

Abstract
Control of enantioselectivity remains a major challenge in radical chemistry. The emergence of metalloradical catalysis (MRC) offers a conceptually new strategy for addressing this and other outstanding issues. Through the employment of D2-symmetric chiral amidoporphyrins as the supporting ligands, Co(II)-based MRC has enabled the development of new catalytic systems for asymmetric radical transformations with a unique profile of reactivity and selectivity. With the support of new-generation HuPhyrin chiral ligands whose cavity environment can be fine-tuned, the Co-centered d-radicals enable to address challenging issues that require exquisite control of fundamental radical processes. As showcased with asymmetric 1,5-C−H amination of sulfamoyl azides, the enantiocontrol of which has proven difficult, the judicious use of HuPhyrin ligand by tuning the bridge length and other remote nonchiral elements allows for controlling both the degree and sense of asymmetric induction in a systematic manner. This effort leads to successful development of new Co(II)-based catalytic systems that are highly effective for enantiodivergent radical 1,5-C−H amination, producing both enantiomers of the strained five-membered cyclic sulfamides with excellent enantioselectivities. Detailed deuterium-labeling studies, together with DFT computation, have revealed an unprecedented mode of asymmetric induction that consists of enantiodifferentiative H-atom abstraction and stereoretentive radical substitution.
Graphical Abstract
1. INTRODUCTION
Aminyl radicals have been increasingly explored as synthetic intermediates for the formation of C−N bonds.1 Among important applications, the N-centered radicals have been commonly employed in intramolecular fashion for construction of N-heterocyclic structures, primarily via radical addition (RA) to unsaturated π-bonds in combination with H-atom abstraction (HAA) (Scheme 1a).2 Although mechanistically appealing, the alternative heterocyclization that is based on the association of H-atom abstraction (HAA) and radical substitution (RS) has been seldom documented (Scheme 1b). While abstraction of C−H bonds by aminyl radicals via intramolecular H-atom abstraction is typically facile, the subsequent C−N bond formation via homolytic radical substitution at the nitrogen center by the resulting alkyl radicals has not been well demonstrated and is presumably difficult.3 The well-known Hofmann−Löffler−Freytag (HLF) reaction obviates this inherent challenge to achieve C−N bond formation by switching the substitution step from radical to nucleophilic pathway.4 One potential direct solution to this fundamental problem would be the utilization of α-metalloaminyl radicals R(LnM)N· instead of free aminyl radicals RR′N· (Scheme 1c).5 In view of the lower strength and higher polarity of M−N than C−N bonds, the otherwise difficult radical substitution would become both thermodynamically possible and kinetically feasible, especially with the generation of stable metalloradicals (LnM·). Furthermore, this metalloradical-based approach for radical heterocyclization has the possibility to be turned over catalytically with the promise of controlling enantioselectivity as illustrated with two potential modes of asymmetric induction (Scheme 1d).
Scheme 1.

Heterocyclization Pathways of Aminyl Radicals and Potential Modes of Asymmetric Induction
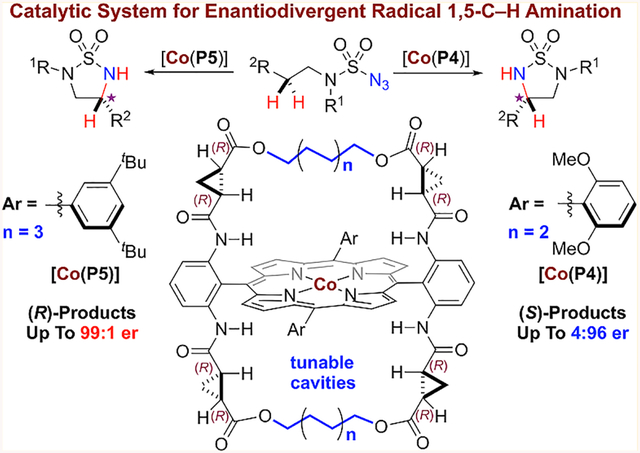
Homolytic radical reactions have vast synthetic potentials and could impact the practice of organic synthesis, which has been dominated by heterolytic ionic reactions. Despite recent advancements, the enduring issue of enantioselectivity remains largely unaddressed for most radical reactions.6 Among recent strategies,7 metalloradical catalysis (MRC) offers a conceptually different approach for achieving stereoselective radical reactions by exploiting metal-centered radicals for catalytic generation of metal-stabilized organic radicals that undergo subsequent radical transformations under the catalyst control.8,9 To this end, Co(II) complexes of porphyrins, as stable 15e metalloradicals, have shown the unusual capability of activating organic azides to generate the fundamentally new αCo(III)-aminyl radicals.10 With the employment of D2-symmetric chiral amidoporphyrins as the supporting ligands, the α-metalloaminyl radicals can undergo radical addition and H-atom abstraction as well as radical substitution, leading to the development of new catalytic processes for enantioselective radical transformations.11 Among them, we recently demonstrated the aforementioned radical heterocyclization strategy for the construction of strained five-membered cyclic sulfamides from sulfamoyl azides, which was difficult by concerned ionic pathway.12 To develop an asymmetric version of the catalytic process (Scheme 2a), we were attracted by the possibility of controlling enantioselectivity via both modes of asymmetric induction (Scheme 1d). In the course of pursuing the common Mode B for asymmetric induction with Co(II) complexes of existing open D2-symmetric chiral amidoporphyrins,11d,13 the recent introduction of new-generation D2-symmetric chiral amidoporphyrins HuPhyrin,14 which contain bridges across two chiral amide units on both sides of the porphyrin plane where the metal-centered d-radical is encapsulated inside a chiral cavity, prompted us to explore the rare Mode A for asymmetric induction. Under the support of HuPhyrin ligand with a proper cavity environment, we imagined that the corresponding α-Co(III)-aminyl radical I could be governed for enantiodifferentiative H-atom abstraction (HAA) of either pro-(R) or pro-(S) hydrogen (Scheme 2b).15 If the newly created facial chirality in the resulting εCo(III)-alkyl radical II could be conformationally stabilized by the geometric constraints inside the confined space,16 the subsequent intramolecular radical substitution (RS) could be enabled stereoretentive, forming five-membered cyclic sulfamides with the enantioselectivity that is predetermined in the HAA step (Scheme 2b). Catalyst engineering by fine-tuning the length of the distal bridge in combination with the remote meso-aryl substituents might allow achieving enantiodivergence for the catalytic radical process,17,18 which would be desirable considering that both enantiomers of a chiral catalyst are not always accessible from available chiral sources. If this type of unprecedented radical amination could be realized, it would be fundamentally appealing and practically useful as the resulting five-membered cyclic sulfamides and related vicinal diamines in both (R)- and (S)-configurations are common structural motifs in biologically important molecules (see Figure S1 in the Supporting Information).
