

Abstract
Human neutrophil elastase (HNE) is a potent protease that plays an important physiological role in many processes and is considered to be a multifunctional enzyme. HNE is also involved in a variety of pathologies affecting the respiratory system. Thus, compounds able to inhibit HNE proteolytic activity could represent effective therapeutics. We present here a new series of pyrrolo[2,3-b]pyridine derivatives of our previously reported potent HNE inhibitors. Our results show that position 2 of the pyrrolo[2,3-b]pyridine scaffold must be unsubstituted, and modifications of this position resulted in loss of HNE inhibitory activity. Conversely, the introduction of certain substituents at position 5 was tolerated, with retention of HNE inhibitory activity (IC50 = 15–51 nM) after most substitutions, indicating that bulky and/or lipophilic substituents at position 5 probably interact with the large pocket of the enzyme site and allow Michaelis complex formation. The possibility of Michaelis complex formation between Ser195 and the ligand carbonyl group was assessed by molecular docking, and it was found that highly active HNE inhibitors are characterized by geometries favorable for Michaelis complex formation and by relatively short lengths of the proton transfer channel via the catalytic triad.
Keywords: Pyrrolo[2,3-b]pyridine; human neutrophil elastase inhibitors; molecular docking
INTRODUCTION
Polymorphonuclear neutrophils represent a large percentage of the circulating leukocyte population and are the most abundant type of white blood cells in human blood. They are also known as granulocytes due to the numerous granules present in their cytoplasm and are characterized by their nuclear morphology, which is segmented into three to five lobes joined together by a thin membrane. These cells play a fundamental role in immune defense against pathogenic organisms and are the first mediators in the inflammatory response [Korkmaz et al., 2010]. Neutrophil granules are classified into primary or azurophil, secondary or specific, tertiary or gelatinase, and secretory [Pham, 2006]. Specifically, the azurophil granules contain myeloperoxidase, some bactericidal proteins, and three serine proteases: proteinase 3, cathepsin G, and human neutrophil elastase (HNE), which is a globular glycoprotein of about 30 kDa belonging to the chymotrypsin family. HNE consists of a single polypeptide chain of 218 amino acids, with two asparagine-linked carbohydrate side chains at Asn95 and Asn144 [Lucas et al., 2011]. HNE is stabilized by four disulfide bridges and has 19 arginine residues, resulting in an isoelectric point of 10–11. As with other neutrophil serine proteases, HNE plays an important physiological role and is considered a multifunctional enzyme that contributes to killing of pathogens, inflammatory processes, and maintenance of tissue homeostasis [Pham, 2006]. HNE proteolytic activity depends on the catalytic triad consisting of Ser195-His57-Asp102 [Fujinaga et al., 1996].
The proteolytic activity of serine proteases, which is essential for the maintenance of important host functions, can also be harmful if not properly regulated, and a number of regulatory mechanisms are present [Von Nussbaum et al., 2016]. For example, these proteases are stored as inactive precursors in specialized compartments, such as azurophil granules for serine proteases. Once active, serine protease activity can be regulated by endogenous inhibitors, such as α1-antitripsin [also known as α1-proteinase inhibitor (α1-PI)], elafin, and secretory leukocyte protease inhibitor, which inhibit protease activity [Zhong et al., 2017; Von Nussbaum et al., 2015]. The involvement of HNE and other neutrophil serine proteases in the development of chronic pulmonary inflammatory diseases, such as chronic obstructive pulmonary disease (COPD), acute lung injury, acute respiratory distress syndrome (ARDS), and cystic fibrosis (CF) is widely documented and correlated with an imbalance between HNE and endogenous inhibitor activity [Polverino et al., 2017].
Recently, we identified and characterized a number of potent HNE inhibitors with differing structural scaffolds [Schepetkin et al., 2007; Crocetti et al., 2011; Crocetti et al., 2013; Giovannoni et al., 2016; Crocetti et al., 2016; Vergelli et al., 2017; Giovannoni et al., 2018; Crocetti et al., 2018]. In the most recent studies, we identified HNE inhibitors with a pyrrolo[2,3-b]pyridine scaffold [Crocetti et al., 2018] and found that the most potent compounds exhibited IC50 values in the low nanomolar range (14–87 nM) (see Figure 1). Based on these studies, we performed further modifications of the pyrrolo[2,3-b]pyridine scaffold. While keeping the best substituents identified for N-1 (m-toluoyl) and position 3 (CN group), we introduced substituents with different features at position 5 to screen for more potent HNE inhibitors. Additionally, we investigated substitutions at position 2 of the scaffold by shifting CN from the 3 to the 2 position or inserting other groups.
Figure 1.
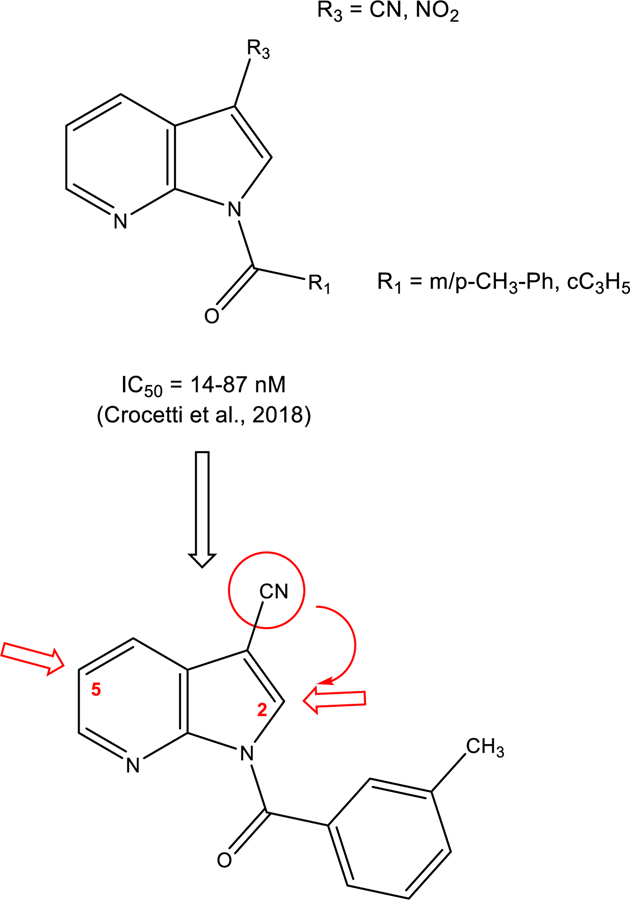
Further modifications of the pyrrolo[2,3-b]pyridine scaffold.
MATERIALS AND METHODS
Chemistry
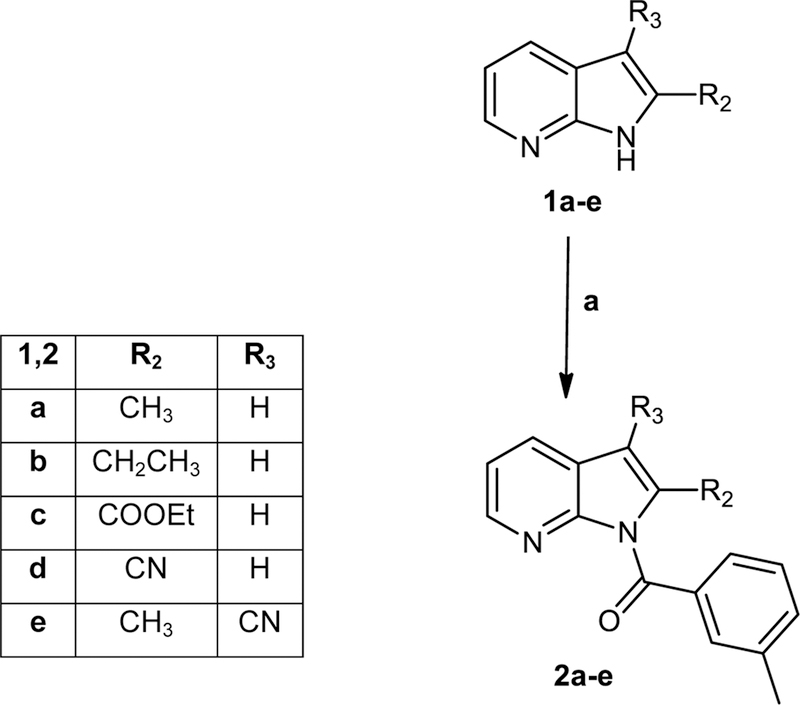
All final compounds were synthesized as reported in Figures 2-4, and the structures were confirmed on the basis of analytical and spectral data. To obtain the 2- or 2,3-disubstituted pyrrolo[2,3-b]pyridines (2a-e), we followed the procedures shown in Figure 2. Starting from the previously synthesized compounds 1a-e [Sandham et al., 2009; Pires et al., 2016; Baltus et al., 2016; Bahekar et al., 2007], we performed benzoylation with m-toluoyl choride and triethylamine in anhydrous dichloromethane, resulting in final compounds 2a-e. The synthesis of compounds with different substitutions at position 5 is shown in Figures 3 and 4. Figure 3 shows the synthesis of pyrrolo[2,3-b]pyridines substituted with a bromine, chlorine, or nitro group at position 5. The formyl group of intermediates 4a-c [Xi and Li, 2014; Nirogi et al., 2012; Ermoli et al., 2009], obtained starting from the corresponding 5-substituted-pyrrolo[2,3-b]pyridines 3a-c [Joydev et al., 2017; Chen et al., 2015; Bhat et al., 2015] by the Duff reaction, was transformed into the corresponding oxime by treatment with hydroxylamine hydrochloride and NaHCO3 at high temperature, resulting in compounds 5a-c [5b, Bahekar et al., 2007]. Further reaction with POCl3 resulted in the 3-carbonitrile derivatives 6a-c [6a, Graczyk et al., 2004], which were benzoylated at position 1 with m-toluoyl chloride and sodium hydride in anhydrous tetrahydrofuran to obtain the 5-substituted pyrrolo[2,3-b]pyridines 7a-c (Figure 3).
Figure 2. Reagents and conditions:
a) m-Toluoyl chloride, Et3N, dry DCM, 0°C, 2h; r.t., 2h.
Figure 4. Reagents and conditions:

a) Ph-SO2Cl, Et3N, dry DCM, 0°C, 2h then r.t., 2h; b) R5-B(OH)2, Pd(PPh3)4, Na2CO3 2M, anhydrous toluene, reflux, 4–5h; c) TBAF, dry THF, reflux, 2–6h; d) HMTA, CH3COOH, reflux, 6h; e) NH2OH∙HCl, H2O, 60°C, 30 min; NaHCO3, 100°C, 4h; f) POCl3, reflux, 2h; g) m-Toluoyl chloride, NaH, dry THF, r.t., 24h.
Figure 3. Reagents and conditions:

a) HTMA, CH3COOH, reflux, 4h; b) NH2OH·HCl, H2O, 60°C, 30 min; NaHCO3, reflux, 4h; c) POCl3, reflux, 1h; d) m-Toluoyl chloride, NaH, dry THF, r.t., 24h.
