

Abstract
Background
Lynch‐like syndrome (LLS) represents around 50% of the patients fulfilling the Amsterdam Criteria II/revised Bethesda Guidelines, characterized by a strong family history of Lynch Syndrome (LS) associated cancer, where a causative variant was not identified during genetic testing for LS.
Methods
Using data extracted from a larger gene panel, we have analyzed next‐generation sequencing data from 22 mismatch repair (MMR) genes (MSH3, PMS1, MLH3, EXO1, POLD1, POLD3 RFC1, RFC2, RFC3, RFC4, RFC5, PCNA, LIG1, RPA1, RPA2, RPA3, POLD2, POLD4, MLH1, MSH2, MSH6, and PMS2) in 274 LLS patients. Detected variants were annotated and filtered using ANNOVAR and FILTUS software.
Results
Thirteen variants were revealed in MLH1, MSH2, and MSH6, all genes previously linked to LS. Five additional genes (EXO1, POLD1, RFC1, RPA1, and MLH3) were found to harbor 11 variants of unknown significance in our sample cohort, two of them being frameshift variants.
Conclusion
We have shown that other genes associated with the process of DNA MMR have a high probability of being associated with LLS families. These findings indicate that the spectrum of genes that should be tested when considering an entity like Lynch‐like syndrome should be expanded so that a more inclusive definition of this entity can be developed.
Keywords: Genetics, germline mutation, high‐throughput sequencing, Lynch syndrome, MMR gene panel
1. INTRODUCTION
Lynch Syndrome (LS) is an autosomal dominantly inherited predisposition to colorectal cancer (CRC) and other epithelial malignancies and accounts for approximately 2%–3% of all CRC patients diagnosed annually (de la Chapelle, 2004; Hampel et al., 2005). LS, also known as hereditary nonpolyposis colorectal cancer (HNPCC), is defined by the presence of germline mutations in one of four genes involved in DNA mismatch repair (MMR); MLH1 (OMIM: 120436), MSH2 (OMIM: 609309), MSH6 (OMIM: 600678), and PMS2 (OMIM: 600259) (Lynch & de la Chapelle, 1999). Deletions in EPCAM are also implicated in LS that are associated with epigenetic silencing of MSH2 (Kuiper et al., 2011). It is important to identify LS mutation carriers, in order to offer them regular surveillance programs like colonoscopy to improve early detection of cancer.
LS tumor spectrum is quite wide, involving in most cases CRC and, for women, a high risk of endometrial cancer (Lek et al., 2016). In addition, MMR mutations in LS causes an increased risk of ovarian, gastric, urologic tract, kidney, ureter, small bowel, and hepatobiliary tract tumors (Samadder et al., 2017; Watson & Riley, 2005).
LS is an entity that has been diagnosed using the Amsterdam criteria (AC) and Bethesda guidelines (BG), or variants of it (the AC II or revised BG) using the patient's pedigree and family history of cancer (Rodriguez‐Bigas et al., 1997; Umar et al., 2004; Vasen, Mecklin, Khan, & Lynch, 1991; Vasen, Watson, Mecklin, & Lynch, 1999). Genetic screening by Sanger sequencing to identify causative variants in MMR genes has been the gold standard to diagnose patients at risk of LS. Individuals fulfilling the AC II or revised BG without a molecular diagnosis are now termed Lynch‐like syndrome (LLS) families (Carethers, 2014; Giardiello et al., 2014).
Families that fulfill the AC, where probands have tumors displaying microsatellite instability (MSI) or a loss of MMR genes expression (as judged by immunohistochemistry), are offered screening for pathogenic variants in MMR genes (usually by DNA sequencing) to identify a causative genetic variant. Using this approach, approximately 50% of LS patients remain without a molecular diagnosis after screening the common MMR genes (Bonis et al., 2007; Lindor et al., 2005; Steinke et al., 2014). Early detection and management provide the best likelihood of survival, thus identifying high‐risk individuals who could benefit from early detection is a priority. The 50% of patients where pathogenic variants cannot be detected are commonly termed LLS families or familial colorectal cancer type X (FCCTX) as disease segregation is suggestive of an inherited disease but in the absence of any identifiable causative variant. This group appears to have a later age of disease onset compared to LS, suggesting that these families have lower levels of disease penetrance (Lipkin & Afrasiabi, 2007). While the definitions of LLS and FCCTX mostly overlap, LLS is defined by patients with MSI‐High tumors but no loss of MMR immunohistochemistry staining (for the four main MMR genes) (Carethers, 2014). On the other hand, FCCTX describes patients fulfilling the AC I but no causative pathogenic variants has been found, and are mostly microsatellite‐stable (MSS) (Lipkin & Afrasiabi, 2007).