Scheme 2.

Toward Enantiodivergence for Intramolecular Radical 1,5-C−H Amination by Co(II) Complexes of Bridged D2-Symmetric Chiral Amidoporphyrins
Intramolecular C−H amination via metal-catalyzed nitrene transfer represents a general approach for stereoselective construction of chiral N-heterocycles through direct functionalization of ubiquitous C−H bonds in organic molecules. Despite recent progresses,19,20 the development of chiral catalysts for asymmetric intramolecular amination is still limited. In particular, catalytic asymmetric intramolecular 1,5C−H amination to synthesize optically active five-membered cyclic sulfamides remains to be developed. As the first demonstration of the aforementioned Mode A for asymmetric induction, we herein wish to report the development of Co(II)-based metalloradical system with the support of the new type of bridged D2-symmetric chiral amidoporphyrins HuPhyrin for asymmetric intramolecular 1,5-C−H amination of sulfamoyl azides. Using HuPhyrin with varied bridge length and different substituents, both the degree and sense of asymmetric induction in the Co(II)-catalyzed amination can be effectively controlled in a systematic way. Two optimal catalysts, which differ only by the remote elements, have emerged from this study that can catalyze the efficient formation of the strained five-membered cyclic sulfamides as the opposite enantiomers, respectively, with high enantioselectivity. This enantiodivergent process is applicable to a broad scope of substrates. Our mechanistic studies support an unprecedented mode of asymmetric induction that consists of enantiodifferentiative HAA and stereoretentive RS.
2. RESULTS AND DISCUSSION
At the outset of this project, sulfamoyl azide 1a containing benzylic C−H bonds was selected as a test substrate for Co(II)-catalyzed radical 1,5-C−H amination. To explore the ligand effect on asymmetric induction during the Co(II)catalyzed radical amination, we employed two series of HuPhyrin ligands that are based on 3,5-di-tert-butylphenyl and 2,6-dimethoxyphenyl groups as 5,15-aryl substituents, respectively (Scheme 3). Both series of HuPhyrin contain the identical chiral amide element and the same type of alkyl bridge; they differ only by the bridge length varying from 4 to 6 to 8 to 10 methylene units. As illustrated in Scheme 3, the length of the distal alkyl bridge in HuPhyrin could significantly affect the asymmetric induction of the Co(II)-catalyzed C−H amination of azide 1a. Variation of the bridge length by two methylene units each time resulted in systematic alteration in enantioselectivity and even led to the switch in the sense of asymmetric induction for the formation of five-membered cyclic sulfamide 2a (Scheme 3). For [Co(3,5-DitBu-HuPhyrin)] catalyst series, C4-bridged [Co(P1)] slightly favored the formation of (R)-2a (52:48 er) while C6-bridged [Co(P3)] produced (S)-2a as the major enantiomer (30:70 er). When the alkyl bridge was further extended to C8-linker in [Co(P5)], the sense of asymmetric induction switched back again, forming (R)-2a in high enantioselectivity (97:3 er). The effect continued for C10-bridged [Co(P7)], which still produced (R)-2a as the major enantiomer but with decreased enantioselectivity (87:13 er). Interestingly, a parallel trend in asymmetric induction was also observed for the reaction by [Co(2,6-DiMeO-HuPhyrin)] catalyst series, generating 2a with (R):(S) enantiomer ratio varying from 32:68 by C4bridged [Co(P2)] to 6:94 by C6-bridged [Co(P4)] to 62:38 by C8-bridged [Co(P6)] to 36:64 by C10-bridged [Co(P8)]. Consequently, the five-membered cyclic chiral sulfamide could be enantiodivergently constructed through C−H amination by the use of C6-bridged [Co(P4)] and C8-bridged [Co(P5)] as the catalysts, producing highly enantioenriched (S)-2a and (R)-2a, respectively (Scheme 3). Considering that the only differences between [Co(P4)] and [Co(P5)] are the distal alkyl bridges and the remote nonchiral substituents, it is remarkable that such enantiodivergence could be realized under the same catalytic conditions.
Scheme 3.

Systematic Control of Degree and Sense of Asymmetric Induction in Intramolecular Radical 1,5-C−H Amination of Sulfamoyl Azide by [Co(HuPhyrin)]a,b,c,d
a Reactions were performed on a 0.10 mmol scale of sulfamoyl azide 1a using 2 mol % of [Co(HuPhyrin)] in 1.0 mL of methyl tert-butyl ether (MTBE) at 40 °C; [1a] = 0.10 M. b Yields in the Supporting Information. c Absolute configuration determined by X-ray crystal structural analysis. d Enantiomeric ratios (er) determined by chiral HPLC analysis.