Figure 4 shows the synthetic steps for insertion of (hetero)aromatic rings at position 5 to obtain final compounds 14a-g. In the first step, the nitrogen at position 1 of intermediate 3a [Joydev et al., 2017] was protected with benzensulfonyl chloride to obtain compound 8 [Liu et al., 2016], which subsequently was treated with tetrakis(triphenylphosphine)palladium(0), 2M sodium carbonate solution, and the appropriate boronic acid in hot anhydrous toluene to obtain the corresponding 5-pyrrolo[2,3-b]pyridine derivatives 9a-g. The protecting group at position N-1 was then removed with tetrabutylammonium fluoride (TBAF) in hot anhydrous tetrahydrofuran, resulting in pyrrolo[2,3-b]pyridines 10a-g [10a, Laha et al., 2017; 10c and 10d, Ibrahim et al., 2007; 10f and 10g, Singh et al., 2017]. Reaction of these compounds with hexamethylenetetramine (HMTA) in acetic acid at reflux resulted in the 3-formyl derivatives 11a-g [11g, Ibrahim et al., 2007], which, when treated with hydroxylamine hydrochloride (12a-g), dehydrated with POCl3 (13a-g), and benzoylated at position 1 with m-toluoyl chloride, led to final compounds 14a-g.
Experimental
All melting points were determined on a Büchi apparatus (New Castle, DE) and are uncorrected. Extracts were dried over Na2SO4, and the solvents were removed under reduced pressure. Merck F-254 commercial plates (Merck, Durham, NC) were used for analytical TLC to follow the course of reactions. Silica gel 60 (Merck 70–230 mesh, Merck, Durham, NC) was used for column chromatography. 1H NMR and 13C NMR spectra were recorded on an Avance 400 instrument (Bruker Biospin Version 002 with SGU, Bruker Inc., Billerica, MA). Chemical shifts (δ) are reported in ppm to the nearest 0.01 ppm using the solvent as an internal standard. Coupling constants (J values) are given in Hz and were calculated using TopSpin 1.3 software (Nicolet Instrument Corp., Madison, WI) and are rounded to the nearest 0.1 vHz. Mass spectra (m/z) were recorded on an ESI-TOF mass spectrometer (Brucker Micro TOF, Bruker Inc., Billerica, MA), and reported mass values are within the error limits of ±5 ppm mass units. Microanalyses indicated by the symbols of the elements were performed with a Perkin–Elmer 260 elemental analyzer (PerkinElmer, Inc., Waltham, MA) for C, H, and N, and the results were within ±0.4% of the theoretical values, unless otherwise stated. Reagents and starting materials were commercially available.
General procedure for compounds 2a-e.
To a cooled (0°C) suspension of the appropriate substrate 1a-e [Sandham et al., 2009; Pires et al., 2016; Baltus et al., 2016; Bahekar et al., 2007] (0.56 mmol) in anhydrous CH2Cl2 (2 mL), 0.72 mmol of Et3N, and 1.67 mmol of m-toluoyl chloride were added. The mixture was stirred at 0°C for 2 h and then at room temperature for an additional 2 h. The solvent was evaporated, cold water was added, and the mixture was neutralized with 0.5 N NaOH. The reaction mixture was extracted with CH2Cl2 (3 × 15 mL), and the solvent was dried over sodium sulfate and evaporated in vacuum. The final compounds 2a-e were purified by column chromatography using toluene/ethyl acetate 9.5:0.5 (for 2a,b) or cyclohexane/ethyl acetate 2:1 (for 2c,d) or 5:1 (for 2e) as eluents.
(2-Methyl-1H-pyrrolo[2,3-b]pyridin-1-yl)(m-tolyl)methanone (2a).
Yield = 67%; oil. 1H-NMR (CDCl3-d1) δ 2.39 (s, 3H, m-CH3-Ph), 2.56 (s, 3H, CH3), 7.02–7.07 (m, 1H, Ar), 6.38 (s, 1H, Ar), 7.30 (t, 1H, Ar, J = 8.0 Hz), 7.41 (d, 1H, Ar, J = 8.0 Hz), 7.50 (d, 1H, Ar, J = 8.0 Hz), 7.66 (s, 1H, Ar), 7.76 (d, 1H, Ar, J = 8.0 Hz), 8.05 (d, 1H, Ar, J = 4.4 Hz). 13C-NMR (CDCl3-d1) δ 15.15 (CH3), 21.33 (CH3), 104.52 (CH), 117.37 (CH), 121.44 (C), 127.52 (CH), 128.04 (CH), 128.16 (CH), 131.13 (CH), 134.19 (CH), 134.77 (C), 138.16 (C), 138.90 (C), 142.62 (CH), 149.53 (C), 169.70 (CO). ESI-MS calcd. for C16H14N2O, 250.30; found: m/z 251.11 [M+H]+. Anal. C16H14N2O (C, H, N).
(2-Ethyl-1H-pyrrolo[2,3-b]pyridin-1-yl)(m-tolyl)methanone (2b).
Yield = 17%; oil. 1H-NMR (CDCl3-d1) δ 1.30 (t, 3H, CH2CH3, J = 7.2 Hz), 2.39 (s, 3H, m-CH3-Ph), 2.98 (q, 2H, CH2CH3, J = 7.2 Hz), 6.43 (s, 1H, Ar), 7.03–7.08 (m, 1H, Ar), 7.30 (t, 1H, Ar, J = 7.6 Hz), 7.41 (d, 1H, Ar, J = 7.6 Hz), 7.49 (d, 1H, Ar, J = 7.6 Hz), 7.66 (s, 1H, Ar), 7.78 (d, 1H, Ar, J = 7.6 Hz), 8.05 (d, 1H, Ar, J = 4.4 Hz). 13C-NMR (CDCl3-d1) δ 12.82 (CH3), 21.44 (CH3), 21.81 (CH2), 102.58 (CH), 118.02 (CH), 121.33 (C), 128.08 (CH), 129.30 (CH), 131.13 (CH), 134.29 (CH), 134.78 (C), 138.20 (C), 142.76 (CH), 145.09 (C), 149.74 (C), 169.93 (CO). ESI-MS calcd. for C17H16N2O, 264.32; found: m/z 265.13 [M+H]+. Anal. C17H16N2O (C, H, N).
Ethyl 1-(3-methylbenzoyl)-1H-pyrrolo[2,3-b]pyridine-2-carboxylate (2c).
Yield = 34%; oil. 1H-NMR (CDCl3-d1) δ 1.45 (t, 3H, OCH2CH3, J = 7.2 Hz), 2.38 (s, 3H, m-CH3-Ph), 4.21 (q, 2H, OCH2CH3, J = 7.2 Hz), 7.16–7.21 (m, 1H, Ar), 7.29–7.34 (m, 2H, Ar), 7.42 (d, 1H, Ar, J = 7.6 Hz), 7.56 (d, 1H, Ar, J = 7.6 Hz), 7.71 (s, 1H, Ar), 8.04 (d, 1H, Ar, J = 8.0 Hz), 8.41 (d, 1H, Ar, J = 4.4 Hz). 13C-NMR (CDCl3-d1) δ 13.94 (CH3), 21.29 (CH3), 61.59 (CH2), 111.04 (CH), 118.64 (CH), 119.46 (C), 127.93 (CH), 128.56 (CH), 130.65 (C), 131.02 (CH), 131.18 (CH), 134.03 (C), 135.01 (CH), 138.61 (C), 147.70 (CH), 150.05 (C), 160.65 (C), 169.00 (CO). ESI-MS calcd. for C18H16N2O3, 308.34; found: m/z 309.12 [M+H]+. Anal. C18H16N2O3 (C, H, N).
1-(3-Methylbenzoyl)-1H-pyrrolo[2,3-b]pyridine-2-carbonitrile (2d).
Yield = 41%; oil. 1H-NMR (CDCl3-d1) δ 2.41 (s, 3H, m-CH3-Ph), 7.25–7.33 (m, 1H, Ar), 7.34–7.40 (m, 2H, Ar), 7.48 (d, 1H, Ar, J = 7.6 Hz), 7.58 (d, 1H, Ar, J = 7.6 Hz) 7.69 (s, 1H, Ar), 8.03 (dd, 1H, Ar, J = 8.0 Hz and J = 1.2 Hz), 8.38 (d, 1H, Ar, J = 3.6 Hz). 13C-NMR (CDCl3-d1) δ 21.53 (CH3), 110.31 (C), 112.22 (C), 118.23 (CH), 119.60 (C), 120.03 (CH), 128.32 (CH), 128.59 (CH), 131.13 (CH), 131.61 (CH), 132.56 (C), 135.23 (CH), 138.52 (C), 148.37 (CH), 166.85 (CO). ESI-MS calcd. for C16H11N3O, 261.28; found: m/z 262.09 [M+H]+. Anal. C16H11N3O (C, H, N).
2-Methyl-1-(3-methylbenzoyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (2e).
Yield = 10%; oil. 1H-NMR (CDCl3-d1) δ 2.40 (s, 3H, m-CH3-Ph), 2.74 (s, 3H, CH3), 7.21–7.26 (m, 1H, Ar), 7.35 (d, 1H, Ar), 7.48 (d, 2H, Ar, J = 7.6 Hz), 7.64 (s, 1H, Ar), 7.98 (d, 1H, Ar, J = 7.2 Hz), 8.19 (d, 1H, Ar, J = 4.0 Hz). 13C-NMR (CDCl3-d1) δ 9.43 (CH3), 20.91 (CH3), 101.37 (C), 115.65 (CH), 115.92 (C), 121.15 (CH), 128.10 (CH), 128.36 (CH), 129.14 (CH), 130.17 (CH), 130.49 (CH), 134.86 (CH), 137.33 (C), 138.91 (C), 142.4 (CH), 146.36 (C), 167.70 (CO). ESI-MS calcd. for C17H13N3O, 275.30; found: m/z 276.11 [M+H]+. Anal. C17H13N3O (C, H, N).
General procedure for compounds 5a and 5c.
A mixture of the appropriate 1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde 4a [Xi and Li, 2014] or 4c [Ermoli et al., 2009] (0.78 mmol) and hydroxylamine hydrochloride (2.35 mmol) in 2 mL of water was heated at 60°C for 30 min. NaHCO3 (2.35 mmol) was then added, and the reaction mixture was heated under reflux for 4 h at 100°C. After cooling at room temperature, the solid was filtered, washed with the excess of ice-cold water and dried. The crude product was recrystallized from hexane to obtain the pure product.
(E)-5-Bromo-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde oxime (5a).
Yield = 94%; mp = 235–237°C (hexane). 1H-NMR (DMSO-d6) δ 7.81 (s, 1H, CH=N-OH), 8.21 (s, 1H, Ar), 8.33 (s, 1H, Ar), 8.38 (s, 1H, Ar), 10.75 (exch br s, 1H, CH=N-OH), 12.16 (exch br s, 1H, NH). ESI-MS calcd. for C8H6BrN3O, 240.06; found: m/z 240.97 [M+H]+. Anal. C8H6BrN3O (C, H, N).