DNA MMR involves the coordinated response of at least 22 proteins (KEGG pathways (Kanehisa, Furumichi, Tanabe, Sato, & Morishima, 2017), Figure 1) that are involved in mismatch recognition, protein recruitment to the lesion, removal of the mismatch and replacement of the incorrect base with the correct one (Fishel, 2015). Thus, the possibility exists that other defects in the DNA MMR pathway may be associated with cancer risk, which manifests as an entity similar to LS. Evidence to support this comes from studies that have examined MLH3 where it has been proposed to be a candidate gene implicated in LS (Liu et al., 2003). This is supported by evidence that demonstrates MSH3 variants appear to confer a low risk of disease (associated with various phenotypes (Carethers, Koi, & Tseng‐Rogenski, 2015) including a phenotype of polyposis (Adam et al., 2016)) and have a synergistic effect when accompanied by MSH2 variants (Duraturo et al., 2011). Previously, POLD1 variants have shown to be associated with an increased risk of CRC that results in a phenotypic disease spectrum, which includes phenotypes, observed in both LS and a polyposis (Buchanan et al., 2017).
Figure 1.
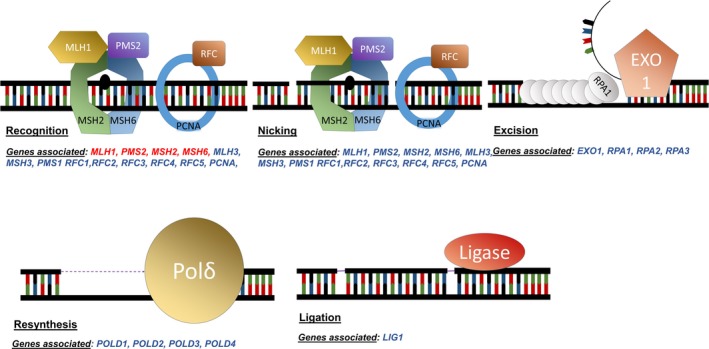
Mismatch repair pathway major steps with genes associated. Genes in red are the one usually screened for mutations in a clinical setting. MLH1, PMS2, MSH2, and MSH6 are all involved in the recognition of DNA damage. PMS2 has an endonuclease function in nicking around the damaged region. EXO1 will then remove the DNA strand containing the error and RPA (Replication Protein A) will protect the remaining single strand of DNA. The DNA polymerase Polδ resynthesises the new DNA strand which is then ligated with a ligase (based on Hsieh & Yamane, 2008)
To assess the involvement of other MMR genes in LS, we investigated the presence of potentially pathogenic variants in 22 MMR genes (MSH3, PMS1, MLH3 (OMIM: 604395), EXO1 (OMIM: 606063), POLD1 (OMIM: 174761), POLD3, RFC1 (OMIM: 102579), RFC2, RFC3, RFC4, RFC5, PCNA, LIG1, RPA1 (OMIM: 179835), RPA2, RPA3, POLD2, POLD4, MLH1, MSH2, MSH6, and PMS2), using next‐generation sequencing (NGS) in patients with a clinical diagnosis of LS. All of whom fulfilled the ACII or the revised BG but lacked a causative variant for the standard MMR gene(s) after genetic testing.
2. METHODS
2.1. Ethics approval
Ethics approval was obtained from the Hunter New England Human Research Ethics Committee (04/03/10/3.11) and the University of Newcastle Human Research Ethics Committee (H‐2008–0337).