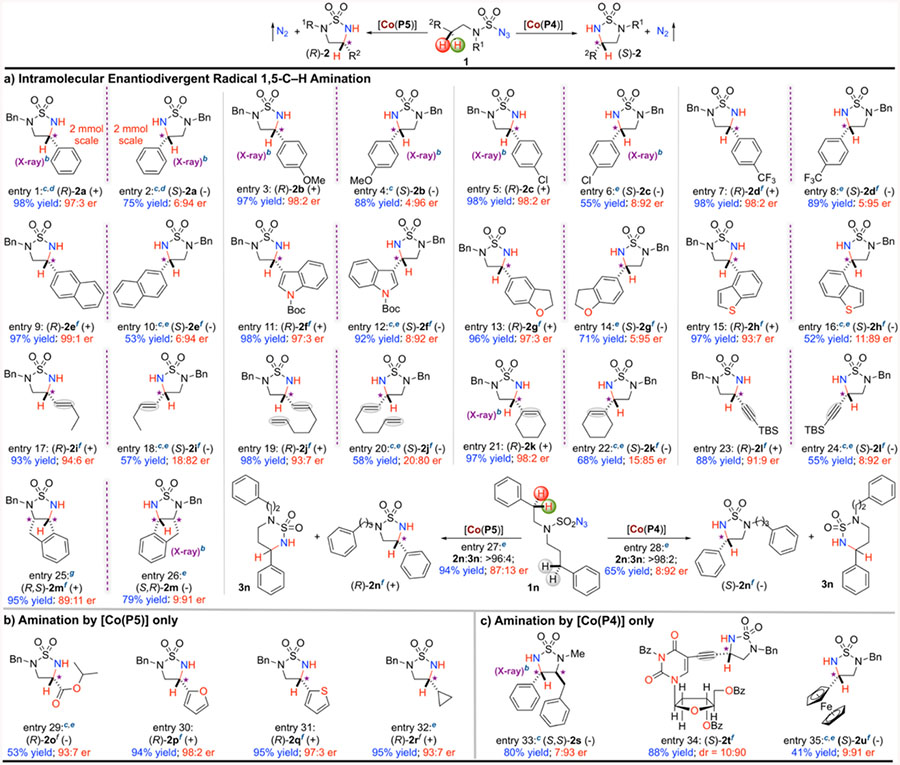
The enantiodivergent radical 1,5-C−H amination exerted by the pair of metalloradical catalysts [Co(P4)] and [Co(P5)] was found general and could be broadly applicable to various sulfamoyl azides with different types of C−H bonds (Table 1a). For the catalytic amination reactions of azide 1a by [Co(P4)] and [Co(P5)], they could be scaled up 20 times (from 0.1 to 2.0 mmol) under the standard conditions, producing (S)-2a and (R)-2a, respectively, in similarly high yields with the same excellent enantioselectivities (entries 1 and 2). Other benzylic C−H bonds with varied electronic properties could also be aminated in high yields with excellent enantioselectivities as shown for formation of both (R)- and (S)-cyclic sulfamides 2b−2d (entries 3−8). In addition, highly enantiodivergent stereocontrol was observed for amination of benzylic C−H bonds in both polyaromatic and heteroaromatic systems, as demonstrated for the productive formation of naphthalene-based 2e (entries 9 and 10), indole-based 2f (entries 11 and 12), dihydrobenzofuran-based 2g (entries 13 and 14), and benzothiophen-based 2h (entries 15 and 16). Furthermore, this enantiodivergent system exhibited excellent chemoselectivity toward radical amination of allylic C−H bonds to afford allylic 1,2-diamine derivatives without affecting the C=C double bonds, including monoene (2i), diene (2j), and cyclic ene (2k) (entries 17−22). Similarly, propargylic C− H bonds could be chemoselectively aminated as exemplified by enantiodivergent formation of propargylic 1,2-diamine derivative 2l without the involvement of the electron-rich C≡C bond (entries 23 and 24).21 Additionally, the Co(II)-based enantiodivergent system could be successfully applied for the desymmetrization of 2-indane-derived sulfamoyl azide to form the cis-fused tricyclic sulfamide 2m with effective control of the two newly generated stereogenic centers (entries 25 and 26). Moreover, the cavity environment of [Co(P4)] and [Co(P5)] enabled the enantiodivergent system with uncommon regioselectivity. This was nicely illustrated by the reactions of sulfamoyl azide 1n bearing sterically and electronically similar benzylic C−H bonds where both 1,5- and 1,6-C−H amination could potentially occur (entries 27 and 28).11d,12 Remarkably, five-membered cyclic sulfamides 2n were regioselectively constructed in high enantiodivergent selectivities over the less-strained six-membered cyclic sulfamides 3n.11d,12
Table 1.
Substrate Scope of Intramolecular Radical 1,5-C−H Amination of Sulfamoyl Azides via Co(II)-Based MRCa
|
Reactions were performed on a 0.10 mmol scale of sulfamoyl azide 1 using 2 mol % [Co(Por*)] in 1.0 mL of methyl tert-butyl ether (MTBE) at room temperature; enantiomeric ratios (er) determined by chiral HPLC analysis.
Absolute configuration determined by X-ray crystal structural analysis.
At 40 °C.
Performed at 2.0 mmol scale with 2 mol % of [Co(Por*)].
5 mol % [Co(Por*)].
Absolute configuration assigned by analogy.
When N-Bn was replaced by N-iPr: 61% yield; 94:6 er.
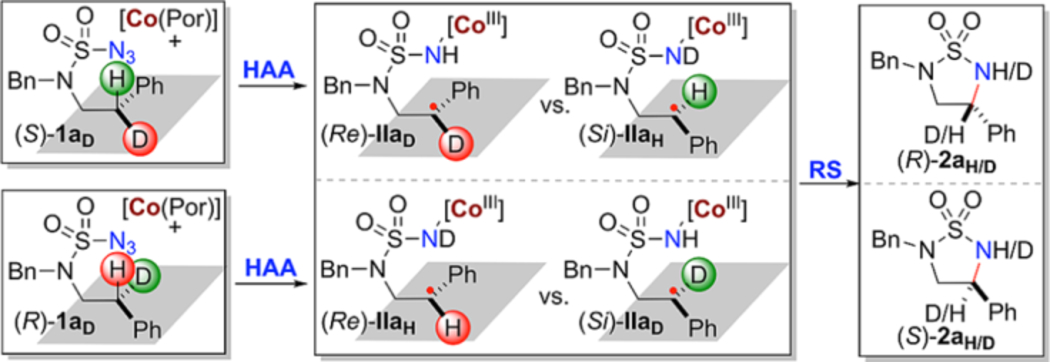
A set of mechanistic experiments were performed to obtain direct evidence for the proposed stepwise radical pathway of the Co(II)-catalyzed 1,5-C−H amination.10a–c As detailed in the Supporting Information, the corresponding α-Co(III)aminyl radical I from the reaction of azide 1a could be directly detected by EPR and HRMS (Figure S6 and S7). In the presence of excess TEMPO (10 equiv), the resulting ε-Co(III)-alkyl radical II after 1,5-H-atom abstraction could be successfully trapped, generating a TEMPO-substituted product along with the amination product 2a (see the Supporting Information). To determine the mode of asymmetric induction in the enantiodivergent system by [Co(P4)] and [Co(P5)], isotopomeric sulfamoyl azides in optically pure form (S)-1aD and (R)-1aD were prepared as substrates to study kinetic isotope effects (KIE) on C−H radical amination (Table 2). With the achiral nonbridged catalyst [Co(P9)] (P9 = 3,5 DitBu-IbuPhyrin), the intramolecular KIE was measured to have the same high value of 23.0 for reactions of both azides (S)-1aD and (R)-1aD (entries 1 and 4), which suggests significant tunneling that might be related to the high-strained transition state of the HAA process. When chiral catalysts are employed, alteration of this intrinsic KIE will be expected because of chirality match and mismatch between the catalyst and substrate. Accordingly, the use of chiral catalyst [Co(P4)] resulted in a lower KIE value of 2.0 for (S)-1aD (entry 2) while a higher KIE value of 61.0 for (R)-1aD (entry 5) was obtained, indicating enantiodifferentiative abstraction of pro-(S) H-atom by [Co(P4)]. On the other hand, chiral catalyst [Co(P5)] raised the KIE to a value of 96.0 for (S)-1aD (entry 3) but lowered it to a value of 0.8 for (R)-1aD (entry 6), suggesting that [Co(P5)] abstracted pro-(R) H-atom enantiodifferentiatively. Based on the measured KIE values, the ratios of the initially established two chiral faces (Re)-IIa to (Si)-IIa could be deduced. And in turn, it permits the calculation of predicted enantiomeric excess (ee) of the amination products 2a based on the assumption that there is no racemization of the face chirality during the subsequent RS step. The fact that the measured ee of product 2a by HPLC for the amination reaction of (S)-1aD by [Co(P5)] agreed near completely with the predicted value revealed stereoretentive RS during the catalytic process (entry 3). Likewise, the RS step for catalytic amination reaction of (R)-1aD by [Co(P4)] was also concluded to be stereoretentive (entry 5). In contrast to [Co(P4)] and [Co(P5)], however, the data suggested that the RS in the catalytic process by the nonbridged catalyst [Co(P9)] is nonstereoretentive (entries 1 and 4). Presumably due to less geometric constraints exerted by the open catalyst [Co(P9)], the highly enantioenriched facial chirality established in the first HAA step from both reactions of (S)-1aD (entry 1) and (R)-1aD (entry 4) was near completely racemized through low-barrier rotation of the α-C−C bond in the corresponding radical intermediate IIa. Clearly, the cavity environment in [Co(P4)] and [Co(P5)] created by bridging is primarily contributed to the realization of this unprecedented mode of asymmetric induction in the catalytic radical process by combining highly enantiodifferentiative HAA with completely retentive RS (Scheme 1d, Mode A).
