

SUMMARY
An autoinhibitory network of hydrogen bonds located at the kinase hinge (referred to as the ‘molecular brake’) regulates the activity of several receptor tyrosine kinases. The mechanism whereby mutational disengagement of the brake allosterically activates the kinase in human disease is incompletely understood. We used a combination of NMR, bioinformatics, and molecular dynamics simulation to show that mutational disruption of the molecular brake triggers localized conformational perturbations that propagate to the active site. This entails changes in interactions of an isoleucine, one of three hydrophobic residues that lock the phenylalanine of the DFG motif in an inactive conformation. Structural analysis of tyrosine kinases provides evidence that this allosteric control mechanism is shared across the tyrosine kinase family. We also show that highly activating mutations at the brake diminish the enzyme’s thermostability, thereby explaining why these mutations cause milder skeletal syndromes compared to less activating mutations in the activation loop.
Keywords: FGF Receptor, Tyrosine Kinases, Allostery, Pathogenic Mutations, NMR Spectroscopy
Graphical Abstract
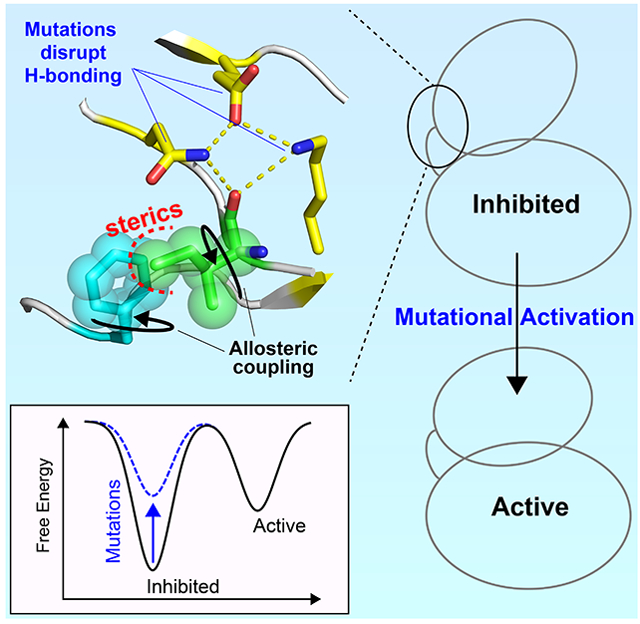
eTOC BLURP
Marsiglia et al. reveal that the kinase molecular brake is coupled to the DFG-motif through an allosteric pathway involving an isoleucine residue which locks the phenylalanine of the DFG-motif activation loop in an inactive conformation. Pathogenic mutations that target the molecular brake activate the enzyme by perturbing this allosteric network.
INTRODUCTION
Allostery plays a fundamental role in enzyme regulation and in the mediation of protein-protein, protein-ligand, and protein-nucleic acid interactions. Receptor tyrosine kinases (RTKs) constitute an essential class of allosteric enzymes involved in cell signaling pathways. Ligand binding on the extracellular domain of the RTK results in receptor dimerization, which enables intracellular transphosphorylation of activation-loop (A-loop) tyrosines within the kinase domain. These reactions activate the kinase and trigger the propagation of intracellular signals.
Naturally occurring human gain-of-function mutations within RTKs are common, and cause a multitude of diseases by enhancing the intrinsic kinase activity and thereby bypassing the need for A-loop tyrosine transphosphorylation (i.e., by uncoupling activation from extracellular ligand binding) (Katoh, 2019; Sharma et al., 2007). These mutations act by hijacking allosteric mechanisms that underlie physiological kinase activation (Chen et al., 2007; Chen et al., 2017). In doing so, these pathogenic mutations shift the kinase equilibrium toward the activated state (Chen et al., 2013; Chen et al., 2017). To map the allosteric pathway that controls kinase activation, we exploited a series of pathogenic mutations occurring at a triad of residues comprised of asparagine, glutamic acid, and lysine. The side chains of these three residues form a network of hydrogen bonds termed the ‘molecular brake’ (Chen et al., 2007), which is located distally to the nucleotide and substrate binding pockets. The molecular brake is conserved in FGFR, VEGFR, PDGFR, c-KIT, TIE, TEK, CSF1R, and FLT3 (Chen et al., 2007). Mutations at any of the three molecular brake residues within these RTKs lead to human growth disorders and cancers (Table S1) (Byron et al., 2013). Here we show that these mutations alter the composition of a regulatory hydrogen bonding network at the molecular brake. This in turn leads to changes in the side chain orientation of a phenylalanine residue in the highly conserved DFG motif known to be a critical determinant of kinase activation (Chen et al., 2017; Kornev et al., 2006). These coupled conformational events provide a mechanistic understanding of an allosteric shift toward the catalytically competent active state (Ahuja et al., 2017; Chen et al., 2013; Saleh et al., 2017; Srivastava et al., 2014).