2.2. Samples
This study used DNA obtained from 82 Norwegian and 192 Australian LLS patients (n = 274, see Table 1) previously described (Hansen et al., 2017). In brief, all patients fulfilled the AC II criteria or revised BG and had no pathogenic variant detected during routine genetic screening for the MMR gene(s) tested (MLH1, MSH2, MSH6, and/or PMS2). All patients were previously screened for one or more MMR genes as per their practician recommendations.
Table 1.
Cohort characteristics and screening results for the 274 samples included in the current study
| Total Cohort (N = 274) | |
|---|---|
| Nationality | |
| Norwegian | 82 |
| Australian | 192 |
| Female | 183 |
| Male | 91 |
| Median age at first cancera | 51.5 [21–86] |
| Cancer historyb | |
| CRC | 229 |
| Other cancers | 28 |
| Only adenomas | 14 |
| Multiple primary cancers | 64 |
| Amsterdam Criteria II | |
| Positive | 262 |
| Negativec | 12 |
| Microsatellite instability statusd | |
| MSS | 38 |
| MSI‐L | 6 |
| MSI‐H | 27 |
| IHCe | |
| Loss of MMR protein staining | 83 |
| Normal staining | 56 |
Data missing for six patients.
Data missing for three patients.
Revised Bethesda Guidelines (BG) positive.
Only available for the Norwegian patients. Data missing for 203 patients.
Data available for 68 Norwegian and 71 Australian samples. Data missing for 135 patients.
The sample cohort consisted of unrelated (Australian) and unrelated/related (Norwegian) individuals; eight families with two to three individuals per family were present in the Norwegian cohort (Hansen et al., 2017).
DNA samples from all patients were sequenced as part of the health‐care system and all patients have given written informed consent for their samples to be used for research. Ethics approval was obtained from relevant committees.
2.3. Gene panel sequencing
Sequencing data were generated (See Figure S1) from a 124 multigene panel study described in (Hansen et al., 2017), which contained 22 MMR genes (MLH1, MSH2, MSH6, PMS2, MSH3, PMS1, MLH3, EXO1, POLD1, POLD3, RFC1, RFC2, RFC3, RFC4, FRC5, PCNA, LIG1, RPA1, RPA2, RPA3, POLD2, and POLD4).
A custom Haloplex design (Agilent Technologies, Santa Clara, CA) was used for library preparation. Description of both the Haloplex design and the sequencing protocols (HiSeq 2500 and NextSeq, Illumina) have been reported previously (Hansen et al., 2017).
2.4. Data analysis
The previous (Hansen et al., 2017) study analyzed only 10 MMR genes, (MLH1, MSH2, MSH6, PMS2, MSH3, PMS1, MLH3, EXO1, POLD1, and POLD3). In this current study, we included data from those genes as well as data from the remaining 12 MMR genes (RFC1, RFC2, RFC3, RFC4, FRC5, PCNA, LIG1, RPA1, RPA2, RPA3, POLD2, and POLD4) to create a complete MMR gene panel (See Figure S1 for a full flowchart of the study's design).
Burrows‐Wheeler Aligner (BWA) (Li & Durbin, 2009) was utilized to align the paired end reads to the human genome (hg19, UCSC assembly, February 2009). BAM files were converted with SAMtools (Li et al., 2009). Variant calling was performed according to GATK Best Practice recommendations using GATK version 3.1 (McKenna et al., 2010) including local realignment around insertion/deletions (indels) and recalibration of quality scores. The variant caller HaplotypeCaller was utilized. Quality control of the called variants was performed using GATK variant filtration with parameter settings according to the recommendations in SEQanswers exome sequencing analysis guide (Van der Auwera et al., 2013). In short, variant quality score recalibration (VQSR) was applied using the recommended set of known variants for both indels and SNP. The tranche threshold of 99.0 was used to select variants. ANNOVAR (Wang, Li, & Hakonarson, 2010) was used to annotate detected variants and filtering of variants was done using the filtering tool FILTUS version 1.0.4 (Vigeland, Gjotterud, & Selmer, 2016).
2.5. Filtering of variants
FILTUS, a desktop software for fast and efficient detection of disease‐causing variants was used on the annotated files (Vigeland et al., 2016). The 10 MMR genes belonging to the gene panel previously analyzed by Hansen et al. (2017 were also included in the current study due to different filtering strategies and for comparative purposes.