Table 2.
 | ||||||
|---|---|---|---|---|---|---|
| entry | azide | catalyst | KIEb | Re:Si of IIac | ee%cal,d | ee%exp,e |
| 1 | (S)-1aD | [Co(P9)] | 23.0 | 96:4 | 92 (R) | 4 (R) |
| 2f | (S)-1aD | [Co(P4)] | 2.0 | 67:33 | 34 (R) | −4 (S) |
| 3 | (S)-1aD | [Co(P5)] | 96.0 | 99:1 | 98 (R) | 94 (R) |
| 4 | (R)-1aD | [Co(P9)] | 23.0 | 4:96 | −92 (S) | −4 (S) |
| 5f | (R)-1aD | [Co(P4)] | 61.0 | 2:98 | −96 (S) | −94 (S) |
| 6 | (R)-1aD | [Co(P5)] | 0.8 | 57:43 | 14 (R) | 32 (R) |
Reactions were performed on a 0.10 mmol scale of sulfamoyl azide (R)-1aD or (S)-1aD using 2 mol % of [Co(Por)] in 1.0 mL of MTBE at 40 °C; yields in the Supporting Information.
Ratio of H:D determined by 1H NMR spectroscopy.
Calculated based on the ratio of H:D.
ee of 2a calculated on the basis of stereoretentive RS.
ee of 2a determined by chiral HPLC analysis, which offered no separation of (R)-2aH from (R)-2aD and (S)-2aH from (S)-2aD.
5 mol % [Co(P4)].
To shed light on the origin of this new mode of asymmetric induction, the transition states (TS) of the HAA step in the reaction of azide 1a by C6-bridged [Co(P4)] and C8-bridged [Co(P5)] catalysts catalysts were calculated with density functional theory (DFT) at the ω- B97XD/Def2TZVPP//M06L/Def2SVPbenzene(SMD) level (see the Supporting Information for details). The DFT calculations revealed two possible lowenergy conformers Iunder and Inear for α-Co(III)-aminyl radical intermediate I, where phenyl ring A is located “underneaththe-bridge” and “nearby-the-bridge”, respectively (Scheme 4, top panel). Accordingly, two major TS were identified for HAA: the most stable underneath-the-bridge TS(I)under and the next stable nearby-the-bridge TS(I)near, enabling pro-(R) HAA to generate ε-Co(III)-alkyl radical intermediate (Re)IIunder and pro-(S) HAA to form ε-Co(III)-alkyl radical intermediate (Si)-IInear, respectively (Scheme 4). In both TS, the chiral cyclopropane unit on the left serves as a primary source for asymmetric induction by forcing phenyl ring B into the front right quadrant. Consistent with the experimental observations, the bridge lengths (C6 vs C8) as well as the identity of the nonchiral substituents (2,6-DiMeO vs 3,5DitBu) appear to be pivotal to reach specific TS. The larger cavity created by the C8-bridge in [Co(P5)] significantly favors C8-TS(I)under (−6.6 kcal/mol below C8-TS(I)near), where the pro-(R) benzylic hydrogen is oriented in closer proximity to the nitrogen radical for HAA so as to avoid the steric interaction of phenyl ring B with the distal 3,5-DitBu groups. In the smaller [Co(P4)] system, C6-TS(I)under is also lower in energy than C6-TS(I)near, albeit to a lesser degree (−1.8 kcal/mol). Considering that the C6-bridge is too small to allow isomerization from C6-Inear to C6-Iunder, we propose that access to C6-TS(I)under is kinetically difficult. This is well illustrated by the DFT-optimized ball-and-stick models of C6-TS(I) and C8TS(I), which are consistent with the structural details of P4 and P5 provided by single-crystal X-ray diffraction analysis (Scheme 4). Consequently, [Co(P4)] is governed to adopt C6TS(I)near, in which the pro-(S) benzylic hydrogen is better positioned to approach the nitrogen radical for HAA in order to prevent steric clash between the protruding phenyl ring B and the proximal 2,6-DiMeO moieties. Collectively, the DFT studies allowed us to establish agreeable stereochemical models where productive HAA can be achieved enantiodifferentiatively through TS(I)near or TS(I)under, depending on the cavity size underneath the distal bridge, which turns on and off the isomerization from intermediate Inear to Iunder, eventually leading to the opposite asymmetric induction. The low barriers associated with radical substitution (TS(II)near and TS(II)under) together with the hindered racemization of the chiral face in εCo(III)-alkyl radical intermediate II are consistent with a stereoretentive RS step.
Scheme 4.

DFT-Optimized Stereochemical Models of Enantiodifferentiative H-Atom Abstraction by [Co(P4)] and [Co(P5)]a
a See the Supporting Information for movies of the major pathways leading to the enantiodifferentiative HAA and stereorentive RS by [Co(P4)] and [Co(P5)].