(E)-5-Nitro-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde oxime (5c).
Yield = 40%; mp = 193–195°C (hexane). 1H-NMR (DMSO-d6) δ 8.00 (s, 1H, Ar), 8.27 (s, 1H, CH=N-OH), 9.04 (s, 1H, Ar), 9.13 (s, 1H, Ar), 11.02 (exch br s, 1H, CH=N-OH), 12.70 (exch br s, 1H, NH). ESI-MS calcd. for C8H6N4O3, 206.16; found: m/z 207.05 [M+H]+. Anal. C8H6N4O3 (C, H, N).
General procedure for compounds 6b and 6c.
A suspension of intermediate 5b [Bahekar et al., 2007] or 5c (0.92 mmol) in 4 mL of POCl3 was stirred at reflux for 2 h. After cooling, ice-cold water (20 mL) was slowly added, and the precipitate was filtered under vacuum and washed with abundant cold water to obtain the desired compound, which was recrystallized from ethanol.
5-Chloro-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (6b).
Yield = 90%; mp = 291–294°C (EtOH). 1H-NMR (DMSO-d6) δ 8.27 (d, 1H, Ar, J = 2.0 Hz), 8.40 (d, 1H, Ar, J = 2.4 Hz), 8.51 (d, 1H, Ar, J = 3.2 Hz), 13.05 (exch br s, 1H, NH). IR ѵ (cm−1): 2240 (CN). ESI-MS calcd. for C8H4ClN3, 177.59; found: m/z 179.01 [M+H]+. Anal. C8H4ClN3 (C, H, N).
5-Nitro-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (6c).
Yield = 81%; mp = 248–250°C (EtOH). 1H-NMR (DMSO-d6) δ 8.71 (s, 1H, Ar), 8.91 (s, 1H, Ar), 9.21 (s, 1H, Ar), 13.56 (exch br s, 1H, NH). IR ѵ (cm−1): 2220 (CN). ESI-MS calcd. for C8H4N4O2, 188.14; found: m/z 189.04 [M+H]+. Anal. C8H4N4O2 (C, H, N).
General procedure for compounds 7a-c.
To a suspension of the substrate 6a-c [6a, Graczyk et al., 2004] (0.53 mmol) in 10 mL of anhydrous THF, 1.06 mmol of sodium hydride and 0.64 mmol of m-toluoyl chloride were added. The mixture was stirred at room temperature overnight. The solvent was concentrated in vacuo to obtain a residue that was purified by column chromatography using cyclohexane/ethyl acetate 6:1 (for 7a,b) or 5:1 (for 7c) as eluents.
5-Bromo-1-(3-methylbenzoyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (7a).
Yield = 30%; mp = 156–159°C. 1H-NMR (CDCl3-d1) δ 2.44 (s, 3H, m-CH3-Ph), 7.41 (t, 1H, Ar, J = 7.6 Hz), 7.50–7.55 (m, 2H, Ar), 7.62 (s, 1H, Ar), 8.20–8.25 (m, 2H, Ar), 8.46 (s, 1H, Ar). 13C-NMR (CDCl3-d1) δ 21.52 (CH3), 90.31 (C), 127.78 (CH), 128.67 (CH), 131.11 (CH), 131.77 (C), 135.21 (CH), 136.23 (CH), 138.87 (C), 145.41 (C), 166.46 (CO). IR ѵ (cm−1): 2224 (CN). ESI-MS calcd. for C16H10BrN3O, 340.17; found: m/z 341.00 [M+H]+. Anal. C16H10BrN3O (C, H, N).
5-Chloro-1-(3-methylbenzoyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (7b).
Yield = 10%; mp = 150–152°C. 1H-NMR (CDCl3-d1) δ 2.44 (s, 3H, m-CH3-Ph), 7.41 (t, 1H, Ar, J = 7.2 Hz), 7.50–7.55 (m, 2H, Ar), 7.62 (s, 1H, Ar), 8.10 (s, 1H, Ar), 8.24 (s, 1H, Ar), 8.38 (s, 1H, Ar). 13C-NMR (CDCl3-d1) δ 21.32 (CH3), 112.77 (C), 127.79 (CH), 127.96 (CH), 128.48 (CH), 128.71 (C), 130.99 (CH), 131.01 (C), 135.06 (CH), 136.04 (CH), 138.82 (C), 145.11 (C), 145.63 (CH), 166.33 (CO). IR ѵ (cm−1): 2223 (CN). ESI-MS calcd. for C16H10ClN3O, 295.72; found: m/z 297.05 [M+H]+. Anal. C16H10ClN3O (C, H, N).
1-(3-Methylbenzoyl)-5-nitro-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (7c).
Yield = 35%; mp = 152–154°C. 1H-NMR (CDCl3-d1) δ 2.45 (s, 3H, m-CH3-Ph), 7.43 (t, 1H, Ar, J = 7.6 Hz), 7.55 (d, 2H, Ar, J = 7.6 Hz), 7.64 (s, 1H, Ar), 8.43 (s, 1H, Ar), 8.96 (d, 1H, Ar, J = 2.4 Hz), 9.27 (d, 1H, Ar, J = 2.4 Hz). 13C-NMR (CDCl3-d1) δ 20.93 (CH3), 101.34 (C), 115.51 (C), 122.13 (C), 123.68 (CH), 128.16 (CH), 129.19 (CH), 130.42 (C), 130.57 (CH), 131.62 (C), 134.11 (CH), 137.65 (CH), 138.94 (C), 152.78 (C), 167.74 (CO). IR ѵ (cm−1): 2220 (CN). ESI-MS calcd. for C16H10N4O3, 306.28; found: m/z 307.08 [M+H]+. Anal. C16H10N4O3 (C, H, N).
General procedure for compounds 9a-g.
To a suspension of intermediate 8 [Liu et al., 2016] (0.34 mmol) in 3 mL of toluene, 0.051 mmol of tetrakis(triphenylphosphine)palladium(0), 3 mL of Na2CO3 2M solution, and 0.68 mmol of the appropriate hetero(phenyl)-boronic acid were added. The mixture was stirred at reflux for 4 h. After cooling, ice-cold water (20 mL) was added, the suspension was extracted with CH2Cl2 (3 × 15 mL), dried over sodium sulfate, and the solvent was evaporated in vacuo to obtain compounds 9a-g, which were purified by column chromatography using hexane/ethyl acetate 3:1 (for 9b,e), hexane/acetone 4:1 (for 9a,c,d,f), or cyclohexane/ethyl acetate 1:5 (for 9g) as eluents.
5-Phenyl-1-(phenylsulfonyl)-1H-pyrrolo[2,3-b]pyridine (9a).
Yield = 79%; mp = 123–125°C. 1H-NMR (CDCl3-d1) δ 6.63 (d, 1H, Ar, J = 4.0 Hz), 7.36 (t, 1H, Ar, J = 7.2 Hz), 7.42–7.49 (m, 4H, Ar), 7.52–7.57 (m, 3H, Ar), 7.75 (d, 1H, Ar, J = 4.0 Hz), 7.98 (d, 1H, Ar, J = 2.0 Hz), 8.22 (d, 2H, Ar, J = 7.2 Hz), 8.65 (d, 1H, Ar, J = 2.0 Hz). ESI-MS calcd. for C19H14N2O2S, 334.39; found: m/z 335.08 [M+H]+. Anal. C19H14N2O2S (C, H, N).
1-(Phenylsulfonyl)-5-(3-(trifluoromethyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (9b).
Yield = 79%; mp = 145–148°C. 1H-NMR (CDCl3-d1) δ 6.66 (d, 1H, Ar, J = 4.0 Hz), 7.50 (t, 2H, Ar, J = 8.0 Hz), 7.56–7.61 (m, 2H, Ar), 7.63 (d, 1H, Ar, J = 7.6 Hz), 7.72 (d, 1H, Ar, J = 7.6 Hz), 7.78 (d, 2H, Ar, J = 4.0 Hz), 8.02 (d, 1H, Ar, J = 2.0 Hz), 8.22 (d, 2H, Ar, J = 7.2 Hz), 8.63 (d, 1H, Ar, J = 2.0 Hz). ESI-MS calcd. for C20H13F3N2O2S, 402.39; found: m/z 403.07 [M+H]+. Anal. C20H13F3N2O2S (C, H, N).
1-(Phenylsulfonyl)-5-(m-tolyl)-1H-pyrrolo[2,3-b]pyridine (9c).
Yield = 78%; mp = 140–143°C. 1H-NMR (CDCl3-d1) δ 2.41 (s, 3H, m-CH3-Ph), 6.63 (d, 1H, Ar, J = 4.0 Hz), 7.19 (s, 1H, Ar), 7.30–7.35 (m, 3H, Ar), 7.48 (t, 1H, Ar, J = 8.0 Hz), 7.56 (t, 2H, Ar, J = 7.6 Hz), 7.75 (d, 1H, Ar, J = 4.0 Hz), 7.98 (d, 1H, Ar, J = 2.0 Hz), 8.22 (d, 2H, Ar, J = 7.6 Hz), 8.64 (d, 1H, Ar, J = 2.0 Hz). ESI-MS calcd. for C20H16N2O2S, 348.42; found: m/z 349.10 [M+H]+. Anal. C20H16N2O2S (C, H, N).
5-(3-Methoxyphenyl)-1-(phenylsulfonyl)-1H-pyrrolo[2,3-b]pyridine (9d).
Yield = 78%; mp = 106–109°C. 1H-NMR (CDCl3-d1) δ 3.85 (s, 3H, OCH3), 6.64 (d, 1H, Ar, J = 4.0 Hz), 6.92 (dd, 1H, Ar, J = 2.0 Hz and J = 8.4 Hz), 7.07 (s, 1H, Ar), 7.12 (d, 1H, Ar, J = 7.6 Hz), 7.37 (t, 1H, Ar, J = 7.6 Hz), 7.50 (t, 2H, Ar, J = 8.0 Hz), 7.58 (t, 1H, Ar, J = 7.6 Hz), 7.75 (d, 1H, Ar, J = 4.0 Hz), 8.00 (d, 1H, Ar, J = 2.0 Hz), 8.22 (d, 2H, Ar, J = 7.6 Hz), 8.65 (d, 1H, Ar, J = 2.0 Hz). ESI-MS calcd. for C20H16N2O3S, 364.42; found: m/z 365.09 [M+H]+. Anal. C20H16N2O3S (C, H, N).
5-(6-Methoxynaphthalen-2-yl)-1-(phenylsulfonyl)-1H-pyrrolo[2,3-b]pyridine (9e).