RESULTS AND DISCUSSION
Pathogenic Mutants at the Molecular Brake Destabilize the Kinase Fold
Previous work from our group used tyrosine kinases from the fibroblast growth factor receptor (FGFR) family of RTKs to identify four regions that form a long-range allosteric network which relays conformational changes between the A-loop to the kinase hinge located between the N- and C-lobes of the enzyme. These regions are: 1) the A-loop plug, 2) the αC tether, 3) the DFG latch, and 4) the molecular brake (Chen et al., 2017). Disruption of hydrogen bonds among N549, E565, and K641 at the molecular brake of FGFR2 kinase (FGFR2K) causes a conformational shift from an autoinhibited to an activated state (Fig. 1a) (Chen et al., 2007). To understand the mechanism driving this population shift (Chen et al., 2013; Chen et al., 2007; Chen et al., 2017; Huang et al., 2013; Nussinov et al., 2014), we used differential scanning calorimetry to assess the thermal stability of FGFR2K mutants harboring pathogenic mutations at each of the three molecular brake residues (i.e., N549K, E565A, K641R). Unphosphorylated and A-loop phosphorylated wild-type FGFR2K and the FGFR2K K659E mutant were used as controls. The latter mutation, which maps onto the kinase A-loop, introduces intramolecular hydrogen bonds that stabilize the kinase active state, thereby mimicking the action of A-loop tyrosine phosphorylation (Chen et al., 2013). Thermogram data showed that N549K, E565A, K641R mutants all had diminished melting temperatures, implying a mutation-induced destabilizing effect (Fig. 1b). Similar to N549K of FGFR2K, we also observed a reduction in the melting temperature for the analogous mutation (N535K) of FGFR4K (Fig. S1), which supports the conclusion that mutations at the molecular brake for other FGFRK isoforms induce a similar effect. In contrast to the reduced thermal stability for molecular brake mutants, the pathogenic K659E mutant and the A-loop phosphorylated FGFR2K displayed melting temperatures similar to that of the unphosphorylated kinase (Fig. S1). We conclude that in contrast to the K659E mutation, N549K, E565A, and K641R elevate the lowest energy conformation within the kinase free energy landscape, thereby facilitating the shift in equilibrium toward the active conformation (Fig. 1c). Importantly, the diminished thermal stability of the E565A enzyme explains why this mutation, in spite of imparting greater kinase activation compared to A-loop mutants, leads to a clinically milder form of skeletal disorder (Chen et al., 2017).
Figure 1. Gain-of-function mutations at the molecular brake destabilize the inhibited conformation.

(a) Left: structural view depicting engaged molecular brake as observed in the inhibited FGFR1 kinase (PDB: 3KY2); note that numbering follows that of FGFR2K. Right: disengaged molecular brake as seen in the A-loop tyrosine phosphorylated activated FGFR2K (PDB: 2PVF). Kinase molecule is shown as gray cartoon; αC-β4 loop, kinase hinge, and β8 strand contributing residues to the molecular brake are highlighted in red, blue and green, respectively. The triad of residues (i.e., N549, E565 and K641) comprising the molecular brake are shown as green sticks. Oxygen and nitrogen atoms are colored red and blue, respectively; hydrogen bonds are denoted by dashed yellow lines. (b) Differential scanning calorimetry thermogram data comparing thermal stabilities of wild-type FGFR2K and molecular brake mutants (i.e., N549K, E565A, and K641R). Note that molecular brake mutants have reduced melting temperatures relative to the wild-type FGFR2 kinase. A complete list of melting temperatures is shown in Table 1. (c) Schematic of free energy landscape in wild-type (black) and molecular brake mutants (dashed red). The red arrow highlights that molecular brake mutations elevate the basal free energy of the kinase; i.e., they destabilize the inhibited state.
Molecular Brake Mutation Increases Conformational Disorder Near the DFG Phenylalanine
To delve deeper into the mechanism whereby molecular brake mutations destabilize the inhibited conformation, we used NMR spectroscopy, a technique capable of detecting site-specific protein dynamics under native conditions (Boehr et al., 2006; Kay, 2016; Kumar et al., 2018; Manley and Loria, 2012). Specifically, we carried out 1H-1H dipolar cross-correlated relaxation rate (η) experiments in order to calculate generalized order parameters (S2) for regions spanning throughout the kinase structure (Sun et al., 2011). The S2 value ranges from zero to one and reflects fully disordered or ordered regions, respectively. To perform these measurements, we isotopically enriched wild-type FGFR2K and the most enzymatically active molecular brake mutant (i.e., E565A) with 13C at isoleucine, leucine, and valine in their methyl groups (ILV labeling (Rosen et al., 1996)). The S2 and η values for both samples are presented in Fig. 2a and Table S2. Analysis of these values shows that these measurements could distinguish flexible loops from the rigid core of the kinase (Fig. S2). For example, the majority of N- and C-lobe residues involved in core hydrophobic packing such as V562 (in the β5 sheet), I696 (in the αF helix), and I760 (in the αI helix) displayed S2 values above 0.6, indicative of order on a nanosecond timescale (Fig. 2a). On the other hand, loop residues such as I503 (in the β2-β3 loop), I541 (in the αC-β4 loop), and I651 and I654 (both in the A-loop) showed reduced order (i.e., S2 values < 0.4), thus validating the suitability of our NMR approach for gauging differences in conformational dynamics induced upon mutation at the molecular brake.
Figure 2. The E565A mutation increases localized conformational dynamics and induces allosteric changes to residues surrounding the phenylalanine within the DFG motif.