Variants were filtered in the 22 MMR genes individually (gene lookup in FILTUS), with function collapse = lists all samples that have same variant together and saved as individual files on gene name, before being combined into one file. Variants with a frequency of more than 0.05 in public databases (ExaC or gNomad (Lek et al., 2016)) were first excluded. Then variants were excluded if detected in more than five unrelated individuals in our cohort (not likely to be pathogenic due to their high frequency) and intronic variants were ignored if they had no variant prediction. Nonsynonymous single‐nucleotide variants (SNVs) and indel variants were included in further filtering.
After in silico filtering, using FILTUS, we performed some manual filtering and variant interpretation to remove artifacts and only selecting variants most likely to be causative. We checked detected variants against results reported by Hansen et al. (2017. Further, variant interpretation was performed utilizing Alamut software (Interactive Biosoftware, Rouen, France) and evaluating available literature.
After the initial filtering step of each gene individually using FILTUS, we identified 29 potential pathogenic exonic indels and 170 exonic nonsynonymous SNVs. We applied the following nonsynonymous variant categorical predictions: AVSIFT score <0.05 = deleterious, LJB PhyloP prediction = conserved (>0.95), LJB SIFT = damaging (>0.99), LJB PolyPhen2 = damaging (>0.85), LJB LRT = deleterious (>0.99), MutationTaster = damaging (>0.59), GERP++ (>3.0 – except one with a score of 1.84) which resulted in 50 exonic nonsynonymous SNVs being identified that were predicted to be deleterious/damaging by in silico analysis. All of the variants identified in this study were heterozygous. After additional manual filtering, 25 possible causative variants remained in our list (see Table 2).
Table 2.
List of variants identified in LS screened MMR genes (MLH1, MSH2, and MSH6) and other MMR genes (EXO1, POLD1, RFC1, and RPA1)
| Gene | Hansen et al. (Hansen et al., 2017) | Reference sequence | DNA change | AA change | Domain | Rs IDa | gnomad AFa | Classification | Type | Sample ID |
|---|---|---|---|---|---|---|---|---|---|---|
| MLH1 | b | NM_000249.3 | c.514G > A | p.Glu172Lys | NR | NR | VUS | Missense | 250 | |
| MLH1 | NM_001167619.2 | c.1130A > C | Lys377Thr | rs63750449 | 0.004564 | VUS | Missense | 187 and 214 | ||
| MLH1 | b | NM_000249.3 | c.2103 + 1G>T | N/A | rs267607888 | N/A | Pathogenic | Missense | 116 | |
| MLH1 | NM_000249.3 | c.1039−31_1039−29delATA | N/A | rs778381149 | 0.0003969 | VUS | Intronic | 295 | ||
| MSH2 | b | NM_000251.2 | c.138C > G | p.His46Gln | rs33946261 | 0.0003619 | Likely Pathogenic | Missense | 9 | |
| MSH2 | b | NM_000251.2 | c.1045C > G | p.Pro349Ala | rs267607939 | 0.00009148 | Likely Pathogenic | Missense | 281 | |
| MSH6 | NM_000179.2 | c.431G > T | p.Ser144Ile | DNA binding PWWP domain | rs3211299 | 0.001187 | Benign | Missense | 201 | |
| MSH6 | NM_001281492.1 | c.892A > G | p.Lys298Glu | rs761822293 | 0.000003979 | VUS | Missense | 169 | ||
| MSH6 | NM_001281492.1 | c.1054C > T | p.Arg352X | rs63750909 | 0.00003186 | Pathogenic | Nonsense | 133 | ||
| MSH6 | NM_001281492.1 | c.1118C > G | p.Ser373Cys | rs63750897 | 0.001165 | Likely Benign | Missense | 225 | ||
| MSH6 | b | NM_000179.2 | c.1282A > G | p.Lys428Glu | rs761822293 | 0.000003979 | VUS | Missense | 169 | |
| MSH6 | b | NM_000179.2 | c.2079dup | p.Cys694Metfs*4 | rs267608083 | NR | Pathogenic | Frameshift | 183 | |