Yield = 91%; mp = 173–176°C dec. 1H-NMR (CDCl3-d1) δ 3.93 (s, 3H, OCH3), 6.65 (d, 1H, Ar, J = 3.6 Hz), 7.15–7.21 (m, 2H, Ar), 7.49 (t, 2H, Ar, J = 8.0 Hz), 7.56 (d, 1H, Ar, J = 7.2 Hz), 7.64 (d, 1H, Ar, J = 8.0 Hz), 7.75–7.83 (m, 3H, Ar), 7.92 (s, 1H, Ar), 8.08 (s, 1H, Ar), 8.24 (d, 2H, Ar, J = 7.6 Hz), 8.75 (s, 1H, Ar). ESI-MS calcd. for C24H18N2O3S, 414.48; found: m/z 415.11 [M+H]+. Anal. C24H18N2O3S (C, H, N).
1-(Phenylsulfonyl)-5-(thiophen-3-yl)-1H-pyrrolo[2,3-b]pyridine (9f).
Yield = 55%; mp = 186–189°C. 1H-NMR (CDCl3-d1) δ 6.59–6.63 (m, 1H, Ar), 7.22–7.27 (m, 1H, Ar), 7.35–7.50 (m, 4H, Ar), 7.53–7.58 (m, 1H, Ar), 7.70–7.75 (m, 1H, Ar), 8.00 (s, 1H, Ar), 8.18–8.23 (m, 2H, Ar), 8.68 (s, 1H, Ar). ESI-MS calcd. for C17H12N2O2S2, 340.43; found: m/z 341.04 [M+H]+. Anal. C17H12N2O2S2 (C, H, N).
1-(Phenylsulfonyl)-5-(pyridin-3-yl)-1H-pyrrolo[2,3-b]pyridine (9g).
Yield = 98%; mp = 148–152°C. 1H-NMR (CDCl3-d1) δ 6.68 (d, 1H, Ar, J = 3.6 Hz), 7.49–7.62 (m, 4H, Ar), 7.82 (s, 1H, Ar), 8.03 (s, 2H, Ar), 8.23 (d, 2H, Ar, J = 7.6 Hz), 8.62–8.67 (m, 2H, Ar), 8.85 (s, 1H, Ar). ESI-MS calcd. for C18H13N3O2S, 335.38; found: m/z 336.08 [M+H]+. Anal. C18H13N3O2S (C, H, N).
General procedure for compounds 10b and 10e.
To a solution of intermediate 9b or 9e (0.29 mmol) in anhydrous THF (3 mL), 0.87 mmol of TBAF was added, and the mixture was stirred at reflux for 4 h. After cooling, the solvent was evaporated, and cold-ice water (20 mL) was added. The precipitate was recovered by vacuum filtration and purified by crystallization from ethanol to obtain the desired compound 10b or by column chromatography using cyclohexane/ethyl acetate 1:1 as eluent for 10e.
5-(3-(Trifluoromethyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (10b).
Yield = 97%; mp = 158–161°C (EtOH). 1H-NMR (CDCl3-d1) δ 6.65 (d, 1H, Ar, J = 3.2 Hz), 7.47 (d, 1H, Ar, J = 3.2 Hz), 7.59–7.67 (m, 2H, Ar), 7.80 (d, 1H, Ar, J = 7.2 Hz), 7.86 (s, 1H, Ar), 8.27 (d, 1H, Ar, J = 1.2 Hz), 8.54 (s, 1H, Ar), 10.12 (exch br s, 1H, NH). ESI-MS calcd. for C14H9F3N2, 262.23; found: m/z 263.08 [M+H]+. Anal. C14H9F3N2 (C, H, N).
5-(6-Methoxynaphthalen-2-yl)-1H-pyrrolo[2,3-b]pyridine (10e).
Yield = 45%; mp = 247–250°C dec. 1H-NMR (CDCl3-d1) δ 3.87 (s, 3H, OCH3), 6.50 (d, 1H, Ar, J = 1.6 Hz), 7.18 (dd, 1H, Ar, J = 2.4 Hz and J = 8.4 Hz), 7.33 (s, 1H, Ar), 7.50 (d, 1H, Ar, J = 2.4 Hz), 7.82–7.90 (m, 3H, Ar), 8.16 (d, 1H, Ar, J = 4.0 Hz), 8.29 (d, 1H, Ar, J = 3.6 Hz), 8.62 (d, 1H, Ar, J = 3.2 Hz), 11.71 (exch br s, 1H, NH). ESI-MS calcd. for C18H14N2O, 274.32; found: m/z 275.11 [M+H]+. Anal. C18H14N2O (C, H, N).
General procedure for compounds 11a-f.
A mixture of the appropriate 1H-pyrrolo[2,3-b]pyridine derivative 10a-f (0.95 mmol) [10a, Laha et al., 2017; 10c and 10d, Ibrahim et al., 2007; 10f, Singh et al. 2017] and HMTA (1.43 mmol) in glacial acetic acid (3 mL) was heated under reflux for 6 h. The reaction mixture was cooled at room temperature and diluted with cold water. The solid obtained was filtered, washed with an excess of ice-cold water, and dried. The crude compound was purified by crystallization from hexane (11b,e) or by column chromatography using cyclohexane/ethyl acetate 1:3 (for 11a,d), dichloromethane/methanol 95:5 (for 11c), or cyclohexane/ethyl acetate 1:5 (for 11f) as eluents.
5-Phenyl-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde (11a).
Yield = 56%; mp = 212–215°C. 1H-NMR (CDCl3-d1) δ 7.42 (t, 1H, Ar, J = 7.2 Hz), 7.51 (t, 2H, Ar, J = 7.6 Hz), 7.67 (d, 2H, Ar, J = 7.2 Hz), 8.10 (s, 1H, Ar), 8.68 (s, 1H, Ar), 8.89 (s, 1H, Ar), 10.08 (s, 1H, CHO), 11.82 (exch br s, 1H, NH). ESI-MS calcd. for C14H10N2O, 222.24; found: m/z 223.08 [M+H]+. Anal. C14H10N2O (C, H, N).
5-(3-(Trifluoromethyl)phenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde (11b).
Yield = 80%; mp = 193–196°C (hexane). 1H-NMR (DMSO-d6) δ 7.23 (d, 2H, Ar, J = 7.2 Hz), 8.04 (d, 2H, Ar, J = 8.4 Hz), 8.52 (s, 1H, Ar), 8.63 (d, 1H, Ar, J = 2.0 Hz), 8.71 (d, 1H, Ar, J = 2.0 Hz), 9.45 (s, 1H, CHO), 12.65 (exch br s, 1H, NH). ESI-MS calcd. for C15H9F3N2O, 290.24; found: m/z 291.07 [M+H]+. Anal. C15H9F3N2O (C, H, N).
5-(m-Tolyl)-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde (11c).
Yield = 62%; mp = 162–165°C. 1H-NMR (DMSO-d6) δ 2.38 (s, 3H, CH3), 7.19 (d, 1H, Ar, J = 7.6 Hz), 7.36 (t, 1H, Ar, J = 7.2 Hz), 7.48 (d, 1H, Ar, J = 7.6 Hz), 7.52 (s, 1H, Ar), 8.48 (s, 1H, Ar), 8.55 (d, 1H, Ar, J = 2.0 Hz), 8.62 (d, 1H, Ar, J = 2.0 Hz), 9.93 (s, 1H, CHO), 12.75 (exch br s, 1H, NH). ESI-MS calcd. for C15H12N2O, 236.27; found: m/z 237.10 [M+H]+. Anal. C15H12N2O (C, H, N).
5-(3-Methoxyphenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde (11d).
Yield = 37%; mp = 200–203°C. 1H-NMR (DMSO-d6) δ 3.90 (s, 3H, OCH3), 6.97 (d, 1H, Ar, J = 8.4 Hz), 7.17 (s, 1H, Ar), 7.23 (s, 1H, Ar), 7.42 (t, 1H, Ar, J = 8.0 Hz), 8.07 (s, 1H, Ar), 8.64 (s, 1H, Ar), 8.92 (s, 1H, Ar), 10.08 (s, 1H, CHO), 12.70 (exch br s, 1H, NH). ESI-MS calcd. for C15H12N2O2, 252.27; found: m/z 253.09 [M+H]+. Anal. C15H12N2O2 (C, H, N).
5-(6-methoxynaphthalen-2-yl)-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde (11e).
Yield = 84%; mp = 145–147°C (hexane). 1H-NMR (DMSO-d6) δ 3.88 (s, 3H, OCH3), 7.20 (d, 1H, Ar, J = 8.8 Hz), 7.36 (s, 1H, Ar), 7.84–7.95 (m, 3H, Ar), 8.21 (s, 1H, Ar), 8.52 (s, 1H, Ar), 8.69 (s, 1H, Ar), 8.78 (s, 1H, Ar), 9.97 (s, 1H, CHO), 12.57 (exch br s, 1H, NH). ESI-MS calcd. for C19H14N2O2, 302.33; found: m/z 303.11 [M+H]+. Anal. C19H14N2O2 (C, H, N).
5-(Thiophen-3-yl)-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde (11f).
Yield = 37%; mp = 194–196°C. 1H-NMR (DMSO-d6) δ 7.60–7.66 (m, 2H, Ar), 7.95 (s, 1H, Ar), 8.46 (s, 1H, Ar), 8.60 (s, 1H, Ar), 8.74 (s, 1H, Ar), 9.93 (s, 1H, CHO), 12.71 (exch br s, 1H, NH). ESI-MS calcd. for C12H8N2OS, 228.27; found: m/z 229.04 [M+H]+. Anal. C12H8N2OS (C, H, N).
General procedure for compounds 12a-g.
Compounds 12a-g were obtained following the same procedure reported for compounds 5a,c, but starting with intermediates 11a-g [11g, Ibrahim et al., 2007]. The crude products were purified by crystallization from ethanol (12c,d,g) or by column chromatography using cyclohexane/ethyl acetate 1:5 (for 12a,b), cyclohexane/ethyl acetate 1:2 (for 12e), or cyclohexane/ethyl acetate 1:3 (for 12f) as eluents.
5-Phenyl-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde oxime (12a).
Yield = 42%; mp = 224–226°C. 1H-NMR (CDCl3-d1) δ 7.40 (d, 1H, Ar, J = 7.6 Hz), 7.46–7.52 (m, 3H, 2H Ar + 1H CH=N-OH), 7.63 (d, 2H, Ar, J = 7.6 Hz), 8.33 (s, 1H, Ar), 8.59 (s, 1H, Ar), 8.69 (s, 1H, Ar), 10.31 (exch br s, 1H, CH=N-OH), 11.95 (exch br s, 1H, NH). ESI-MS calcd. for C14H11N3O, 237.26; found: m/z 238.09 [M+H]+. Anal. C14H11N3O (C, H, N).
5-(3-(Trifluoromethyl)phenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde oxime (12b).
Yield = 52%; mp = 247–250°C. 1H-NMR (DMSO-d6) δ 7.72 (d, 3H, Ar, J = 4.0 Hz), 7.82 (s, 1H, CH=N-OH), 7.96 (s, 1H, Ar), 8.28 (s, 1H, Ar), 8.51 (d, 1H, Ar, J = 2.0 Hz), 8.64 (d, 1H, Ar, J = 1.6 Hz), 10.74 (exch br s, 1H, CH=N-OH), 12.10 (exch br s, 1H, NH). ESI-MS calcd. for C15H10F3N3O, 305.25; found: m/z 306.08 [M+H]+. Anal. C15H10F3N3O (C, H, N).