(a) Comparison of generalized order parameters (S2; left axis) and 1H-1H cross-correlated relaxation rates (η; right axis) for wild-type (blue circles) and E565A (red squares). Error bars denote the error of fitted η values used to calculate S2. (b) Difference plot of S2 values (ΔS2) from panel (a) calculated by subtracting E565A values from those of wild-type FGFR2K. Note that compared to the C-lobe, several N-lobe residues display ΔS2 values significantly greater than zero. (c) ΔS2 values are mapped on corresponding methyl carbon atoms as spheres onto the inhibited structure of FGFRK (PDB ID:3KY2) represented in gray cartoon. The inset shows a zoomed-in view of the DFG latch. The range of ΔS2 is denoted by a colored boxed bar. Note the decrease in order for the methyl groups of I541 and L617 in E565E which are directly surrounding the phenylalanine from the DFG latch motif.
To visualize changes in intrinsic dynamics between wild-type FGFR2K and the E565A mutant, differences in S2 values (ΔS2) were mapped onto the inhibited FGFR1K crystal structure (PDB ID: 3KY2) (Fig. 2b, c). This plot revealed that the majority of residues in the C-lobe showed no significant differences, implying that the C-lobe is unaffected by the molecular brake mutation. However, the S2 values for several sites within the N-lobe were significantly reduced in E565A relative to the wild-type enzyme. Specifically, we observed that residues in close vicinity to the E565A mutation had ΔS2 values that were significantly reduced (e.g., V512, L551, V564), suggesting an increase in local dynamics on the nanosecond timescale. In addition, we also observed that two constituents surrounding the phenylalanine from the hydrophobic DFG latch, namely I541 and L617, incurred significant reductions in S2 values (Fig. 2c). These residues, together with I547, are involved in locking the phenylalanine within the DFG motif in an inactive conformation (Chen et al., 2017). We conclude that the loss of hydrogen bonding seen in the E565A crystal structure leads to increased conformational flexibility around the DFG motif phenylalanine residue as well as a reduction in thermal stability observed using DSC (Fig. 1b).
To map the allosteric trajectory, we further investigated high-resolution crystal structures of active and inhibited FGFRKs (PDB IDs: 3KY2-inhibited and 2PVF-active). Our analysis revealed that I547 undergoes two major changes in its interaction patterns. First, hydrogen bonding distances between the carbonyl oxygen of I547 with both the side chains of N549 and K641 increase upon transition to the active conformation: N549 to I547 is 3.0 Å in the inhibited conformation and 6.9 Å in the active conformation, while K641 to I547 is 3.3 Å in the inhibited conformation and 4.6 Å in the active conformation (Fig. 3a). Second, the chi1 angle of the I547 side chain switches from a positive gauche conformation (59°) in the inhibited state to a negative gauche conformation (−68°) in the active form. These changes are accompanied by a difference in the F645 chi1 dihedral angle from 56° in the inhibited state to −69° in the activated state. Based on these coupled changes between molecular brake residues and I547 and the changes in I547 and F645 chi1 angles, we infer that I547 acts as a key intermediary of allosteric communication between the molecular brake and the DFG latch.
Figure 3. Molecular dynamics simulations support the existence of allostery between the molecular brake and the DFG latch mediated by I547.

(a) Cartoon representation of crystal structures of FGFRKs in the inhibited (left; PDB ID: 3KY2) and activated forms (right; PDB ID: 2PVF) showing that hydrogen bonding (dotted lines) between side chains of N549 and K641 with the carbonyl oxygen of I547 in the inhibited form becomes disrupted upon activation. These structural views also display the change in the DFG phenylalanine (F645) side chain orientation between the inhibited and active forms of the enzyme that correlates with the hydrogen bonding at the molecular brake. (b, c) Analysis of chi1 dihedral angles and hydrogen bonds from molecular dynamics simulations carried out on wild-type FGFR1K (b) and E565A (c). The left panels in (b) and (c) display chi1 dihedral angles for F645 as a function of time for each of the three replica simulations. Dotted lines correspond to dihedral angles for the inhibited (~60°) and active forms (~−60°) of the enzyme and match those of the structural views in panel (a). The upper right panel within (b) and (c) shows calculated chi1 dihedral angles for F645 and I547 throughout the molecular dynamics simulations for wild-type FGFR1K and E565A. Darker regions of the plot indicate a greater population of configurations present within the simulations. In panel (c), the green circles correspond to the dihedral angles of the inhibited (PDB ID: 3KY2) and active form of FGFRK (PDB ID: 2PVF). The bottom right panel of (b) and (c) shows the number of hydrogen bonds formed by the side chains of K641 and N549 with the backbone carbonyl of I547 for structural snapshots of the molecular dynamics simulation that correspond to the inhibited, active, or either of the intermediate forms. Colors of red, green, and blue correspond to zero, one, and two hydrogen bonds, where a hydrogen bond is defined at a distance of ≤ 3.5 Å.