| MSH6 | b | NM_000179.2 | c.3261 dup | p.Phe1088Leufs*5 | rs748452299 | 0.0018 | Pathogenic | Frameshift | 41 | |
| EXO1 | NM_003686.4 | c.1928T > A | p.Leu643X | MLH1 and MSH2 interaction domain removed | NR | N/A | VUS | Nonsense | 165 | |
| EXO1 | NM_003686.4 | c.2009A > G | p.Glu670Gly | MSH2 interaction domain | rs1776148 | 0.78 | Benign | Missense | 166 | |
| EXO1 | NM_006027 0.4 | c.2485G > T | p.Glu829X | MLH1 interaction domain removed | rs757677420 | 0.00000292 | VUS | Nonsense | 154 | |
| POLD1 | NM_001256849.1 | c.1249A > G | p.Thr417Ala | DNA‐directed DNA polymerase, family B exonuclease domain | NR | N/A | VUS | Missense | 226 | |
| POLD1 | NM_001256849.1 | c.1558insG | p. | DNA‐directed DNA polymerase, family B, multifunctional domain | NR | N/A | VUS | Frameshift | 109 | |
| POLD1 | NM_001256849.1 | c. 2510G > C | p.Gly811Ala | DNA‐directed DNA polymerase, family B multifunctional domain | NR | N/A | VUS | Missense | 277 | |
| RFC1 | NM_001204747.1 | c.2017G > A | p.Val673Met | ATPase domain | rs28903096 | 0.0006994 | VUS | Missense | 174 | |
| RFC1 | NM_001204747.1 | c.2276A > G | p.Lys759Arg | ATPase domain | NR | N/A | VUS | Missense | 227 | |
| RPA1 | NM_002945.4 | c.856G > T | p.Val286Phe | rs55800538 | 0.002942 | VUS | Missense | 161 | ||
| RPA1 | NM_002945.4 | c.1160G > A | p.Gly387Asp | OB domain | NR | N/A | VUS | Missense | 239 | |
| RPA1 | NM_002945.4 | c.1165C > T | p.Arg389W | OB domain | rs202068855 | 0.0005468 | VUS | Missense | 221 | |
| MLH3 | b | NM_001040108.1 | c.885del | p.His296Thrfs*12 | NR | N/A | VUS | Frameshift | 14 |
Reported here are the gene symbol, Refseq reference sequence, DNA and amino acid (AA) change, protein domain, the rs ID of the variant if known, then gnomad allele frequency and the classification according to the ACMG 2015 guidelines (Richards et al., 2015).
Not reported (NR) ‐ previously unreported variant.
Variant previously identified in Hansen et al. (2017).
2.6. Validation of detected variants and segregation analysis by Sanger sequencing
Sanger sequencing was used to confirm possible pathogenic variants remaining after applying filtering steps described above. The fragments were amplified using AmpliTaq Gold® 360 MasterMix and 360 GC Enhancer (Life Technologies). Cycle sequencing reaction was performed with BigDye® Terminator v3.1 (Life Technologies) and subsequent capillary electrophoresis was performed on the ABI 3130xl or ABI 3730 (Life Technologies). Sanger sequencing data were analyzed using SeqScape Software v3.0 (Life Technologies).
3. RESULTS
After the final filtering, 25 possible causative variants remained (see Table 2). Of these 25 variants, six were predicted to be pathogenic (24%), all located in either MLH1 or MSH6.
3.1. Variants identified in MMR genes already linked to LS
We identified 13 variants in MLH1, MSH2, and MSH6, seven previously reported by Hansen et al. (2017 will not be discussed further. Two nonsynonymous variants were identified in MLH1; c.1130A > C and an intronic ATA deletion (rs778381149) upstream of exon 12 in a highly variable region of DNA. Two unrelated individuals were identified with the c.1130A > C variant.
In MSH6, four separate variants were found (each in different individuals); c.1054C > T, c.431G > T, c.1118C > G, and c.892A > G. The c.1054C > T variant is a nonsense variant resulting in a truncated protein p.R352X previously described as pathogenic (ClinVar variation ID: 89194). The c.431G > T and c.1118C > G variants are classified as benign and likely benign respectively, while c.892A > G is of unknown significance.