5-(m-Tolyl)-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde oxime (12c).
Yield = 51%; mp = 208–211°C (EtOH). 1H-NMR (DMSO-d6) δ 2.37 (s, 3H, CH3), 7.16 (s, 1H, CH=N-OH), 7.35 (t, 1H, Ar, J = 7.6 Hz), 7.44 (t, 1H, Ar, J = 7.6 Hz), 7.50–7.56 (m, 1H, Ar), 7.78 (d, 1H, Ar, J = 2.0 Hz), 8.25–8.30 (m, 1H, Ar), 8.46 (d, 1H, Ar, J = 2.0 Hz), 8.54–8.59 (m, 1H, Ar), 10.71 (exch br s, 1H, CH=N-OH), 12.00 (exch br s, 1H, NH). ESI-MS calcd. for C15H13N3O, 251.28; found: m/z 252.11 [M+H]+. Anal. C15H13N3O (C, H, N).
5-(3-Methoxyphenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde oxime (12d).
Yield = 84%; mp = 191–194°C (EtOH). 1H-NMR (DMSO-d6) δ 3.81 (s, 3H, OCH3), 6.91–6.96 (m, 1H, Ar), 7.17–7.22 (m, 2H, Ar), 7.35–7.40 (m, 2H, 1H Ar + 1H CH=N-OH), 7.78 (s, 1H, Ar), 8.26 (s, 1H, Ar), 8.64 (s, 1H, Ar), 10.72 (exch br s, 1H, CH=N-OH), 12.03 (exch br s, 1H, NH). ESI-MS calcd. for C15H13N3O2, 267.28; found: m/z 268.10 [M+H]+. Anal. C15H13N3O2 (C, H, N).
5-(6-Methoxynaphthalen-2-yl)-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde oxime (12e).
Yield = 37%; mp = 204–207°C dec. 1H-NMR (DMSO-d6) δ 3.88 (s, 3H, OCH3), 7.19 (d, 1H, Ar, J = 8.4 Hz), 7.35 (s, 1H, Ar), 7.74–7.79 (m, 1H, Ar), 7.89–7.94 (m, 3H, Ar), 8.12 (s, 1H, CH=N-OH), 8.29 (d, 1H, Ar, J = 6.8 Hz), 8.57 (s, 1H, Ar), 8.66–8.71 (m, 1H, Ar), 10.73 (exch br s, 1H, CH=N-OH), 12.01 (exch br s, 1H, NH). ESI-MS calcd. for C19H15N3O2, 317.34; found: m/z 318.12 [M+H]+. Anal. C19H15N3O2 (C, H, N).
5-(Thiophen-3-yl)-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde oxime (12f).
Yield = 54%; mp = 239–242°C. 1H-NMR (DMSO-d6) δ 7.50 (d, 1H, Ar, J = 4.8 Hz), 7.66 (s, 1H, Ar), 7.77 (d, 1H, Ar, J = 7.6 Hz), 8.25 (s, 1H, CH=N-OH), 8.45 (s, 1H, Ar), 8.62–8.67 (m, 2H, Ar), 10.71 (exch br s, 1H, CH=N-OH), 11.99 (exch br s, 1H, NH). ESI-MS calcd. for C12H9N3OS, 243.28; found: m/z 244.05 [M+H]+. Anal. C12H9N3OS (C, H, N).
5-(pyridin-3-yl)-1H-pyrrolo[2,3-b]pyridine-3-carbaldehyde oxime (12g).
Yield = 67%; mp = 241–243°C (EtOH). 1H-NMR (DMSO-d6) δ 7.50 (d, 1H, Ar, J = 4.8 Hz), 7.79 (s, 1H, Ar), 8.07 (d, 1H, Ar, J = 6.4 Hz), 8.27 (s, 1H, CH=N-OH), 8.49–8.65 (m, 3H, Ar), 8.81 (s, 1H, Ar), 10.74 (exch br s, 1H, CH=N-OH), 12.10 (exch br s, 1H, NH). ESI-MS calcd. for C13H10N4O, 238.24; found: m/z 239.09 [M+H]+. Anal. C13H10N4O (C, H, N).
General procedure for compounds 13a-g.
Compounds 13a-g were obtained following the same procedure reported for compounds 6b,c, but starting with intermediates 12a-g. The crude products were purified by crystallization from ethanol.
5-Phenyl-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (13a).
Yield = 40%; mp = 199–202°C (EtOH). 1H-NMR (CDCl3-d1) δ 7.47 (d, 1H, Ar, J = 7.2 Hz), 7.53 (t, 2H, Ar, J = 7.2 Hz), 7.63 (d, 2H, Ar, J = 7.6 Hz), 7.97 (s, 1H, Ar), 8.48 (s, 1H, Ar), 8.71 (s, 1H, Ar), 12.65 (exch br s, 1H, NH). IR ѵ (cm−1): 2220 (CN). ESI-MS calcd. for C14H9N3, 219.24; found: m/z 220.08 [M+H]+. Anal. C14H9N3 (C, H, N).
5-(3-(Trifluoromethyl)phenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (13b).
Yield = 85%; mp = 240–242°C (EtOH). 1H-NMR (CDCl3-d1) δ 7.70 (t, 1H, Ar, J = 7.6 Hz), 7.76 (d, 1H, Ar, J = 7.6 Hz), 7.82 (d, 1H, Ar, J = 7.6 Hz), 7.88 (s, 1H, Ar), 8.09 (s, 1H, Ar), 8.62 (s, 1H, Ar), 8.71 (s, 1H, Ar), 12.75 (exch br s, 1H, NH). IR ѵ (cm−1): 2222 (CN). ESI-MS calcd. for C15H8F3N3, 287.24; found: m/z 288.07 [M+H]+. Anal. C15H8F3N3 (C, H, N).
5-(m-Tolyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (13c).
Yield = 74%; mp = 194–197°C (EtOH). 1H-NMR (DMSO-d6) δ 2.38 (s, 3H, CH3), 7.19 (d, 1H, Ar, J = 7.6 Hz), 7.36 (t, 1H, Ar, J = 7.6 Hz), 7.55 (d, 1H, Ar, J = 7.6 Hz), 7.61 (s, 1H, Ar), 8.29 (d, 1H, Ar, J = 1.6 Hz), 8.46 (d, 1H, Ar, J = 2.4 Hz), 8.67 (d, 1H, Ar, J = 2.0 Hz), 12.87 (exch br s, 1H, NH). IR ѵ (cm−1): 2220 (CN). ESI-MS calcd. for C15H11N3, 233.27; found: m/z 234.10 [M+H]+. Anal. C15H11N3 (C, H, N).
5-(3-Methoxyphenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (13d).
Yield = 94%; mp = 201–204°C (EtOH). 1H-NMR (CDCl3-d1) δ 3.83 (s, 3H, OCH3), 6.94 (d, 1H, Ar, J = 7.6 Hz), 7.30–7.35 (m, 2H, Ar), 7.38 (t, 1H, Ar, J = 7.2 Hz), 8.33 (d, 1H, Ar, J = 1.6 Hz), 8.47 (s, 1H, Ar), 8.69 (d, 1H, Ar, J = 1.6 Hz), 12.91 (exch br s, 1H, NH). IR ѵ (cm−1): 2220 (CN). ESI-MS calcd. for C15H11N3O, 249.27; found: m/z 250.09 [M+H]+. Anal. C15H11N3O (C, H, N).
5-(6-Methoxynaphthalen-2-yl)-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (13e).
Yield = 63%; mp = 193–196°C (EtOH). 1H-NMR (DMSO-d6) δ 3.88 (s, 3H, OCH3), 7.14–7.19 (m, 1H, Ar), 7.35 (s, 1H, Ar), 7.91–7.99 (m, 3H, Ar), 8.28 (s, 1H, Ar), 8.43–8-50 (m, 2H, Ar), 8.82 (s, 1H, Ar), 12.89 (exch br s, 1H, NH). IR ѵ (cm−1): 2221 (CN). ESI-MS calcd. for C19H13N3O, 299.33; found: m/z 300.11 [M+H]+. Anal. C19H13N3O (C, H, N).
5-(thiophen-3-yl)-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (13f).
Yield = 88%; mp = 165–167°C (EtOH). 1H-NMR (DMSO-d6) δ 7.67–7.73 (m, 2H, Ar), 8.05 (s, 1H, Ar), 8.39–8.44 (m, 2H, Ar), 8.80 (s, 1H, Ar), 12.86 (exch br s, 1H, NH). IR ѵ (cm−1): 2222 (CN). ESI-MS calcd. for C12H7N3S, 225.27; found: m/z 226.04 [M+H]+. Anal. C12H7N3S (C, H, N).
5-(pyridin-3-yl)-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (13g).
Yield = 71%; mp = 285–287°C dec (EtOH). (DMSO-d6) δ 7.44–7.49 (m, 1H, Ar), 8.18 (d, 1H, Ar, J = 6.0 Hz), 8.40 (s, 1H, Ar), 8.47 (s, 1H, Ar), 8.57 (s, 1H, Ar), 8.73 (s, 1H, Ar), 8.97 (s, 1H, Ar), 12.96 (exch br s, 1H, NH). IR ѵ (cm−1): 2220 (CN). ESI-MS calcd. for C13H8N4, 220.23; found: m/z 221.08 [M+H]+. Anal. C13H8N4 (C, H, N).
General procedure for compounds 14a-g.
Compounds 14a-g were obtained following the same procedure reported for compounds 7a-c, but starting with intermediates 13a-g. The crude products were purified by column chromatography using cyclohexane/ethyl acetate 5:1 (for 14a-e), cyclohexane/ethyl acetate 4:1 (for 14f), or cyclohexane/ethyl acetate 1:3 (for 14g) as eluents.
1-(3-Methylbenzoyl)-5-phenyl-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (14a).
Yield = 17%; mp = 160–163°C. 1H-NMR (CDCl3-d1) δ 2.45 (s, 3H, m-CH3-Ph), 7.40–7.45 (m, 2H, Ar), 7.51 (t, 3H, Ar, J = 7.2 Hz), 7.61 (t, 3H, Ar, J = 7.6 Hz), 7.68 (s, 1H, Ar), 8.24 (s, 1H, Ar), 8.26 (d, 1H, Ar, J = 2.0 Hz), 8.68 (d, 1H, Ar, J = 2.0 Hz). 13C-NMR (CDCl3-d1) δ 20.93 (CH3), 101.18 (C), 115.32 (C), 123.82 (CH), 127.54 (CH), 128.17 (CH), 130.86 (CH), 134.56 (CH), 136.41 (C), 138.83 (C), 144.65 (C), 167.76 (CO). IR ѵ (cm−1): 2220 (CN). ESI-MS calcd. for C22H15N3O, 337.37; found: m/z 338.12 [M+H]+. Anal. C22H15N3O (C, H, N).