Allosteric Coupling Involves a Hydrogen Bond Interaction Pathway
To gain further insight into the coupling mechanism between the molecular brake and the DFG latch, we utilized all-atom molecular dynamics simulations starting from an inhibited FGFR1K crystal structure bound to ATP and Mg2+ (PDB ID: 3KY2). Simulations were also carried out for E565A (Chen et al., 2017). We selected the FGFR1 isoform structure because we previously found that the A-loop conformation in the inhibited FGFR2K crystal structure was influenced by crystal packing, and thus did not represent an authentically inhibited kinase(Chen et al., 2017). Molecular dynamics were simulated for 200 nsec using three separate replicates, giving a total sampling time of 600 nsec for wild-type and the E565A mutant. The root mean squared deviation (RMSD) for heavy atoms (CA, C’, N, O) within structured regions of the kinase indicated stable trajectories for all replica runs (Fig. S3a). To correlate simulation data with the X-ray crystallographic and NMR data, we evaluated conformational fluctuations around the DFG latch by calculating the chi1 angle of the DFG phenylalanine throughout each trajectory (Fig. 3b and 3c). These plots show that E565A had a greater number of chi1 rotamer switches compared to the wild-type kinase. Specifically, E565A underwent dihedral angle changes between approximately 60° and −60°, in line with our analysis of crystal structures corresponding to inhibited and active states of positive gauche and negative gauche, respectively. These findings are consistent with our NMR spectroscopic data showing a decrease in order parameters around the DFG latch, thus reinforcing the existence of allosteric coupling between the molecular brake and DFG latch.
To further characterize the coupling mechanism between the molecular brake and the DFG latch regions, we quantified the number of hydrogen bonds formed by K641 and N549 with the carbonyl backbone of I547 and analyzed the chi1 angles of I547 in addition to those detailed above for F645. The simulation results for the wild-type enzyme revealed that the chi1 angles of F645 and I547 predominantly sampled the inhibited rotamer conformation (78.7%), whereas the active rotamers constituted only a minor population (3.7%). The remaining population (intermediates 1 and 2) showed mixed characteristics in which either the F645 rotamer resembled that of the inhibited state and I547 resembled that of the active state (15.3%), or F645 in the active state and I547 in the inhibited state (2.3%) (Fig. 3b). The extent of hydrogen-bonding indicated that the molecular brake was primarily intact, as evidenced by the high probability of two hydrogen bonds between the carbonyl oxygen of I547 and the side chains of N549 and K641 (Fig. 3b).
In stark contrast to the case of the wild-type enzyme, the corresponding populations of the E565A mutant for the inhibited and active forms were reversed: 7.8% populated the inhibited conformation and 49.8% populated the active state (Fig. 3c). Intermediate states were also present in which active F645 and inhibited I547 chi1 angles comprised 16.3%, while the opposite conformational states had a population of 26.1%. The higher percentage of intermediate rotamer states implies greater flexibility of the E565A mutant at the molecular brake region relative to the wild-type kinase. The E565A mutation also imparted larger perturbations on the hydrogen bond network of the molecular brake, as anticipated from loss of the salt-bridge involving this glutamic acid in the wild-type kinase (Fig. 3c). We conclude that E565A weakens hydrogen bonding interactions at the molecular brake, thereby lowering the free energy barrier for chi1 rotameric changes of I547 and F645 (Fig. 1c).
It is noteworthy that the overall RMSD of structured and unstructured regions in both wild-type and E565A simulations remained relatively small over the total simulation time of 600 nsec (Fig. S3a). While we observed local conformational fluctuations around the DFG latch and at the molecular brake (Fig. S3b), we found no evidence for transitioning of the A-loop to an active configuration (Fig. S3c). It has been reported that simulations on the millisecond timescale are required to observe A-loop conformational changes in EGFR kinase (Shan et al., 2013). For this reason, conformational changes of I547 and F645 were considered to be active-like transitions and may not represent a global transition to the active state. This is consistent with our previously reported activity data showing subtle cooperativity between the DFG-latch with the A-loop or the molecular brake with the A-loop (Chen et al., 2017). Namely, double mutations at the DFG-latch and A-loop or at the molecular brake and A-loop produced an additive effect of individual mutations (Chen et al., 2017). In contrast, combined mutations at the DFG-latch and molecular brake produce no greater activity than each mutation alone. Taken together, these observations support conformational coupling via a defined allosteric pathway involving hydrogen bonding of I547 to the molecular brake, which directly influences the orientation of the phenylalanine from the DFG latch.
Conservation of Allosteric Pathway in Other Tyrosine Kinases
To determine whether our allosteric coupling pathway between activated and inhibited kinase conformations in the FGFR family is applicable to other kinases, we analyzed crystal structures for tyrosine kinases containing isoleucine and phenylalanine residues at the analogous positions to I547 and F645 within FGFR2K. Because of the known influence of ATP-competitive inhibitors on the DFG conformation (DFGin or DFGout) (Muller et al., 2015), we focused on drug-free kinase structures. In addition, we included only those tyrosine kinases with glutamic acid and lysine residues corresponding to E565 and K641 of FGFR2K; note that these account for >85% of all tyrosine kinases (Fig. S4a). The calculated chi1 angle plots for phenylalanine and isoleucine residues showed clustering of structures with inhibited and activated side chain dihedral angles similar to those in the FGFRK family (Fig. 4). Specifically, out of 101 structures analyzed, 14 had chi1 angles that clustered in the inhibited conformation, while 80 were found within the active conformation. The remaining seven kinase structures had mixed characteristics in which the chi1 angle for the phenylalanine of the DFG motif was observed in an active conformation and the isoleucine was in an inhibited conformation. Notably, in these latter structures, the phenylalanine conformation may be biased because of the tightly bound sulfate ion by A-loop residues.
