Lett 2006;397:219–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gauchan P, Andoh T, Ikeda K, Fujita M, Sasaki A, Kato A, Kuraishi Y. Mechanical allodynia induced by paclitaxel, oxaliplatin and vincristine: different effectiveness of gabapentin and different expression of voltage-dependent calcium channel alpha(2)delta-1 subunit. Biol Pharm Bull 2009;32:732–4. [DOI] [PubMed] [Google Scholar]
- [17].George SZ, Wittmer VT, Fillingim RB, Robinson ME. Sex and pain-related psychological variables are associated with thermal pain sensitivity for patients with chronic low back pain. J Pain 2007;8:2–10. [DOI] [PubMed] [Google Scholar]
- [18].Hahm TS, Ahn HJ, Ryu S, Gwak MS, Choi SJ, Kim JK, Yu JM. Combined carbamazepine and pregabalin therapy in a rat model of neuropathic pain. Br J Anaesth 2012;109:968–74. [DOI] [PubMed] [Google Scholar]
- [19].Han FY, Kuo A, Nicholson JR, Corradinni L, Smith MT. Comparative analgesic efficacy of pregabalin administered according to either a prevention protocol or an intervention protocol in rats with cisplatin-induced peripheral neuropathy. Clin Exp Pharmacol Physiol 2018;45:1067–75. [DOI] [PubMed] [Google Scholar]
- [20].Haroutounian S. Somatosensory phenotyping for better translation in neuropathic pain? PAIN 2016;157:995–6. [DOI] [PubMed] [Google Scholar]
- [21].Haroutounian S, Nikolajsen L, Bendtsen TF, Finnerup NB, Kristensen AD, Hasselstrøm JB, Jensen TS. Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. PAIN 2014;155:1272–9. [DOI] [PubMed] [Google Scholar]
- [22].Hershman DL, Weimer LH, Wang A, Kranwinkel G, Brafman L, Fuentes D, Awad D, Crew KD. Association between patient reported outcomes and quantitative sensory tests for measuring long-term neurotoxicity in breast cancer survivors treated with adjuvant paclitaxel chemotherapy. Breast Cancer Res Treat 2011;125:767–74. [DOI] [PubMed] [Google Scholar]
- [23].Karlsson P, Haroutounian S, Polydefkis M, Nyengaard JR, Jensen TS. Structural and functional characterization of nerve fibres in polyneuropathy and healthy subjects. Scand J Pain 2016;10:28–35. [DOI] [PubMed] [Google Scholar]
- [24].Karlsson P, Hincker AM, Jensen TS, Freeman R, Haroutounian S. Structural, functional and symptom relations in painful distal symmetric polyneuropathies: a systematic review. PAIN 2018;160:286–97. [DOI] [PubMed] [Google Scholar]
- [25].Kautio AL, Haanpää M, Saarto T, Kalso E. Amitriptyline in the treatment of chemotherapy-induced neuropathic symptoms. J Pain Symptom Manage 2008;35:31–9. [DOI] [PubMed] [Google Scholar]
- [26].Lauria G, Hsieh ST, Johansson O, Kennedy WR, Leger JM, Mellgren SI, Nolano M, Merkies IS, Polydefkis M, Smith AG, Sommer C, Valls-Solé J; Societies EFoN, Society PN. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on the use of skin biopsy in the diagnosis of small fiber neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society. Eur J Neurol 2010;17:903–12, e944–909. [DOI] [PubMed] [Google Scholar]
- [27].Ling B, Authier N, Balayssac D, Eschalier A, Coudore F. Behavioral and pharmacological description of oxaliplatin-induced painful neuropathy in rat. PAIN 2007;128:225–34. [DOI] [PubMed] [Google Scholar]
- [28].Lynch JJ, Wade CL, Zhong CM, Mikusa JP, Honore P. Attenuation of mechanical allodynia by clinically utilized drugs in a rat chemotherapy-induced neuropathic pain model. PAIN 2004;110:56–63. [DOI] [PubMed] [Google Scholar]