1-(3-Methylbenzoyl)-5-(3-(trifluoromethyl)phenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (14b).
Yield = 10%; mp = 155–157°C. 1H-NMR (CDCl3-d1) δ 2.46 (s, 3H, m-CH3-Ph), 7.43 (t, 1H, Ar, J = 7.6 Hz), 7.52 (d, 1H, Ar, J = 8.0 Hz), 7.58–7.71 (m, 4H, Ar), 7.80 (d, 1H, Ar, J = 7.6 Hz), 7.85 (s, 1H, Ar), 8.28 (s, 2H, Ar), 8.67 (d, 1H, Ar, J = 2.0 Hz). 13C-NMR (CDCl3-d1) δ 21.31 (CH3), 103.41 (C), 115.62 (C), 121.42 (CH), 124.75 (CH), 125.18 (CH), 126.74 (CH), 128.96 (CH), 129.56 (CH), 130.48 (CH), 130.92 (CH), 136.72 (C), 138.92 (C), 144.67 (C), 167.48 (CO). IR ѵ (cm−1): 2220 (CN). ESI-MS calcd. for C23H14F3N3O, 405.37; found: m/z 406.11 [M+H]+. Anal. C23H14F3N3O (C, H, N).
1-(3-Methylbenzoyl)-5-(m-tolyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (14c).
Yield = 10%; mp = 115–118°C. 1H-NMR (CDCl3-d1) δ 2.45 (s, 6H, 2 x m-CH3-Ph), 7.24 (s, 1H, Ar), 7.38–7.43 (m, 4H, Ar), 7.51 (d, 1H, Ar, J = 7.2 Hz), 7.59 (d, 1H, Ar, J = 7.2 Hz), 7.67 (s, 1H, Ar), 8.23 (s, 1H, Ar), 8.25 (d, 1H, Ar, J = 2.0 Hz), 8.67 (d, 1H, Ar, J = 2.0 Hz). 13C-NMR (CDCl3-d1) δ 21.39 (CH3), 21.56 (CH3), 90.72 (C), 113.51 (C), 121.17 (C), 124.55 (CH), 126.66 (CH), 127.26 (CH), 127.83 (CH), 128.22 (CH), 128.45 (CH), 129.06 (CH), 129.18 (CH), 131.03 (CH), 132.07 (C), 134.31 (CH), 134.88 (CH), 137.30 (C), 138.75 (C), 139.03 (C), 146.04 (CH), 146.35 (C), 166.72 (CO). IR ѵ (cm−1): 2220 (CN). ESI-MS calcd. for C23H17N3O, 351.40; found: m/z 352.14 [M+H]+. Anal. C23H17N3O (C, H, N).
5-(3-Methoxyphenyl)-1-(3-methylbenzoyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (14d).
Yield = 10%; mp = 152–155°C. 1H-NMR (CDCl3-d1) δ 2.45 (s, 3H, m-CH3-Ph), 3.88 (s, 3H, OCH3), 6.97 (d, 1H, Ar, J = 8.0 Hz), 7.13–7.20 (m, 2H, Ar), 7.41 (d, 2H, Ar, J = 7.6 Hz), 7.51 (d, 1H, Ar, J = 7.2 Hz), 7.60 (d, 1H, Ar, J = 7.2 Hz), 7.67 (s, 1H, Ar), 8.25 (d, 2H, Ar, J = 7.6 Hz), 8.67 (s, 1H, Ar). 13C-NMR (CDCl3-d1) δ 21.18 (CH3), 55.76 (CH3), 102.47 (C), 113.48 (CH), 114.39 (CH), 115.49 (C), 119.82 (CH), 121.43 (C), 123.64 (CH), 126.12 (CH), 128.17 (CH), 129.32 (CH), 130.14 (CH), 131.18 (C), 134.56 (CH), 138.97 (C), 144.65 (C), 161.18 (C), 168.71 (CO). IR ѵ (cm−1): 2220 (CN). ESI-MS calcd. for C23H17N3O2, 367.40; found: m/z 368.14 [M+H]+. Anal. C23H17N3O2 (C, H, N).
5-(6-Methoxynaphthalen-2-yl)-1-(3-methylbenzoyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (14e).
Yield = 22%; oil. 1H-NMR (CDCl3-d1) δ 2.46 (s, 3H, m-CH3-Ph), 3.95 (s, 3H, OCH3), 7.18–7.25 (m, 2H, Ar), 7.43 (t, 1H, Ar, J = 7.6 Hz), 7.51 (d, 1H, Ar, J = 7.2 Hz), 7.61 (d, 1H, Ar, J = 7.2 Hz), 7.68–7.73 (m, 2H, Ar), 7.81–7.88 (m, 2H, Ar), 8.00 (s, 1H, Ar), 8.25 (s, 1H, Ar), 8.36 (s, 1H, Ar), 8.79 (s, 1H, Ar). 13C-NMR (CDCl3-d1) δ 21.19 (CH3), 55.85 (CH3), 101.06 (C), 105.92 (CH), 113.49 (CH), 114.39 (C), 115.41 (C), 119.87 (CH), 121.43 (C), 123.68 (CH), 125.44 (CH), 126.15 (CH), 128.17 (CH), 129.35 (CH), 129.60 (CH), 130.14 (CH), 131.11 (C), 134.56 (CH), 134.82 (C), 138.91 (C), 144.62 (C), 157.28 (C), 167.70 (CO). IR ѵ (cm−1): 2220 (CN). ESI-MS calcd. for C27H19N3O2, 417.46; found: m/z 418.15 [M+H]+. Anal. C27H19N3O2 (C, H, N).
1-(3-methylbenzoyl)-5-(thiophen-3-yl)-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (14f).
Yield = 40%; mp = 171–173°C. 1H-NMR (CDCl3-d1) δ 2.45 (s, 3H, m-CH3-Ph), 7.42 (d, 2H, Ar, J = 7.2 Hz), 7.47–7.52 (m, 2H, Ar), 7.54–7.59 (m, 2H, Ar), 7.66 (s, 1H, Ar), 8.22 (s, 1H, Ar), 8.25 (d, 1H, Ar, J = 2.0 Hz), 8.71 (d, 1H, Ar, J = 2.0 Hz). 13C-NMR (CDCl3-d1) δ 21.35(CH3), 90.66 (C), 113.42 (C), 121.22 (C), 121.75 (CH), 125.68 (CH), 126.07 (CH), 127.36 (CH), 127.80 (CH), 128.41 (CH), 129.05 (C), 130.99 (CH), 132.08 (C), 132.08 (CH), 135.33 (CH), 138.25 (C), 138.72 (C), 145.36 (CH), 146.09 (C), 166.64 (CO). IR ѵ (cm−1): 2222 (CN). ESI-MS calcd. for C20H13N3OS, 343.40; found: m/z 344.08 [M+H]+. Anal. C20H13N3OS (C, H, N).
1-(3-methylbenzoyl)-5-(pyridin-3-yl)-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (14g).
Yield = 32%; mp = 155–157°C. 1H-NMR (CDCl3-d1) δ 2.44 (s, 3H, m-CH3-Ph), 7.40–7.67 (m, 5H, Ar), 7.93 (d, 1H, Ar, J = 6.0 Hz), 8.27 (s, 2H, Ar), 8.67 (d, 2H, Ar, J = 7.6 Hz), 8.88 (s, 1H, Ar). 13C-NMR (CDCl3-d1) δ 21.32 (CH3), 90.71 (C), 113.22 (C), 121.28 (C), 123.57 (CH), 126.84 (CH), 126.98 (CH), 127.85 (CH), 128.47 (CH), 130.83 (C), 131.02 (CH), 131.92 (C), 133.19 (C), 134.83 (CH), 135.02 (CH), 138.81 (C), 145.54 (CH), 145.77 (CH), 146.76 (C), 149.27 (CH), 166.56 (CO). IR ѵ (cm−1): 2220 (CN). ESI-MS calcd. for C21H14N4O, 338.36; found: m/z 339.12 [M+H]+. Anal. C21H14N4O (C, H, N).
Pharmacology
Compounds were dissolved in 100% DMSO at 5 mM stock concentrations. The final concentration of DMSO in the reactions was 1%, and this level of DMSO had no effect on HNE activity. The HNE inhibition assay was performed in black flat-bottom 96-well microtiter plates. Briefly, a buffer solution containing 200 mM Tris–HCl, pH 7.5, 0.01% bovine serum albumin, 0.05% Tween-20, and 20 mU/mL HNE (Calbiochem) was added to wells containing different concentrations of each compound. The reactions were initiated by addition of 25 µM elastase substrate (N-methylsuccinyl-Ala-Ala-Pro-Val-7-amino-4-methylcoumarin, Calbiochem) in a final reaction volume of 100 µL/well. Kinetic measurements were obtained every 30 s for 10 min at 25 °C using a Fluoroskan Ascent FL fluorescence microplate reader (Thermo Electron, MA) with excitation and emission wavelengths at 355 and 460 nm, respectively. For all compounds tested, the concentration of inhibitor that caused 50% inhibition of the enzymatic reaction (IC50) was calculated by plotting % inhibition versus logarithm of inhibitor concentration (at least six points). The data are presented as the mean values of at least three independent experiments with relative standard deviations of <15%.
Molecular modeling
RESULTS AND DISCUSSION
All new products were tested for their ability to inhibit HNE, and the experiments were conducted in triplicate using N-methylsuccinyl-Ala-Ala-Pro-Val-7-amino-4-cumarin as the substrate. The results are reported in the Tables 1–2 and are compared with the Sivelestat and 1-(3-methylbenzoyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (compound X), which we previously found to be a potent HNE inhibitor [Crocetti et al., 2018]. As shown in Table 1, the introduction of substituents at position 2 of the pyrrolo[2,3-b]pyridine scaffold was not favorable for HNE inhibitory activity. In fact, introduction of an alkyl group (compounds 2a and 2b) or a carbethoxy group (2c) led to products completely devoid of activity. In addition, shift of the carbonitrile group from position 3 of compound X to position 2 (2d) also led to a significant loss in HNE inhibitory activity (two orders of magnitude lower). Likewise, the introduction of a methyl group at position 2 of compound X was detrimental for activity, resulting in 2e with an IC50 of 10.8 µM, further confirming that substituents at position 2 are not tolerated without significant loss in HNE inhibitory activity.
Table 1:
HNE inhibitory activity of 1H-pyrrolo[2,3-b]pyridine derivatives 2a-e.