Figure 4. A conserved allosteric mechanism connecting the molecular brake and DFG latch.

Plot of chi1 angles calculated for apo or nucleotide bound tyrosine kinase coordinates (total of 101) taken from the PDB. These kinases share the analogous I547 and F645 positions present in FGFR2K. Green circles indicate that the analogous K641 sidechain-I547 carbonyl bond distance is less than 3.5 Å. Red circles indicate that the analogous K641 sidechain-I547 carbonyl bond distance is less than 3.5 Å. Large circles highlight clustering of structures in inhibited (total = 14) and active (total = 80) conformations. A tabulated list of kinases and corresponding PDB IDs is given in Table S3.
We also measured bond distances between the carbonyl oxygen of the analogous I547 residue and the side chain nitrogen at the analogous K641 position to further explore correspondence of the DFG latch orientation with hydrogen bonding at the molecular brake: longer bond distances would imply disrupted hydrogen bonding at the molecular brake, while shorter distances would be indicative of intact hydrogen bonding. Remarkably, we found that 12 of 14 inhibited structures had bond distances less than 3.5 Å, while 73 of 80 structures had bond distances greater than 3.5 Å. This dichotomy suggests a general activation mechanism that connects hydrogen bonding at the molecular brake to the rotamer orientation of the phenylalanine within the DFG motif. We note that some of the tyrosine kinases in this analysis do not share the asparagine position of the molecular brake conserved in FGFR, VEGFR, PDGFR, c-KIT, TIE, TEK, CSF1R, and FLT3. However, these kinases do have a hydrogen bonding pattern that resembles those interactions identified as part of the molecular brake (Chen et al., 2007). In particular, their structures show a conserved hydrogen bond from the backbone nitrogen of the analogous N549 position to the side chain carboxylate of E565 (Fig. S4b, c). This observation coupled with the high degree of conservation at the analogous E565 and K641 positions, implies that the molecular brake is a generalized regulatory element within the tyrosine kinase subfamily (Molina-Vila et al., 2014).
Our experimental and computational data lead us to conclude that mutations at the molecular brake impart local disorder, which is transmitted to the DFG latch via a change in the backbone hydrogen bonding distance to I547. The loss of hydrogen bonding alters the I547 side chain orientation which in turn directly influences the side chain conformation of the phenylalanine from the DFG motif. Thus, mutations at the molecular brake operate via an allosteric propagation mechanism in which there is a cooperative conformational transition from the site of mutation to the DFG latch. The timescale of A-loop motion is too slow to be captured using nanosecond MD simulations described in this work. Nevertheless, NMR chemical shift perturbations on A-loop residues I651 and I654 (Fig. S5) suggest that molecular brake mutants can sample the active state of the DFG latch component of the allosteric network implying that they can indeed populate the active state. This is evidenced by the remarkable consistency of active and inhibited A-loop conformations with the corresponding F645 and I547 chi1 angles observed in crystal structures where the A-loop has ample time to sample different conformations during the crystallization process.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by Nathaniel J. Traaseth (traaseth@nyu.edu)
Preparation of FGFR Kinase Samples
cDNA fragments encoding residues P458 to E768 of human FGFR2c (Accession code: P21802-1) and residues L445 to E753 of human FGFR4 (Accession code: P22455-1) were amplified by PCR and sub-cloned into a pET bacterial expression vector with an N-terminal 6x-His-tag. Note that wild-type FGFR2 kinase was mutated at C491 to an alanine; this functional mutant was used for all subsequent site-directed mutants. All pathogenic mutations at the molecular brake were introduced using the QuikChange site-directed mutagenesis kit (Stratagene). The E. coli strain BL21(DE3) cells were transformed with the expression constructs, and kinase expression was induced with 0.1 mM (FGFR2K) or 1.0 mM (FGFR4K) isopropyl-L-thio-B-D-galactopyranoside overnight at 20 °C. The cells were lysed, and the soluble kinase proteins were purified using nickel affinity chromatography as previously described (Chen et al., 2007; Chen et al., 2017). Trace amounts of phosphorylation from the bacterial expression were removed by treating the kinases with FastAP™ Thermosensitive Alkaline Phosphatase (Thermo Scientific) and subsequently purified by anion exchange chromatography (Source 15Q, GE Healthcare Life Sciences) and gel filtration chromatography (Superdex 200 Increase 10/300GL, GE Healthcare Life Sciences).