These stereochemical models were further substantiated by additional experiments that employed substrates with unique steric or electronic properties to probe difference in reactivity and stereoselectivity (Table 1, entries 29−35). First, when azides 1o−1q bearing heteroatoms in proximity to the C−H site were used as the substrates, the catalytic reactions by [Co(P4)] were inefficient (<10% yields even at 40 °C) (see the Supporting Information) while [Co(P5)] could catalyze the high-yielding formation of the desired amination products 2o−2q with high (R)-enantioselectivities (Table 1b, entries 29−31). The negative outcomes of these catalytic reactions by [Co(P4)] are attributed to the high barrier to reach the dominated C6-TS(I)near owing to the lone-pair repulsion between heteroatom in the substrates and the oxygen of 2,6DiMeO groups in the catalyst. Such unfavorable repulsive interactions are absent with the 3,5-DitBu groups in [Co(P5)] through C8-TS(I)under. The productive formation of (R)-2o (entry 29) is particularly noteworthy both fundamentally and practically in view of the difficulty associated with amination of the electron-deficient α-C−H bonds of ester by C−H insertion via electrophilic metallonitrenes as well as the importance of the resulting α,β-diamino acid ester as a recurring core unit in a variety of bioactive compounds.22 As a probe for steric effect, azide 1r carrying a less sterically hindered cyclopropyl moiety adjacent to the C−H site was found to be efficiently aminated by [Co(P5)] to afford the desired 2r with high (R)enantioselectivity (entry 32) whereas the use of [Co(P4)] led to the formation of 2r with moderate (R)-enantioselectivity (58:42 er) (see the Supporting Information). Considering the similar size of the two N-substituents (cyclopropylethyl vs phenylmethyl), such low and anomalous (R)-enantioselectivity by [Co(P4)] agrees well with the general models because of the decreased preference for the cyclopropyl moiety to stay in the front right quadrant to react in the dominated C6-TS(I)near. The high-yielding reaction of substrate 1r (Table 1, entry 32) indicated that the rate of homolytic substitution is faster than the rate of ring-opening of the cyclopropoylcarbinyl radical. On the other hand, azides 1s−1u bearing bulky groups could be productively aminated by [Co(P4)] to produce the desired 2s−2u with high (S)-enantiocontrol (Table 1c, entries 33− 35), indicating that the majority of steric bulkiness in these substrates was effectively directed outside the catalyst cavity in the corresponding C6-TS(I)near. In addition to demonstrating the suitability for large substrates, the productive formation of cyclic sulfamides 2t containing densely functionalized deoxyuridine highlights the remarkable degree of functional group tolerance by the catalytic system (entry 34). It is also notable that [Co(P4)] could effectively desymmetrize the two benzyl groups in azide 1s, leading to trans-(S,S)-2s with effective control of the two newly generated stereogenic centers (entry 33). However, when [Co(P5)] was used as the catalyst, the catalytic reactions of azides 1s−1u afforded the amination products 2s−2u with moderate (R)-enantioselectivity (~58:42 er) (see the Supporting Information). As well, these results are considered to be consistent with the general models in view of the decreased preference for C8-TS(I)under over C8-TS(I)near as a result of the large size associated with these substrates. Furthermore, the observed systematic variation in enantiodivergence described in Scheme 3 appears to be well consistent with the stereochemical models.
3. CONCLUSIONS
In summary, we have demonstrated an unprecedented mode of asymmetric induction in radical process that is based on sequential combination of enantiodifferentiative H-atom abstraction (HAA) and stereoretentive radical substitution (RS). Under Co(II)-based metalloradical catalysis (MRC), this new mode of asymmetric induction has been put in practice for successful development of the first asymmetric system for stereoselective synthesis of the strained five-membered cyclic sulfamides via radical 1,5-C−H amination of sulfamoyl azides. With the support of bridged HuPhyrin ligands, whose chiral cavity can be fine-tuned by varying the combination of the distal alkyl bridges and the remote nonchiral substituents, Co(II)-based metalloradical system has been shown to have an unusual capability of controlling both the degree and sense of asymmetric induction in the catalytic radical C−H amination in a systematic manner. A pair of similar metalloradical catalysts [Co(P4)] and [Co(P5)], which differ only by the length of the distal alkyl bridges and the position of the remote nonchiral substituents, have been identified as effective catalysts for the enantiodivergent radical C−H amination with high enantioselectivity under the same mild conditions. The enantiodivergent system is applicable to a broad scope of substrates with different types of C(sp3)−H bonds and exhibits a remarkable profile of reactivity and selectivity, providing access to both enantiomers of useful five-membered cyclic sulfamides in a highly enantioenriched form. In addition to shedding light on the origin of the asymmetric induction in the current catalytic system, the stereochemical models, established on the basis of the combined experimental and computational studies, may have far-reaching implications for how to design catalysts for enantioselective radical reactions. This study paves the way for further exploration of unrecognized potential of radical reactions in stereoselective organic synthesis, including development of catalytic radical processes for enantiodivergent synthesis of chiral molecules.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for financial support by NIH (R01GM098777) and in part by NSF (CHE-1900375).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b05850.