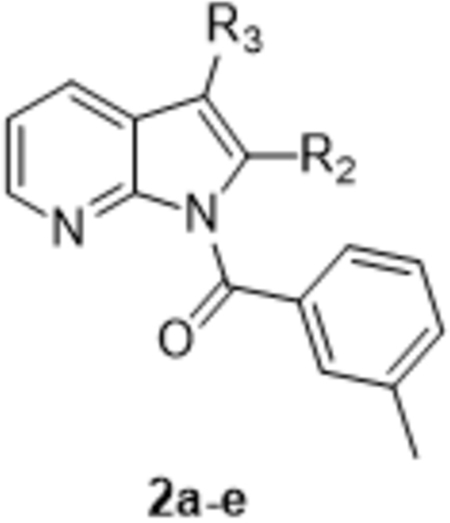 | |||
|---|---|---|---|
| Compound | R2 | R3 | IC50 (µM)a |
| 2a | CH3 | H | N.A.b |
| 2b | CH2CH3 | H | N.A.b |
| 2c | COOEt | H | N.A.b |
| 2d | CN | H | 3.5 ± 1.2 |
| 2e | CH3 | CN | 10.8 ± 2.2 |
| Xc | H | CN | 0.015 ± 0.004 |
| Sivelestat | 0.050 ± 0.020 | ||
IC50 values are presented as the mean ± SD of three independent experiment with a 6 -point dilution series for each experiment.
NA: no inhibitory activity was found at the highest concentration of compound tested (50 µM).
Table 2:
HNE inhibitory activity of 1H-pyrrolo[2,3-b]pyridine derivatives 7a-c and 14a-g.
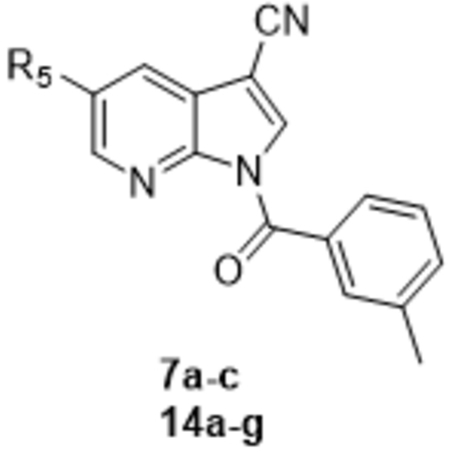 | ||
|---|---|---|
| Compound | R5 | IC50 (µM)a |
| 7a | Br | 0.051 ± 0.014 |
| 7b | Cl | 0.16 ± 0.05 |
| 7c | NO2 | 0.40 ± 0.11 |
| 14a | Ph | 0.050 ± 0.02 |
| 14b | m-CF3Ph | 0.18 ± 0.07 |
| 14c | m-CH3Ph | 0.13 ± 0.04 |
| 14d | m-OCH3Ph | 0.070 ± 0.015 |
| 14e | 6-OCH3-naphthalene | 1.6 ± 0.33 |
| 14f | 3-thienyl | 0.042 ± 0.015 |
| 14g | 3-pyridyl | 0.015 ± 0.004 |
| Xb | H | 0.015 ± 0.004 |
| Sivelestat | - | 0.050 ± 0.020 |
IC50 values are presented as the mean ± SD of three independent experiments with a 6 -point dilution series for each experiment.
In contrast, position 5 was more tolerant to substitutions, as most of the 5-substitued pyrrolo[2,3-b]pyridines exhibited IC50 values in the nanomolar range (0.015–0.070 µM) (see Table 2). The 5-bromine derivative was about 3-fold more potent (7a, IC50= 0.051 µM) than the 5-chloro (7b, IC50= 0.160 µM) derivative, suggesting that lipophilicity (π = 0.86 for Br and 0.71 for Cl), steric bulk (Mr = 8.88 for Br and 6.03 for Cl), or electron withdrawing ability could be important requirements for activity. On the other hand, the introduction of a nitro group (7c), which has an intermediate Mr between bromine and chlorine (Mr = 7.36) but hydrophilic properties (π = −0.28) was about 2-fold less potent than the 5-chloro derivative 7b (IC50= 0.400 and 0.160 µM, respectively).
Insertion of bulky and lipophilic rings was favorable for HNE inhibitory activity within certain limits, as demonstrated by the comparable potency of the 5-(substituted)phenyl derivatives 14a-d (IC50= 0.050–0.180 µM) and the low activity of compound 14e bearing a 6-OCH3-naphtalen at position 5 (IC50= 1.6 μM). Replacement of the phenyl at position 5 in compound 14a with the isostere thienyl or pyridyl rings resulted in the thienyl derivative 14f with comparable potency to 14a (IC50= 0.042 versus 0.050 µM, respectively) and the pyridyl derivative 14g, which was about 3-fold more potent than 14a (IC50= 0.015 versus 0.050 µM, respectively). Thus, the presence of pyridine in compound 14g may be important not only for steric hindrance but also for the formation of an H-bond that involves a nitrogen atom, which is not found in 14a.
Taking into account these biological results and in particular the observation that the most potent 5-substituted compound (14g) exhibited the same IC50 as the 5-unsubstitued lead compound X, we can conclude that the introduction of certain substituents at position 5 maintained HNE inhibitory activity, probably by interacting with the large pocket of the enzyme site. On the other hand, we were not able to improve the HNE inhibitory activity with respect the lead X.
The difference in activities of the test compounds can be explained by the length L of the proton transfer channel, angle α, and length d1. As reported previously, a nitrogen atom in the six-membered pyridine moiety of the lead X forms an H-bond with Asp194 [Crocetti et al., 2018]. Additional H-bonding occurs with Val216 due to the nitrogen atom of the cyano group in lead compound X [Crocetti et al., 2018]. The distance d1 for molecule X is smaller than for other active compounds in Table 3. In general, the geometry of the inhibitor-enzyme complex is favorable for the formation of a Michaelis complex when the oxygen atom of Ser195 attacks the carbonyl carbon atom of compound X.
Table 3:
Geometric parameters of the enzyme-inhibitor complexes predicted by molecular dockinga
| Compound | IC50 (µM) | d1 (Å) | α (degree) | d3 (Å) | d2 (Å) | L (Å)b |
|---|---|---|---|---|---|---|
| Xc | 0.015 | 2.601 | 117.0 | 3.302 | 3.218, 4.390 | 6.520 |
| 2d | 3.5 | 3.562 | 41.1 | 2.600 | 2.477, 3.562 | 5.077 |
| 2e | 10.8 | 3.342 | 117.23 | 3.535 | 5.355, 6.141 | 8.890 |
| 14c | 0.05 | 3.084 | 67.33 | 2.466 | 3.964, 5.421 | 6.430 |
| 14g | 0.015 | 4.364 | 117.9 | 3.007 | 4.143, 2.945 | 5.952 |
The geometric parameters important for formation of a Michaelis complex in the HNE active site are as reported previously [Crocetti et al., 2011], based on the model of synchronous proton transfer from the oxyanion hole in HNE [Groutas et al., 1997]. According to the docking results, a Michaelis complex with Ser195 is formed with participation of the ester carbonyl group.
Length of the channel for proton transfer calculated as d3 + min(d2).
Although compound 2e has distance d1 and angle α suitable for inhibitory activity, arrangement of the amino acid residues of the key triad Ser195-His57-Asp102 becomes unfavorable because the length L of the proton transfer channel is greatly increased (Table 3), which may be the main reason for the low inhibitory activity of compound 2e, despite the formation of H-bonds with Ser195 and Gly193 involving two nitrogen atoms (Figure 5A). Although compound 2d has a short length L, it has strong anchoring due to H-bonding of the carbonyl oxygen with Ser195 of the catalytic triad (not shown). Clearly, significant fixation of the molecule at the binding site leads to unfavorable activity (i.e., angle α is too acute for compound 2d, see Table 3). This may cause difficulties in formation of the Michaelis complex and, therefore, reduced activity of 2d. It should be noted that the cyano group at position 2 does not form H-bonds with the enzyme, in contrast with the same group at position 3 of molecule X.
Figure 5.

Docking poses of inhibitors 2e and 14g based on the pyrrolo[2,3-b]pyridine scaffold in the human neutrophil elastase binding site (1HNE entry of Protein Data Bank). Panel A. Docking pose of molecule 2e. Ser195 is H-bonded to amide nitrogen atom, while the cyano group forms a hydrogen bond with Gly193. Panel B. Docking pose of molecule 14g. The carbonyl oxygen atom forms a hydrogen bond with Val216. Residues within 4 Å from each pose are shown.
As for the highly active HNE inhibitors 14c and 14g, the geometric parameters of the docking poses, as well as the distance L, were favorable for the formation of Michaelis complexes and for proton transfer via the catalytic triad (Table 3). It should be noted that the angle α for compound 14c falls slightly out of the optimal interval 80–120° [Groutas et al., 1997; Vergely et al., 1996], which can explain the reduced inhibitory activity in comparison with inhibitor 14g. Molecules 14c and 14g are fixed within the HNE binding site by H-bonds in the above-mentioned orientations that are favorable for biological activity (Figure 5B). Thus, the geometrical parameters of the docking poses found with flexible side chains of the enzyme allow us to explain the differences in inhibitory activity of the investigated compounds.
CONCLUSIONS
In the present study, we further investigated the pyrrolo[2,3-b]pyridine scaffold, which was previously found to be suitable for the synthesis of HNE inhibitors (IC50=14–87 nM) [Crocetti et al., 2018]. While maintaining the best substituents identified for N-1 and position 3 (m-toluoyl and CN groups, respectively), we introduced substituents with different features at position 5 in order to assess the impact of these modifications on binding and HNE inhibition. Moreover, we also evaluated modification of the substituent at position 2 of the scaffold. The biological results clearly demonstrated that position 2 of the pyrrolo[2,3-b]pyridine scaffold must be unsubstituted, whereas the introduction of certain substituents at position 5 maintained HNE inhibitory activity comparable with that of reference compound X, probably by interacting with the large pocket of the enzyme site. According to our molecular modeling, the most active HNE inhibitors bind the enzyme in conformations suitable for the formation of a Michaelis complex between the ligand C=O group and Ser195. Additionally, for the potent inhibitors, a mutual orientation of the ligand with the catalytic triad is necessary for effective proton transfer from the oxyanion hole.
ACKNOWLEDGEMENTS
This research was supported in part by National Institutes of Health IDeA Program COBRE Grant GM110732; USDA National Institute of Food and Agriculture Hatch project 1009546; Montana University System Research Initiative: 51040-MUSRI2015–03; Tomsk Polytechnic University Competitiveness Enhancement Program; Ministry of Education and Science of the Russian Federation project No. 4.8192.2017/8.9, and the Montana State University Agricultural Experiment Station.