Of the 13 variants in MLH1, MSH2, and MSH6, four are considered pathogenic or likely pathogenic; two of the samples had not been tested for the gene where we found the variant previously (one due to IHC results indicating MLH1/PMS2 when the variant found is in MSH2). The other two were tested in late 1990s or early 2000.
3.2. Variants identified in the other 18 MMR genes
Five additional MMR genes were found to harbor 12 variants in our cohort (see Table 2). A frameshift variant in MLH3 will not be discussed further as it has been previously described (Hansen et al., 2017). EXO1, POLD1, RFC1, and RPA1 were all found to harbor variants, one benign and the remaining variants of unknown significance (VUSs). Moreover, the variants (except RFC1 c.2276A > G and POLD1 c.2510G > C) were nonconservative, suggesting the resultant amino acid is significantly different from the original.
EXO1 harbored three different variants; c.2009A > G is a missense variant leading to a change of amino acid from glutamic acid to glycine, p.(Glu670Gly), the variant affects the interaction domain between MSH2 and EXO1 but is predicted to be benign. Two nonsense variants were also identified in two different patients; c.1928T > A and c.2485G > T both predicted to result in a truncated protein p.L643X and p.E829X. The p.L643X variant misses both MLH1 and MSH2 interaction domains and the p.E829X misses only the MLH1 interaction domain. Failing to recruit the EXO1 protein with the recognition complex would lead to a defective MMR pathway, the accumulation of pathogenic variant and MSI.
POLD1 encodes for the catalytic subunit of the polymerase delta. Three different VUSs were detected in this gene. The c.1558insG is a frameshift variant that would most likely affect POLD1 mRNA, leading to RNA degradation and no protein expression. Two other missense variants were identified; c.1592A > G and c2510G > C both located between protein domains.
RFC1 codes for the subunit 1 of the Replication Factor C. Two different variants were identified; c.2017G > A and c.2276A > G. The two variants of RFC1 are both located on the ATPase functional domain of the protein subunit. RFC acts as a clamp loader for PCNA during DNA replication and damage repair. DNA resynthesis is significantly impaired by the loss of function of both RFC1 and PCNA.
Three variants were found in RPA1, namely c.856G > T, c.1165C > T and c.1160G > A. Two of these, c.1165C > T and c.1160G > A, were located in the oligonucleotide/oligosaccharide‐binding domain (OB) domain. The OB domain allows RPA1 protein to bind ssDNA in a nonsequence‐specific manner and to stabilize the single strand after the damaged strand is excised (Bochkareva, Korolev, Lees‐Miller, & Bochkarev, 2002). The remaining variant c.856G > T did not appear to alter the functional domain of the protein.
4. DISCUSSION
The presence of potentially pathogenic variants in patients diagnosed with LLS shows that there is a clear need to create an exhaustive list of pathogenic or potentially pathogenic genes for inherited CRC in order to identify individuals with a high risk of developing CRC and genes/variants appropriate for functional analysis. MMR genes are good candidates given that they are predicted to be causative in 8% of patients in the current study, a yield comparable to similar studies (Dong et al., 2018; Paulo et al., 2018). In addition, our results suggest that re‐screening the four known LS genes in previously variant‐negative LS patients with the more sensitive approach of NGS should be undertaken to ensure no pathogenic variants have been missed using less sensitive screening methods.
From the 274 patients enrolled in this study and not previously described we revealed 22 potentially causative variants in nine different MMR genes. Included in the study were the four known LS MMR genes (MSH2, MLH1, MSH6, and PMS2) as it is well accepted that older variant detection methods were not as sensitive as approaches that are more contemporary. The number of variants identified in MLH1 and MSH6 reflects the sensitivity of older screening methodologies that may not have revealed the presence of these causative variants. We detected additional variants in MLH1 and MSH6 compared to Hansen et al. (2017 due to less stringent filtering strategies in FILTUS.