Measurement of Order Parameters using NMR Spectroscopy
1H-1H dipolar cross-correlated relaxation rates (η) were calculated from the intensities of the triple (forbidden) and single (allowed) quantum transition experiments as previously described (Sun et al., 2011). The relaxation delays (T) used for the triple and single quantum measurements were 1.25, 2.5, 5, 7.5, 10, 12, 15, and 18 msec. The forbidden experiment was acquired using 24 scans, while the allowed experiment was acquired using 16 scans. To correct for differences in peak heights due to unequal scans between the two experiments, the peak heights obtained from the forbidden experiment were multiplied by a factor of (16/24). Intensity ratios for each relaxation delay (Iforbidden/Iallowed) were fit to determine the 1H-1H dipolar cross-correlated relaxation rate using equation (3):
| (3) |
C is 0.75 and δ compensates for relaxation effects arising from external protons; the latter was a floatable parameter in the fit. The acquisition parameters for 2D experiments utilized direct (1H) and indirect (13C) spectral widths of 10000 and 3018 Hz and acquisition times of 60.0 msec and 19.6 msec, respectively. All measurements were performed in duplicate for FGFR2 kinase and E565A, which were 0.5 mM in concentration and prepared uniformly 15N labeled with –CH3 groups labeled with 13C at Ile (Cδ1), Leu (Cδ1/δ2), and Val (Cγ1/γ2) in a perdeuterated background (referred to as ILV labeling). All experiments were carried out at 10 °C. Data were processed in NMRPipe (Delaglio et al., 1995) and analyzed in Sparky (Goddard and Kneller)
To calculate order parameters (S2) from the fitted η values in equation (3), we measured correlation times for wild-type and E565A kinases using the TRACT method (Lee et al., 2006). Datasets measuring the relaxation rates of the TROSY and anti-TROSY components were acquired in an interleaved fashion with relaxation delays from 0 to 200 msec in in 4 msec intervals. Relaxation curves were fit to an exponential decay to extract the relaxation rate of each TROSY component and the correlation time was determined as previously described (Lee et al., 2006). The correlation time (τC) was determined to be 27 nsec for both wild-type and mutant FGFR2Ks.
Order parameters (S2) were calculated using the following equation (Sun et al., 2011):
| (4) |
where ℏ is the reduced Plank’s constant, rHH is the distance between methyl group protons (1.813 Å), and P2(cosθaxis,HH) was set to −0.5 (Tugarinov et al., 2007). Errors in Figure 2 reflect non-linear least square fitting errors from datasets acquired in duplicate.
Differential Scanning Calorimetry
All measurements were performed on a NanoDSC (TA Instruments) at a scanning rate of 60 °C/hr from 10 °C to 70 °C. Kinase concentrations were 20 or 80 μM in 25 mM HEPES, 150 mM NaCl, pH 7.5. For AMP-PNP measurements, the concentrations of protein, AMPPNP, and magnesium chloride were 80 μM, 5 mM, and 10 mM, respectively. For phosphorylated samples, 1 mM EDTA was included in the DSC buffer to chelate Mg2+. Melting temperatures (Tm) were fit using the NanoAnalyze software (TA instruments).
Computational Details
Molecular dynamics simulations were performed using the AMBER 16.06 package with the ff14SB force field for the protein and TIP3P water model. Force fields developed for ATP(Meagher et al., 2003) and Mg2+ (Allner et al., 2012) were used in the nucleotide bound simulations. The initial structure for molecular dynamics simulations of wild-type enzyme was modeled based on the inhibited FGFR1 kinase structure 3KY2, with missing loops repaired using Modeller (Webb and Sali, 2017). The initial structure for the molecular brake mutant was obtained by mutating E565 into alanine. Wild-type and E565A mutant systems were neutralized using three and two Na+ ions, respectively, and were solvated using a cubic TIP3P water box with a 15 A buffer region between protein and boundary.
Each prepared simulation system was equilibrated using repeated steps of minimization and restrained dynamics. The solvated complex was initially minimized using a conjugate gradient minimization for 2000 steps followed by a 200 psec constant volume simulation at 300K with a 500 kcal/mol restraint on the crystal waters, cofactors, and the heavy atoms of the protein. Another round of conjugate gradient minimization for 2000 steps were performed using the same restraints followed by another round of minimization but with reduced restraints of 50 kcal/mol. A series of 200 psec constant volume simulations were performed with decreasing restraints from 50, 10, and 2 kcal/mol at 300 K. Lastly, two constant pressure simulations were run for 200 psec each at 300K with decreasing restraints of 2 and 0.5 kcal/mol. Finally, unrestrained production runs were initiated from the density equilibrated simulations with snapshots being collected every 10 psec. For all simulations, time steps were set at 2 fsec, with SHAKE constraints, particle mesh-Ewald (Salomon-Ferrer et al., 2013) for electrostatics and a 12 Å cut-off for non-bonded interactions.
Quantification of Statistical Detail
All experimental DSC and NMR order parameter measurements were performed in duplicate. All listed and plotted points represent the average of two experiments; error in the DSC measurements reflect the standard deviation while errors associated with η and S2 values represent the errors of the fit. The fits to equation (3) for the NMR order parameter measurements were performed in Prism 7 (Graphpad).
Supplementary Material
Table 1.
Summary of melting temperatures measured using differential scanning calorimetry in the presence and absence of the ATP mimic AMPPNP. The standard deviations from duplicate measurements are less than or equal to 0.4 °C.
| FGFR Isoform | Form/Mutation | AMPPNP | Tm (°C) |
|---|---|---|---|
| FGFR2 | WT | + | 53.4 |
| N549K | + | 50.0 | |
| K641R | + | 45.5 | |
| E565A | + | 43.9 | |
| K659E | + | 54.2 | |
| WT | − | 51.8 | |
| Y656/Y657a | − | 52.1 | |
| pY656/pY657a | − | 51.1 | |
| N549K | − | 45.7 | |
| K641R | − | 41.9 | |
| E565A | − | 40.1 | |
| K659E | − | 51.8 | |
| FGFR4 | WT | − | 47.7 |
| N535Kb | − | 41.7 |
Y656/Y657 and pY656/pY657 constructs correspond to unphosphorylated and phosphorylated versions of an FGFR2K domain, respectively. These kinases have only two phosphorylatable tyrosines which are located in the A-loop (i.e. a triple mutant of Y466F, Y586L, and Y588S).