Experimental details, supplementary figures, and spectroscopic data (PDF)
Coordinates for DFT-calculated structures (XYZ)
Movie for major C−H amination pathway by [Co(P4)] (MP4)
Movie for major C−H amination pathway by [Co(P5)] (MP4)
Crystallography data for (R)-2a, (S)-2a, (R)-2b, (S)-2b, (R)-2c, (S)-2c, (R)-2k, (S,R)-2m, (S,S)-2s, P4, and P5 (ZIP)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1) (a).Xiong T; Zhang Q New Amination Strategies Based on Nitrogen-Centered Radical Chemistry. Chem. Soc. Rev 2016, 45, 3069–3087. [DOI] [PubMed] [Google Scholar]; (b) Zhao Y; Xia W Recent Advances in Radical-Based C−N Bond Formation via Photo-/Electrochemistry. Chem. Soc. Rev 2018, 47, 2591–2608. [DOI] [PubMed] [Google Scholar]
- (2).Zard SZ Recent Progress in the Generation and Use of Nitrogen-Centred Radicals. Chem. Soc. Rev 2008, 37, 1603–1618. [DOI] [PubMed] [Google Scholar]
- (3).(a) C−N bond formation via homolytic radical susbstiution at the nitrogen center has rarely been reported. For a related study in gas phase, see:Antoniotti P Benzi P; Borocci S; Demaria C; Giordani M; Grandinetti F; Operti L; Rabezzana R Bimolecular Homolytic Substitutions at Nitrogen: an Experimental and Theoretical Study on the Gas-Phase Reactions of Alkyl Radicals with NF3. Chem. - Eur. J 2015, 21, 15826–15834. [DOI] [PubMed] [Google Scholar]; (b) For discussion on challenges of homolytic radical substitution at the carbon center, see:Walton JC Homolytic Substitution: a Molecular Mènage à Trois. Acc. Chem. Res 1998, 31, 99–107. [Google Scholar]
- (4) (a).Wolff ME Cyclization of N-Halogenated Amines (The Hofmann-Löffler reaction). Chem. Rev 1963, 63, 55–64. [Google Scholar]; (b) For a recent example, see:Wappes EA; Nakafuku KM; Nagib DA Directed β C−H Amination of Alcohols via Radical Relay Chaperones. J. Am. Chem. Soc 2017, 139, 10204–10207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) For select review, see:Johnson MD Bimolecular Homolytic Displacement of Transition-Metal Complexes from Carbon. Acc. Chem. Res 1983, 16, 343–349. [Google Scholar]; (b) For related examples of homolytic substitutions with Ti−O bonds at the oxygen center, see:Trost BM; Shen HC; Surivet J-P An Enantioselective Biomimetic Total Synthesis of (−)-Siccanin. Angew. Chem., Int. Ed 2003, 42, 3943–3947. [DOI] [PubMed] [Google Scholar]; (c) Gansäuer A; Fleckhaus A; Lafont MA; Okkel A; Kotsis K; Anoop A; Neese F Catalysis via Homolytic Substitutions with C−O and Ti−O Bonds: Oxidative Additions and Reductive Eliminations in Single Electron Steps. J. Am. Chem. Soc 2009, 131, 16989–16999. [DOI] [PubMed] [Google Scholar]
- (6) (a).Sibi MP; Manyem S; Zimmerman J Enantioselective Radical Processes. Chem. Rev 2003, 103, 3263–3295. [DOI] [PubMed] [Google Scholar]; (b) Studer A; Curran DP Catalysis of Radical Reactions: a Radical Chemistry Perspective. Angew. Chem., Int. Ed 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]; (c) Yan M; Lo JC; Edwards JT; Baran PS Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc 2016, 138, 12692–12714. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lu Q; Glorius F Radical Enantioselective C(sp3)−H Functionalization. Angew. Chem., Int. Ed 2017, 56, 49–51. [DOI] [PubMed] [Google Scholar]
- (7).(a) For select examples, see:Nicewicz DA; MacMillan DWC Merging Photoredox Catalysis with Organocatalysis: the Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322, 77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rono LJ; Yayla HG; Wang DY; Armstrong MF; Knowles RR Enantioselective Photoredox Catalysis Enabled by Proton-Coupled Electron Transfer: Development of an Asymmetric Aza-Pinacol Cyclization. J. Am. Chem. Soc 2013, 135, 17735–17738. [DOI] [PubMed] [Google Scholar]; (c) Hashimoto T; Kawamata Y; Maruoka K An Organic Thiyl Radical Catalyst for Enantioselective Cyclization. Nat. Chem 2014, 6, 702–705. [DOI] [PubMed] [Google Scholar]; (d) Huo H; Shen X; Wang C; Zhang L; Röse P; Chen L-A; Harms K; Marsch M; Hilt G; Meggers E Asymmetric Photoredox Transition-Metal Catalysis Activated by Visible Light. Nature 2014, 515, 100–103. [DOI] [PubMed] [Google Scholar]; (e) Blum TR; Miller ZD; Bates DM; Guzei IA; Yoon TP Enantioselective Photochemistry through Lewis Acid−Catalyzed Triplet Energy Transfer. Science 2016, 354, 1391–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Kainz QM; Matier CD; Bartoszewicz A; Zultanski SL; Peters JC; Fu GC Asymmetric Copper-Catalyzed C−N Cross-Couplings Induced by Visible Light. Science 2016, 351, 681–684. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Lin J-S; Dong X-Y; Li T-T; Jiang N-C; Tan B; Liu X-Y A Dual-Catalytic Strategy to Direct Asymmetric Radical Aminotrifluoromethylation of Alkenes. J. Am. Chem. Soc 2016, 138, 9357–9360. [DOI] [PubMed] [Google Scholar]; (h) Zhang W; Wang F; McCann SD; Wang D; Chen P; Stahl SS; Liu G Enantioselective Cyanation of Benzylic C−H Bonds via CopperCatalyzed Radical Relay. Science 2016, 353, 1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Funken N; Mühlhaus F; Gansäuer A General, Highly Selective Synthesis of 1,3- and 1,4-Difunctionalized Building Blocks by Regiodivergent Epoxide Opening. Angew. Chem., Int. Ed 2016, 55, 12030–12034. [DOI] [PubMed] [Google Scholar]; (j) Kern N; Plesniak MP; McDouall JJW; Procter DJ Enantioselective Cyclizations and Cyclization Cascades of Samarium Ketyl Radicals. Nat. Chem 2017, 9, 1198–1204. [DOI] [PubMed] [Google Scholar]; (k) Morrill C; Jensen C; Just-Baringo X; Grogan G; Turner NJ; Procter DJ Biocatalytic Conversion of Cyclic Ketones Bearing α-Quaternary Stereocenters into Lactones in an Enantioselective Radical Approach to Medium-Sized Carbocycles. Angew. Chem., Int. Ed 2018, 57, 3692–3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) For reviews and highlights on Co(II)-based MRC, see:Demarteau J; Debuigne A; Detrembleur C Organocobalt Complexes as Sources of Carbon-Centered Radicals for Organic and Polymer Chemistries. Chem. Rev 2019, 119, 6906–6955. [DOI] [PubMed] [Google Scholar]; (b) Miyabe H; Kawashima A; Yoshioka E; Kohtani S Progress in Enantioselective Radical Cyclizations. Chem. - Eur. J 2017, 23, 6225–6236. [DOI] [PubMed] [Google Scholar]; (c) Pellissier H; Clavier H Enantioselective CobaltCatalyzed Transformations. Chem. Rev 2014, 114, 2775–2823. [DOI] [PubMed] [Google Scholar]; (d) Lu H; Zhang XP Catalytic C−H Functionalization by Metalloporphyrins: Recent Developments and Future