The identification of 11 potentially pathogenic variants through in silico analysis in the extended MMR gene panel does reveal the extent to which the DNA MMR pathway might be associated with the risk of cancer development in families classified as LLS. We show here that families categorized as LLS harbor potentially pathogenic variants in other MMR genes than those already associated with LS. The genes EXO1, POLD1, RFC1, and RPA1 harbor that variants that were predicted to be pathogenic. None of the variants identified in this study have previously been associated with a cancer phenotype, which is probably due to their extremely low frequency in the general population. However, disrupting the MMR pathway could be a possible cause of cancer development. It is known that RFC1, RPA1, and POLD1 are involved in DNA damage repair mechanisms other than MMR and DNA synthesis during replication (see KEGG orthology: K10754, K02999 and K02327). EXO1 is involved with other DNA repair and maintenance mechanisms (Keijzers, Liu, & Rasmussen, 2016). Interestingly, the two nonsense EXO1 variants identified in our study are predicted to affect the binding to MSH2/MLH1, suggesting that the loss of function would specifically affect EXO1 MMR‐related functions. Moreover, RPA1 and POLD1 have been previously described as deleterious when mutated in cancer (Nicolas, Golemis, & Arora, 2016; Wang et al., 2005). Variants in POLD1 are described in patients presenting a polyposis phenotype termed polymerase‐proofreading associated polyposis (PPAP) (Palles et al., 2013). The pathogenic variants identified in POLD1 in the current study support the notion that the phenotypes of LS and PPAP might overlap, both with a multitumor phenotype.
RPA1 variants have previously been implicated in cancers that are associated with chromosome instability (Hass, Gakhar, & Wold, 2010; Wang et al., 2005). However, a comprehensive genetic study did not show a clear association with CRC (Jokic et al., 2011).
A meta‐analysis supports a significant association between RFC1 p.G80A and plasma cell malignancies (Huang et al., 2016). Moreover, the Cancer Genome Atlas (TCGA data) (Cancer Genome Atlas Research et al., 2013) shows that somatic RFC1 variants occur in 10.2% of uterine cancers and 5.5% of CRCs, which is consistent with tumors identified in LS. RFC1 has been previously described as a member of the BRCA1‐associated genome surveillance complex (BASC) (Wang et al., 2000). This complex is involved in DNA damage and abnormal structure detection and more generally in the maintenance of genomic integrity making it a good candidate gene for LLS.
Pathogenic variants in EXO1 were also found in the current study but its exact role remains to be determined. A previous study suggests that the gene is either associated with low‐disease penetrance or influencing a polygenic risk score (Talseth‐Palmer et al., 2016). Other reports indicate that even in healthy patients, EXO1 variants are present, including those that result in a truncated protein (Jagmohan‐Changur et al., 2003) and consequently loss of function. Notwithstanding, in our study, both EXO1 c.1928T > A and c.2485G > T leads to a truncated protein. The variant c.1928T > A has lost the MLH2 and MLH1 interaction domain, whereas the c.2485G > T variant truncates only the MLH1 interaction domain. Lack of either of these domains could affect EXO1 recruitment at the site of the mismatch or DNA damage, impairing the MMR process (Goellner, Putnam, & Kolodner, 2015).
To assess the link between the variants identified in the current study and the development of CRC in LLS families, larger sample cohorts are needed with detailed analysis of the tumor phenotypes to establish if indeed many of the downstream MMR functions are associated with MSI tumors. Furthermore, detailed segregation analysis is required to determine if the variant segregates with disease. Finally, functional analysis would significantly aid in characterizing their respective pathogenic effects.
Limitations of the current study include the relatively small number of patients tested. A larger sample cohort and functional studies of the identified variants are required to confirm the results of this study. Segregation analysis would provide insights into the pathogenicity of these variants but could not be performed as part of this study.
In conclusion, we have shown that other genes associated with the process of DNA MMR have a high probability of being associated with LLS families. In addition, approximately 8% of families that fulfill the ACII or RB criteria in our sample cohort appear to be accounted for by genes involved in the MMR pathway. These findings indicate that these variants are important as they will guide future research focused on the functional impact of newly discovered variants.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
This work was supported by the Liaison Committee between the Central Norway Regional Health Authority (RHA) and the Norwegian University of Science and Technology (NTNU), Norway; Møre and Romsdal Hospital Trust, Norway; the Hunter Cancer Research Alliance (HCRA) and the Cancer Institute NSW, Australia.