The N535K mutation in FGFR4K is homologous to the N549K mutation in FGFR2K.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. coli BL21(DE3) competent cells | Sigma-Aldrich | CMC0014 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| HEPES | RPI corp | H75030-250.0 |
| Sodium chloride | RPI corp | S23020-5000.0 |
| isopropyl-L-thio-B-D-galactopyranoside | Gold biotech | I2481C50 |
| 4 mm shigemi tubes | Cortecnet | BMS-004B |
| D2O | Cambridge Isotopes | DLM-4-99.8-1L |
| QuikChange | Agilent | 200555 |
| AMP-PNP | Sigma | 10102547001 |
| Magnesium chloride | Sigma | M8266 |
| FastAP™ | Thermo Scientific | FEREF0652 |
| Recombinant DNA | ||
| pETDUET-FGFR2K (WT/N549K/E565A/K641R) (458-768) | Chen, H., Ma, J., Li, W., Eliseenkova, A.V., Xu, C., Neubert, T.A., Miller, W.T., and Mohammadi, M. (2007). A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol Cell 27, 717-730. 10.1016/j.molcel.2007.06.028 | |
| pETDUET-FGFR4K (WT) (445-753) | Huang, Z., Tan, L., Wang, H., Liu, Y., Blais, S., Deng, J., Neubert, T.A., Gray, N.S., Li, X., and Mohammadi, M. (2014). DFG-out Mode of Inhibition by an Irreversible Type-1 Inhibitor Capable of Overcoming Gate-Keeper Mutations in FGF Receptors. ACS Chemical Biology 10, 299-309. 10.1021/cb500674s | |
| Software and Algorithms | ||
| Pymol | pymol.org | |
| AMBER 16.06 package | ambermd.org | |
| Modeller | Webb, B., and Sali, A. (2017). Protein Structure Modeling with MODELLER. Methods Mol Biol 1654, 39-54. 10.1007/978-1-4939-7231-9_4 | |
| Cosmic database | cancer.sanger.ac.uk/cosmic | |
| NanoAnalyze | TA Instruments https://www.tainstruments.com/support/software-downloads-support/downloads/ | |
| Other | ||
| Differential Scanning Calorimeter | TA instruments | NYU shared instrumentation facility |
| 600 MHz NMR instrument with a triple resonance CPTCI-cryoprobe (1H, 13C, 15N) | Bruker | NYU shared instrumentation facility |
| Source 15Q resin | GE Healthcare | 17094701 |
| Superdex 200 Increase 10/300GL | GE Healthcare | 28990944 |
HIGHLIGHTS.
There is an allosteric pathway between the DFG-motif and the molecular brake
An isoleucine near the DFG-motif serves as a conduit of allostery
Pathological mutations at the molecular brake perturb the allosteric network
Molecular brake mutants decrease kinase thermal stability
ACKNOWLEDGMENTS
This work was supported by NIH grants R01GM117118 (to N.J.T. and M.M.), R01DE13686 (to M.M.), and R35GM127040 (to Y.Z.). W.M.M. acknowledges NIH predoctoral funding from an F99/K00 award (F99CA212474). The NMR data collected with a cryoprobe at NYU was supported by an NIH S10 grant (OD016343). Data collected at the New York Structural Biology Center was made possible by a grant from NYSTAR. The authors also acknowledge support from NYU-ITS for providing computational resources and Prof. Donghan Lee for sharing a TRACT pulse sequence.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahuja LG, Kornev AP, McClendon CL, Veglia G, and Taylor SS (2017). Mutation of a kinase allosteric node uncouples dynamics linked to phosphotransfer. Proc Natl Acad Sci U S A 114, E931–E940. 10.1073/pnas.1620667114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allner O, Nilsson L, and Villa A (2012). Magnesium Ion-Water Coordination and Exchange in Biomolecular Simulations. J Chem Theory Comput 8, 1493–1502. 10.1021/ct3000734 [DOI] [PubMed] [Google Scholar]
- Boehr DD, Dyson HJ, and Wright PE (2006). An NMR perspective on enzyme dynamics. Chemical reviews 106, 3055–3079. 10.1021/cr050312q [DOI] [PubMed] [Google Scholar]
- Byron SA, Chen H, Wortmann A, Loch D, Gartside MG, Dehkhoda F, Blais SP, Neubert TA, Mohammadi M, and Pollock PM (2013). The N550K/H mutations in FGFR2 confer differential resistance to PD173074, dovitinib, and ponatinib ATP-competitive inhibitors. Neoplasia 15, 975–988. 10.1593/neo.121106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Huang Z, Dutta K, Blais S, Neubert TA, Li X, Cowburn D, Traaseth NJ, and Mohammadi M (2013). Cracking the molecular origin of intrinsic tyrosine kinase activity through analysis of pathogenic gain-of-function mutations. Cell reports 4, 376–384. 10.1016/j.celrep.2013.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ma J, Li W, Eliseenkova AV, Xu C, Neubert TA, Miller WT, and Mohammadi M (2007). A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol Cell 27, 717–730. 10.1016/j.molcel.2007.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Marsiglia WM, Cho MK, Huang Z, Deng J, Blais SP, Gai W, Bhattacharya S, Neubert TA, Traaseth NJ, et al. (2017). Elucidation of a four-site allosteric network in fibroblast growth factor receptor tyrosine kinases. Elife 6 10.7554/eLife.21137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, and Bax A (1995). NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6, 277–293. 10.1007/BF00197809 [DOI] [PubMed] [Google Scholar]