Directions. Chem. Soc. Rev 2011, 40, 1899–1909. [DOI] [PubMed] [Google Scholar]; (e) Doyle MP Exceptional Selectivity in Cyclopropanation Reactions Catalyzed by Chiral Cobalt(II)−Porphyrin Catalysts. Angew. Chem., Int. Ed 2009, 48, 850–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) For select examples on Ti(III)-based radical processes, see:RajanBabu TV; Nugent WA Intermolecular Addition of Epoxides to Activated Olefins: a New Reaction. J. Am. Chem. Soc 1989, 111, 4525–4527. [Google Scholar]; (b) RajanBabu TV; Nugent WA Selective Generation of Free Radicals from Epoxides Using a Transition-Metal Radical. A Powerful New Tool for Organic Synthesis. J. Am. Chem. Soc 1994, 116, 986–997. [Google Scholar]; (c) Gansäuer A; Lauterbach T; Bluhm H; Noltemeyer M A Catalytic Enantioselective Electron Transfer Reaction: Titanocene-Catalyzed Enantioselective Formation of Radicals from Meso-Epoxides. Angew. Chem., Int. Ed 1999, 38, 2909–2910. [DOI] [PubMed] [Google Scholar]; (d) Gansäuer A; Fan C-A; Keller F; Keil J Titanocene-Catalyzed Regiodivergent Epoxide Openings. J. Am. Chem. Soc 2007, 129, 3484–3485. [DOI] [PubMed] [Google Scholar]; (e) Gansäuer A; Shi L; Otte M Catalytic Enantioselective Radical Cyclization via Regiodivergent Epoxide Opening. J. Am. Chem. Soc 2010, 132, 11858–11859. [DOI] [PubMed] [Google Scholar]; (f) Gansäuer A; Hildebrandt S; Vogelsang E; Flowers RA II Tuning the Redox Properties of the Titanocene(III)/(IV)Couple for Atom-Economical Catalysis in Single Electron Steps. Dalton Trans. 2016, 45, 448–452. [DOI] [PubMed] [Google Scholar]; (g) Zhang Y-Q; Vogelsang E; Qu Z-W; Grimme S; Gansäuer A Titanocene-Catalyzed Radical Opening of N-Acylated Aziridines. Angew. Chem., Int. Ed 2017, 56, 12654–12657. [DOI] [PubMed] [Google Scholar]; (h) Hao W; Wu X; Sun JZ; Siu JC; MacMillan SN; Lin S Radical Redox-Relay Catalysis: Formal [3 + 2] Cycloaddition of N-Acylaziridines and Alkenes. J. Am. Chem. Soc 2017, 139, 12141–12144. [DOI] [PubMed] [Google Scholar]; (i) Hao W; Harenberg JH; Wu X; MacMillan SN; Lin S Diastereo- and Enantioselective Formal [3 + 2] Cycloaddition of Cyclopropyl Ketones and Alkenes via TiCatalyzed Radical Redox Relay. J. Am. Chem. Soc 2018, 140, 3514–3517. [DOI] [PubMed] [Google Scholar]
- (10).(a) For detailed experimental and theoretical studies on the radical mechanism involving α-Co(III)-aminyl radical (also known as Co(III)-nitrene radical) intermediates for olefin aziridination and C−H amination, see:Lyaskovskyy V; Suarez AIO; Lu H; Jiang H; Zhang XP; de Bruin B Mechanism of Cobalt(II) Porphyrin-Catalyzed C−H Amination with Organic Azides: Radical Nature and H-Atom Abstraction Ability of the Key Cobalt(III)−Nitrene Intermediates. J. Am. Chem. Soc 2011, 133, 12264–12273. [DOI] [PubMed] [Google Scholar]; (b) Hopmann KH; Ghosh A Mechanism of Cobalt-Porphyrin−Catalyzed Aziridination. ACS Catal. 2011, 1, 597–600. [Google Scholar]; (c) Goswami M; Lyaskovskyy V; Domingos SR; Buma WJ; Woutersen S; Troeppner O; Ivanović- Burmazović I; Lu H; Cui X; Zhang XP; Reijerse EJ; DeBeer S; van Schooneveld MM; Pfaff FF; Ray K; de Bruin B Characterization of Porphyrin-Co(III)-’Nitrene Radical’ Species Relevant in Catalytic Nitrene Transfer Reactions. J. Am. Chem. Soc 2015, 137, 5468–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) For select examples on nonasymmetric radical C−H amination and radical olefin aziridination via Co(II)-MRC, see:Ruppel JV; Jones JE; Huff CA; Kamble RM; Chen Y; Zhang XP A Highly Effective Cobalt Catalyst for Olefin Aziridination with Azides: Hydrogen Bonding Guided Catalyst Design. Org. Lett 2008, 10, 1995–1998. [DOI] [PubMed] [Google Scholar]; (e) Lu H; Jiang H; Wojtas L; Zhang XP Selective Intramolecular C−H Amination through the Metalloradical Activation of Azides: Synthesis of 1,3-Diamines under Neutral and Nonoxidative Conditions. Angew. Chem., Int. Ed 2010, 49, 10192–10196. [DOI] [PubMed] [Google Scholar]; (f) Villanueva O; Weldy NM; Blakey SB; MacBeth CE Cobalt Catalyzed sp3 C−H Amination Utilizing Aryl Azides. Chem. Sci 2015, 6, 6672–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Jiang H; Lang K; Lu H; Wojtas L; Zhang XP Intramolecular Radical Aziridination of Allylic Sulfamoyl Azides by Cobalt(II)-Based Metalloradical Catalysis: Effective Construction of Strained Heterobicyclic Structures. Angew. Chem., Int. Ed 2016, 55, 11604–11608. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Kuijpers PF; Tiekink MJ; Breukelaar WB; Broere DLJ; van Leest NP; van der Vlugt JI; Reek JNH; de Bruin B Cobalt-Porphyrin-Catalysed Intramolecular Ring-Closing C−H Amination of Aliphatic Azides: a Nitrene-Radical Approach to Saturated Heterocycles. Chem.-Eur. J 2017, 23, 7945–7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) For select examples on asymmetric radical C−H amination and radical olefin aziridination via Co(II)-MRC, see:Subbarayan V; Ruppel JV; Zhu S; Perman JA; Zhang XP Highly Asymmetric Cobalt-Catalyzed Aziridination of Alkenes with Trichloroethoxysulfonyl Azide (TcesN3). Chem. Commun 2009, 4266–4268. [DOI] [PubMed] [Google Scholar]; (b) Jin LM; Xu X; Lu H; Cui X; Wojtas L; Zhang XP Effective Synthesis of Chiral N-Fluoroaryl Aziridines through Enantioselective Aziridination of Alkenes with Fluoroaryl Azides. Angew. Chem., Int. Ed 2013, 52, 5309–5313. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jiang H; Lang K; Lu H; Wojtas L; Zhang XP Asymmetric Radical Bicyclization of Allyl Azidoformates via Cobalt(II)-Based Metalloradical Catalysis. J. Am. Chem. Soc 2017, 139, 9164–9167. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li C; Lang K; Lu H; Hu Y; Cui X; Wojtas L; Zhang XP Catalytic Radical Process for Enantioselective Amination of C(sp3)−H Bonds. Angew. Chem., Int. Ed 2018, 57, 16837–16841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lu H; Lang K; Jiang H; Wojtas L; Zhang XP Intramolecular 1,5-C(sp3)-H Radical Amination via Co(II)-Based Metalloradical Catalysis for Five-Membered Cyclic Sulfamides. Chem. Sci 2016, 7, 6934–6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Our attempts using Co(II)-based catalysts supported by nonbridged form of D2-symmetric chiral amidoporphyrins have so far led to only moderate enantioselectivities for the formation of the strained five-membered ring system (see the Supporting Information) despite our recent success on enantioselective formation of the less strained six-membered system (see ref 11d).