- [29].Ma J, Kavelaars A, Dougherty PM, Heijnen CJ. Beyond symptomatic relief for chemotherapy-induced peripheral neuropathy: targeting the source. Cancer 2018;124:2289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Martinc B, Roškar R, Grabnar I, Vovk T. Simultaneous determination of gabapentin, pregabalin, vigabatrin, and topiramate in plasma by HPLC with fluorescence detection. J Chromatogr B Analyt Technol Biomed Life Sci 2014;962:82–8. [DOI] [PubMed] [Google Scholar]
- [31].Miyazaki R, Yamamoto T. The efficacy of morphine, pregabalin, gabapentin, and duloxetine on mechanical allodynia is different from that on neuroma pain in the rat neuropathic pain model. Anesth Analg 2012;115:182–8. [DOI] [PubMed] [Google Scholar]
- [32].Nihei S, Sato J, Kashiwaba M, Itabashi T, Kudo K, Takahashi K. Efficacy and safety of pregabalin for oxaliplatin- and paclitaxel-induced peripheral neuropathy. Gan To Kagaku Ryoho 2013;40:1189–93. [PubMed] [Google Scholar]
- [33].Ottaiano A, Nappi A, Tafuto S, Nasti G, De Divitiis C, Romano C, Cassata A, Casaretti R, Silvestro L, Avallone A, Capuozzo M, Capozzi M, Maiolino P, Quagliariello V, Scala S, Iaffaioli VR. Diabetes and body mass index are associated with neuropathy and prognosis in colon cancer patients treated with capecitabine and oxaliplatin adjuvant chemotherapy. Oncology 2016;90:36–42. [DOI] [PubMed] [Google Scholar]
- [34].Peng P, Xi Q, Xia S, Zhuang L, Gui Q, Chen Y, Huang Y, Zou M, Rao J, Yu S. Pregabalin attenuates docetaxel-induced neuropathy in rats. J Huazhong Univ Sci Technolog Med Sci 2012;32:586–90. [DOI] [PubMed] [Google Scholar]
- [35].Polomano RC, Bennett GJ. Chemotherapy-evoked painful peripheral neuropathy. Pain Med 2001;2:8–14. [DOI] [PubMed] [Google Scholar]
- [36].Rao RD, Michalak JC, Sloan JA, Loprinzi CL, Soori GS, Nikcevich DA, Warner DO, Novotny P, Kutteh LA, Wong GY; Group NCCT. Efficacy of gabapentin in the management of chemotherapy-induced peripheral neuropathy: a phase 3 randomized, double-blind, placebo-controlled, crossover trial (N00C3). Cancer 2007;110:2110–18. [DOI] [PubMed] [Google Scholar]
- [37].Simpson DM, Schifitto G, Clifford DB, Murphy TK, Durso-De Cruz E, Glue P, Whalen E, Emir B, Scott GN, Freeman R; Group HNS. Pregabalin for painful HIV neuropathy: a randomized, double-blind, placebo-controlled trial. Neurology 2010;74:413–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Smith EM, Pang H, Cirrincione C, Fleishman S, Paskett ED, Ahles T, Bressler LR, Fadul CE, Knox C, Le-Lindqwister N, Gilman PB, Shapiro CL; Oncology AfCTi. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: a randomized clinical trial. JAMA 2013;309:1359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Speck RM, Sammel MD, Farrar JT, Hennessy S, Mao JJ, Stineman MG, DeMichele A. Impact of chemotherapy-induced peripheral neuropathy on treatment delivery in nonmetastatic breast cancer. J Oncol Pract 2013;9:e234–240. [DOI] [PubMed] [Google Scholar]
- [40].Ventzel L, Jensen AB, Jensen AR, Jensen TS, Finnerup NB. Chemotherapy-induced pain and neuropathy: a prospective study in patients treated with adjuvant oxaliplatin or docetaxel. PAIN 2016;157:560–8. [DOI] [PubMed] [Google Scholar]
- [41].Xiao W, Boroujerdi A, Bennett GJ, Luo ZD. Chemotherapy-evoked painful peripheral neuropathy: analgesic effects of gabapentin and effects on expression of the alpha-2-delta type-1 calcium channel subunit. Neuroscience 2007;144:714–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yarnitsky D, Granot M, Nahman-Averbuch H, Khamaisi M, Granovsky Y. Conditioned pain modulation predicts duloxetine efficacy in painful diabetic neuropathy. PAIN 2012;153:1193–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.