REFERENCES
- Bahekar RH, Jain MR, Jadavav PA, Projapati VM, Patel DN, Gupta AA, Sharma A, Tom R, Bandyopadhya D, Modi H. 2007. Synthesis and antidiabetic activity of 2,5-disubstituted-3-imidazol-2-yl-pyrrolo[2,3-b]pyridines. Bioorg Med Chem 15(21):6782–6795. [DOI] [PubMed] [Google Scholar]
- Baltus CB, Jorda R, Marat C, Berka C, Bazgier V, Krystal V, Priè G, Viad-Massuard MC. 2016. Synthesis, biological evaluation and molecular modeling of a novel series of 7-azaindole based tri-heterocyclic compounds as potent CDK2/ cyclin E inhibitors. Eur J Med Chem 108:701–719. [DOI] [PubMed] [Google Scholar]
- Bhat PV, Dere RT, Ravikumar S, Hindupur RM, Pati HN. 2015. Efficient and Scalable Process for Synthesis of 5-Nitro-7-azaindole. Org Process Res Dev 19:1282–1285. [Google Scholar]
- Chen G, Ren H, Zhang N, Lennox W, Turpoff A, Paget S, Li C, Almstead N, Njoroge FG, Gu Z. 2015. (Azaindol-2-yl)pyridine-3-sulfonamides as potent and selective inhibitors targeting hepatitis C virus NS4B. Bioorg Med Chem Lett 25:781–786. [DOI] [PubMed] [Google Scholar]
- Crocetti L, Giovannoni MP, Schepetkin IA, Quinn MT, Khlebnikov AI, Cilibrizzi A, Dal Piaz V, Graziano A, Vergelli C. 2011. Design, synthesis and evaluation of N-benzoylindazole derivatives and analogues as inhibitors of human neutrophil elastase. Bioorg Med Chem 19:4460–4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocetti L, Schepetkin IA, Ciciani G, Giovannoni MP, Guerrini G, Iacovone A, Khlebnikov AI, Kirpotina LN, Quinn MT, Vergelli C. 2016. Synthesis and farmacological evaluation of indole derivatives as deaza analogues of potent human neutrophil elastase inhibitors. Drug Dev Res 77 (6):285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocetti L, Schepetkin IA, Cilibrizzi A, Graziano A, Vergelli C, Giomi D, Khlebnikov AI, Quinn MT, Giovannoni MP. 2013. Optimization of N-benzoylindazole derivatives as inhibitors of human neutrophil elastase. J Med Chem 56:6259–6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocetti L, Giovannoni MP, Schepetkin IA, Quinn MT, Khlebnikov AI, Cantini N, Guerrini G, Iacovone A, Teodori E, Vergelli C. 2018. 1H-pyrrolo[2,3-b]pyridine: A new scaffold for human neutrophil elastase (HNE) inhibitor. Bioorg Med Chem doi.org/10.1016/j.bmc.2018.09.034. [DOI] [PubMed] [Google Scholar]
- Ermoli A, Bargiotti A, Brasca MG, Ciavolella A, Colombo N, Fachin G, Isacchi A, Menichincheri M, Molinari A, Montagnoli A, Pillan A, Rainoldi S, Sirtori FR, Sola F, Thieffine S; Tibolla M, Valsasina B, Volpi D, Santocanale C, Vanotti E. 2009. Cell division cycle 7 kinase inhibitors: 1H-pyrrolo[2,3-b]pyridines, synthesis and structure-activity relationships. Med Chem 52:4380–4390. [DOI] [PubMed] [Google Scholar]
- Fujinaga M, Chernaiaa MM, Halenbeck R, Koths K, James MNG. 1996. The Crystal Structure of PR3, a neutrophil serine proteinase antigen of Wegener’s granulomatosis antibodies. J Mol Biol 261:267–278. [DOI] [PubMed] [Google Scholar]
- Giovannoni MP, Schepetkin IA, Crocetti L, Ciciani G, Cilibrizzi A, Guerrini G, Khelebnikov AI, Quinn MI, Vergelli C. 2016. Cinnoline derivatives as human neutrophil elastase inhibitors. J Enz Inhib Med Chem 31(4):628–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni MP, Schepetkin IA, Quinn MT, Cantini N, Crocetti L, Guerrini G, Iacovone A, Paoli P, Rossi P, Bartolucci G, Menicatti M, Vergelli C. 2018. Synthesis, biological evaluation, and molecular modelling studies of potent human neutrophil elastase (HNE) inhibitors. J Enz Inhib Med Chem 33(1):1108–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graczyk P, Palmer V, Khan A. 2004. Preparation of 3-ethynyl or 3-cyano-1H-pyrrolo[2,3-b]pyridine derivatives as c-Jun N-terminal kinase (JNK) inhibitors. From PCT Int Appl WO 2004101565.
- Groutas WC, Kuang R, Venkataraman R, Epp JB, Ruan S, Prakash O. 1997. Structure-based design of a general class of mechanism-based inhibitors of the serine proteinases employing a novel amino acid-derived heterocyclic scaffold. Biochemistry 36:4739–4750. [DOI] [PubMed] [Google Scholar]
- Ibrahim PN, Artis DR, Bremer R, Habets G, Mamo S, Nespi M, Zhang C, Zhang J, Zhu Y-L, Zuckerman R, Krupka H, Kumar A, West B. 2007. Pyrrolo[2,3-b]pyridine derivatives as protein kinase inhibitors and their preparation, pharmaceutical compositions and use in the treatment of diseases. From PCT Int Appl WO 2007002433.
- Joydev KL, Rohan AB, Mandeep Kaur H. 2017. Intramolecular oxidative arylations in 7-azaindoles and pyrroles: revamping the synthesis of fused N-heterocycle tethered fluorenes. Chem Europ J 23:2044–2050. [DOI] [PubMed] [Google Scholar]
- Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. 2010. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev 62:726–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laha JK, Bhimpuria RA, Hunjan MK. 2017. Intramolecular Oxidative Arylations in 7-Azaindoles and Pyrroles: Revamping the Synthesis of Fused N-Heterocycle Tethered Fluorenes. Chem Europ J DOI: 10.1002/chem.201604192. [DOI] [PubMed] [Google Scholar]
- Liu N, Wang Y, Huang G, Ji C, Fan W, Li H, Cheng Y, Tian H. 2016. Design, synthesis and biological evaluation of 1H-pyrrolo[2,3-b]pyridine and 1H-pyrazolo[3,4-b]pyridine derivatives as c-Met inhibitors. Bioorg Chem 65:146–158. [DOI] [PubMed] [Google Scholar]
- Lucas SD, Costa E, Guedes RC, Moreira R. 2011. Targeting COPD: advances on low-molecular-weight inhibitors of human neutrophil elastase. Med Res Rev 33:E73–E101. [DOI] [PubMed] [Google Scholar]
- Navia MA, McKeever BM, Springer JP, Lin TY, Williams HR, Fluder EM, Dorn CP, Hoogsteen K. 1989. Structure of human neutrophil elastase in complex with a peptide chloromethyl ketone inhibitor at 1.84 Å resolution. Proc Natl Acad Sci USA 86:7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirogi R, Shinde A, Daulatabad A, Kambhampati R, Gudla P, Shaik M, Gampa M, Balasubramaniam S, Gangadasari P, Reballi V. 2012. Design, Synthesis, and Pharmacological Evaluation of Piperidin-4-yl amino aryl sulfonamides: Novel, Potent, Selective, Orally Active, and Brain Penetrant 5-HT6 Receptor Antagonists. J Med Chem 55:9255–9269. [DOI] [PubMed] [Google Scholar]
- Peters MB, Merz KM. 2006. Semiempirical comparative binding energy analysis (SECOMBINE) of a series of trypsin inhibitors. J Chem Theory Comput 2:383–399. [DOI] [PubMed] [Google Scholar]
- Pham CT. 2006. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol 6:541–550. [DOI] [PubMed] [Google Scholar]
- Pires MJD, Poeira DL, Purificacao SI, Marques MMB. 2016. Synthesis of substituted 4-, 5-, 6- and 7-azaindoles from aminopyridines via a cascade C-N cross-coupling/Heck Reaction. Org Lett 18(13):3250–3253. [DOI] [PubMed] [Google Scholar]
- Polverino E, Rosales-Mayor E, Dale GE, Dembowsky K, Torres A. 2017. The role of Neutrophil Elastase Inhibitors in Lung Diseases. Chest 152(2):249–262. [DOI] [PubMed] [Google Scholar]
- Sandham DA, Adcock C, Bala K, Barker L, Brown Z, Dubois G, Budd D, Cox B, Fairhust RA, Furegati M, Leblanc C, Manini J, Profit R, Reilly J, Stringer R, Shmidt A, Turner KL, Watson SJ, Willis J, Williams J, Wilson C. 2009. 7-Azaindole-3-acetic acid derivatives: potent and selective CRTh2 receptor antagonists. Bioorg Med Chem 19:4794–4798. [DOI] [PubMed] [Google Scholar]
- Schepetkin IA, Khlebnikov AI, Quinn MT. 2007. N-benzoylpyrazoles are novel small-molecule inhibitors of human neutrophil elastase. J Med Chem 50:4928–4938. [DOI] [PubMed] [Google Scholar]
- Singh U, Chashoo G, Khan SU, Mahajan P, Singh A, Sharma A, Mintoo MJ, Guru SK, Aruri H, Thatikonda T, Sahu P, Chibber P, Kumar V, Mir SA, Bharate SS, Madishetti S, Nandi U, Singh G, Mondhe DM, Bhushan S, Malik F, Mignani S, Vishwakarma RA, Singh PP. 2017. Design o novel 3-pyrimidinylazaindole CDK2/9 inhibitors with potent in vivo antitumor efficacy in a triple negative breast cancer model. J Med Chem 60(23):9470–9489. [DOI] [PubMed] [Google Scholar]
- Vergelli C, Schepetkin IA, Crocetti L, Iacovone A, Giovannoni MP, Guerrini G, Khlebnikov AI, Ciattini S, Ciciani G, Quinn MT. 2017. Isoxazol-5(2H)-one: a new scaffold for potent human neutrophil elastase (HNE) inhibitors. J Enz Inhib Med Chem 32(1):821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergely I, Laugaa P, Reboud-Ravaux M. 1996. Interaction of human leukocyte elastase with a N-aryl azetidinone suicide substrate: conformational analyses based on the mechanism of action of serine proteinases. J Mol Graph 14(3):158–167. [DOI] [PubMed] [Google Scholar]
- Von Nussbaum F, Li VMJ. 2015. Neutrophil elastase inhibitors for the treatment of (cardio)pulmonary diseases: Into clinical testing with pre-adaptive pharmacophores. Bioorg Med Chem Lett 25:4370–4381. [DOI] [PubMed] [Google Scholar]
- Von Nussbaum F, Li VMJ, Daniel Meibom D, Anlauf S, Bechem M, Delbeck M, Gerisch M, Harrenga A, Karthaus D, Lang D, Lustig K, Mittendorf J, Schäfer M, Schäfer S, Schamberger J. 2016. Potent and selective human neutrophil elastase inhibitors with novel equatorial ring topology: in vivo efficacy of the polar pyrimidopyridazine BAY-8040 in a pulmonary arterial hypertension rat model. ChemMedChem 11:199–206. [DOI] [PubMed] [Google Scholar]
- Xi N, Li X. 2014. Preparation of alkenyl-pyrrolo[2,3-b]pyridine compounds as protein tyrosine kinase modulators useful for treatment of proliferative disorders. From PCT Int Appl WO 2014193647.
- Zhong QQ, Wang X, Li YF, Peng LJ, Jiang ZS. 2017. Secretory leukocyte protease inhibitor promising protective roles in obesity-associated atherosclerosis. Exp Biol Med 242(3):250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]