- (14).Hu Y; Lang K; Tao J; Marshall M; Cheng Q; Cui X; Wojtas L; Zhang XP Next-Generation D2-Symmetric Chiral Porphyrins for Co(II)-Based Metalloradical Catalysis: Catalyst Engineering by Distal Bridging. Angew. Chem., Int. Ed 2019, 58, 2670–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15) (a).Groves JT; Viski P Asymmetric Hydroxylation by a Chiral Iron Porphyrin. J. Am. Chem. Soc 1989, 111, 8537–8538. [Google Scholar]; (b) Burg F; Gicquel M; Breitenlechner S; Pöthig A; Bach T Siteand Enantioselective C−H Oxygenation Catalyzed by a Chiral Manganese Porphyrin Complex with a Remote Binding Site. Angew. Chem., Int. Ed 2018, 57, 2953–2957. [DOI] [PubMed] [Google Scholar]
- (16).Gellman SH Foldamers: a Manifesto. Acc. Chem. Res 1998, 31, 173–180. [Google Scholar]
- (17).Enantiodivergent radical processes have not been previously demonstrated in synthetic systems.
- (18).(a) For an example of possible cavity-controlled regiodivergent reaction, see:Wu B; Parquette JR; RajanBabu TV Regiodivergent Ring Opening of Chiral Aziridines. Science 2009, 326, 1662. [DOI] [PubMed] [Google Scholar]; (b) For an example of in situ switching of the chiral preference of a catalytic system, see:Wang J; Feringa BL Dynamic Control of Chiral Space in a Catalytic Asymmetric Reaction Using a Molecular Motor. Science 2011, 331, 1429–1432. [DOI] [PubMed] [Google Scholar]
- (19).(a) For selective reviews, see:Collet F; Lescot C; Dauban P Catalytic C−H Amination: the Stereoselectivity Issue. Chem. Soc. Rev 2011, 40, 1926–1936. [DOI] [PubMed] [Google Scholar]; (b) Alderson JM; Corbin JR; Schomaker JM Tunable, Chemo-and Site-Selective Nitrene Transfer Reactions through the Rational Design of Silver(I) Catalysts. Acc. Chem. Res 2017, 50, 2147–2158. [DOI] [PubMed] [Google Scholar]; (c) For select examples on non-asymmetric C−H amination, see:Hennessy ET; Betley TA Complex NHeterocycle Synthesis via Iron-Catalyzed, Direct C−H Bond Amination. Science 2013, 340, 591–595. [DOI] [PubMed] [Google Scholar]; (d) Alderson JM; Phelps AM; Scamp RJ; Dolan NS; Schomaker JM LigandControlled, Tunable Silver-Catalyzed C−H Amination. J. Am. Chem. Soc 2014, 136, 16720–16723. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hong SY; Park Y; Hwang Y; Kim YB; Baik M-H; Chang S Selective Formation of γ-Lactams via C−H Amidation Enabled by Tailored Iridium Catalysts. Science 2018, 359, 1016–1021. [DOI] [PubMed] [Google Scholar]
- (20).(a) For selective examples on asymmetric C−H amination, see:Liang J-L; Yuan S-X; Huang J-S; Yu W-Y; Che C-M Highly Diastereo- and Enantioselective Intramolecular Amidation of Saturated C−H Bonds Catalyzed by Ruthenium Porphyrins. Angew. Chem., Int. Ed 2002, 41, 3465–3468. [DOI] [PubMed] [Google Scholar]; (b) Milczek E; Boudet N; Blakey S Enantioselective C−H Amination Using Cationic Ruthenium(II)−Pybox Catalysts. Angew. Chem., Int. Ed 2008, 47, 6825–6828. [DOI] [PubMed] [Google Scholar]; (c) Wang H; Park Y; Bai Z; Chang S; He G; Chen G Iridium-Catalyzed Enantioselective C(sp3)−H Amidation Controlled by Attractive Noncovalent Interactions. J. Am. Chem. Soc 2019, 141, 7194–7201. [DOI] [PubMed] [Google Scholar]; (d) Zhou Z; Chen S; Qin J; Nie X; Zheng X; Harms K; Riedel R; Houk KN; Meggers E Catalytic Enantioselective Intramolecular C(sp3)−H Amination of 2-Azidoacetamides. Angew. Chem., Int. Ed 2019, 58, 1088–1093. [DOI] [PubMed] [Google Scholar]; (e) Qin J; Zhou Z; Cui T; Hemming M; Meggers E Enantioselective Intramolecular C−H Amination of Aliphatic Azides by Dual Ruthenium and Phosphine Catalysis. Chem. Sci 2019, 10, 3202–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Fukagawa S; Kato Y; Tanaka R; Kojima M; Yoshino T; Matsunaga S Enantioselective C(sp3)−H Amidation of Thioamides Catalyzed by a CobaltIII/Chiral Carboxylic Acid Hybrid System. Angew. Chem., Int. Ed 2019, 58, 1153–1157. [DOI] [PubMed] [Google Scholar]; (g) Nasrallah A; Boquet V; Hecker A; Retailleau P; Darses B; Dauban P Catalytic Enantioselective Intermolecular Benzylic C(sp3)−H Amination. Angew. Chem., Int. Ed 2019, 58, 8192–8196. [DOI] [PubMed] [Google Scholar]; (h) Xing Q; Chan C-M; Yeung Y-W; Yu W-Y Ruthenium(II)-Catalyzed Enantioselective γ-Lactams Formation by Intramolecular C−H Amidation of 1,4,2-Dioxazol-5-ones. J. Am. Chem. Soc 2019, 141, 3849–3853. [DOI] [PubMed] [Google Scholar]
- (21).Mace N; Thornton AR; Blakey SB Unveiling Latent α-Iminocarbene Reactivity for Intermolecular Cascade Reactions through Alkyne Oxidative Amination. Angew. Chem., Int. Ed 2013, 52, 5836–5839. [DOI] [PubMed] [Google Scholar]
- (22).Viso A; Fernández de la Pradilla R; García A; Flores A α,β-Diamino Acids: Biological Significance and Synthetic Approaches. Chem. Rev 2005, 105, 3167–3196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.