- Goddard TD, and Kneller DG SPARKY 3. University of California, San Francisco. [Google Scholar]
- Huang Z, Chen H, Blais S, Neubert TA, Li X, and Mohammadi M (2013). Structural mimicry of a-loop tyrosine phosphorylation by a pathogenic FGF receptor 3 mutation. Structure 21, 1889–1896. 10.1016/j.str.2013.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh M (2019). Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat Rev Clin Oncol 16, 105–122. 10.1038/s41571-018-0115-y [DOI] [PubMed] [Google Scholar]
- Kay LE (2016). New Views of Functionally Dynamic Proteins by Solution NMR Spectroscopy. J Mol Biol 428, 323–331. 10.1016/j.jmb.2015.11.028 [DOI] [PubMed] [Google Scholar]
- Kornev AP, Haste NM, Taylor SS, and Eyck LF (2006). Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc Natl Acad Sci U S A 103, 17783–17788. 10.1073/pnas.0607656103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar GS, Clarkson MW, Kunze,Granata D, Wand AJ, Lindorff-Larsen K, Page R, and Peti W (2018). Dynamic activation and regulation of the mitogen-activated protein kinase p38. Proc Natl Acad Sci U S A 115, 4655–4660. 10.1073/pnas.1721441115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Hilty C, Wider G, and Wuthrich K (2006). Effective rotational correlation times of proteins from NMR relaxation interference. Journal of Magnetic Resonance 178, 72–76. 10.1016/j.jmr.2005.08.014 [DOI] [PubMed] [Google Scholar]
- Manley G, and Loria JP (2012). NMR insights into protein allostery. Arch Biochem Biophys 519, 223–231. 10.1016/j.abb.2011.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meagher KL, Redman LT, and Carlson HA (2003). Development of polyphosphate parameters for use with the AMBER force field. J Comput Chem 24, 1016–1025. 10.1002/jcc.10262 [DOI] [PubMed] [Google Scholar]
- Molina-Vila MA, Nabau-Moreto N, Tornador C, Sabnis AJ, Rosell R, Estivill X, Bivona TG, and Marino-Buslje C (2014). Activating mutations cluster in the "molecular brake" regions of protein kinases and do not associate with conserved or catalytic residues. Hum Mutat 35, 318–328. 10.1002/humu.22493 [DOI] [PubMed] [Google Scholar]
- Muller S, Chaikuad A, Gray NS, and Knapp S (2015). The ins and outs of selective kinase inhibitor development. Nature chemical biology 11, 818–821. 10.1038/nchembio.1938 [DOI] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, and Liu J (2014). Principles of allosteric interactions in cell signaling. J Am Chem Soc 136, 17692–17701. 10.1021/ja510028c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen MK, Gardner KH, Willis RC, Parris WE, Pawson T, and Kay LE (1996). Selective methyl group protonation of perdeuterated proteins. Journal of Molecular Biology 263, 627–636. 10.1006/jmbi.1996.0603 [DOI] [PubMed] [Google Scholar]
- Saleh T, Rossi P, and Kalodimos CG (2017). Atomic view of the energy landscape in the allosteric regulation of Abl kinase. Nature structural & molecular biology 24, 893–901. 10.1038/nsmb.3470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon-Ferrer R, Gotz AW, Poole D, Le Grand S, and Walker RC (2013). Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J Chem Theory Comput 9, 3878–3888. 10.1021/ct400314y [DOI] [PubMed] [Google Scholar]
- Shan Y, Arkhipov A, Kim ET, Pan AC, and Shaw DE (2013). Transitions to catalytically inactive conformations in EGFR kinase. Proc Natl Acad Sci U S A 110, 7270–7275. 10.1073/pnas.1220843110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SV, Bell DW, Settleman J, and Haber DA (2007). Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 7, 169–181. 10.1038/nrc2088 [DOI] [PubMed] [Google Scholar]
- Srivastava AK, McDonald LR, Cembran A, Kim J, Masterson LR, McClendon CL, Taylor SS, and Veglia G (2014). Synchronous opening and closing motions are essential for cAMP-dependent protein kinase A signaling. Structure 22, 1735–1743. 10.1016/j.str.2014.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Kay LE, and Tugarinov V (2011). An optimized relaxation-based coherence transfer NMR experiment for the measurement of side-chain order in methyl-protonated, highly deuterated proteins. The journal of physical chemistry. B 115, 14878–14884. 10.1021/jp209049k [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Sprangers R, and Kay LE (2007). Probing side-chain dynamics in the proteasome by relaxation violated coherence transfer NMR spectroscopy. J Am Chem Soc 129, 1743–1750. 10.1021/ja067827z [DOI] [PubMed] [Google Scholar]
- Webb B, and Sali A (2017). Protein Structure Modeling with MODELLER. Methods Mol Biol 1654, 39–54. 10.1007/978-1-4939-7231-9_4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.