Xavier A, Olsen MF, Lavik LA, et al. Comprehensive mismatch repair gene panel identifies variants in patients with Lynch‐like syndrome. Mol Genet Genomic Med. 2019;7:e850 10.1002/mgg3.850
REFERENCES
- Adam, R. , Spier, I. , Zhao, B. , Kloth, M. , Marquez, J. , Hinrichsen, I. , … Aretz, S. (2016). Exome sequencing identifies biallelic MSH3 germline mutations as a recessive subtype of colorectal adenomatous polyposis. American Journal of Human Genetics, 99(2), 337–351. 10.1016/j.ajhg.2016.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkareva, E. , Korolev, S. , Lees‐Miller, S. P. , & Bochkarev, A. (2002). Structure of the RPA trimerization core and its role in the multistep DNA‐binding mechanism of RPA. EMBO Journal, 21(7), 1855–1863. 10.1093/emboj/21.7.1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonis, P. A. , Trikalinos, T. A. , Chung, M. , Chew, P. , Ip, S. , DeVine, D. A. , & Lau, J. (2007). Hereditary nonpolyposis colorectal cancer: Diagnostic strategies and their implications. Evid Rep Technol Assess (Full Rep), (150), 1–180. [PMC free article] [PubMed] [Google Scholar]
- Buchanan, D. D. , Stewart, J. R. , Clendenning, M. , Rosty, C. , Mahmood, K. , Pope, B. J. , … Win, A. K. (2017). Risk of colorectal cancer for carriers of a germ‐line mutation in POLE or POLD1. Genetics in Medicine, 20(8), 890–895. 10.1038/gim.2017.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carethers, J. M. (2014). Differentiating Lynch‐like from Lynch syndrome. Gastroenterology, 146(3), 602–604. 10.1053/j.gastro.2014.01.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carethers, J. M. , Koi, M. , & Tseng‐Rogenski, S. S. (2015). EMAST is a form of microsatellite instability that is initiated by inflammation and modulates colorectal cancer progression. Genes, 6(2), 185–205. 10.3390/genes6020185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Chapelle, A. (2004). Genetic predisposition to colorectal cancer. Nature Reviews Cancer, 4(10), 769–780. 10.1038/nrc1453 [DOI] [PubMed] [Google Scholar]
- Dong, L. I. , Wu, N. , Wang, S. , Cheng, Y. , Han, L. , Zhao, J. , … Hao, X. (2018). Detection of novel germline mutations in six breast cancer predisposition genes by targeted next‐generation sequencing. Human Mutation, 39(10), 1442–1455. 10.1002/humu.23597 [DOI] [PubMed] [Google Scholar]
- Duraturo, F. , Liccardo, R. , Cavallo, A. , De Rosa, M. , Grosso, M. , & Izzo, P. (2011). Association of low‐risk MSH3 and MSH2 variant alleles with Lynch syndrome: Probability of synergistic effects. International Journal of Cancer, 129(7), 1643–1650. 10.1002/ijc.25824 [DOI] [PubMed] [Google Scholar]
- Fishel, R. (2015). Mismatch repair. Journal of Biological Chemistry, 290(44), 26395–26403. 10.1074/jbc.R115.660142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardiello, F. M. , Allen, J. I. , Axilbund, J. E. , Boland, R. C. , Burke, C. A. , Burt, R. W. , … Rex, D. K. (2014). Guidelines on genetic evaluation and management of Lynch syndrome: A consensus statement by the US Multi‐society Task Force on colorectal cancer. American Journal of Gastroenterology, 109(8), 1159–1179. 10.1038/ajg.2014.186 [DOI] [PubMed] [Google Scholar]
- Goellner, E. M. , Putnam, C. D. , & Kolodner, R. D. (2015). Exonuclease 1‐dependent and independent mismatch repair. DNA Repair, 32, 24–32. 10.1016/j.dnarep.2015.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel, H. , Frankel, W. L. , Martin, E. , Arnold, M. , Khanduja, K. , Kuebler, P. , … de la Chapelle, A. (2005). Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). New England Journal of Medicine, 352(18), 1851–1860. 10.1056/NEJMoa043146 [DOI] [PubMed] [Google Scholar]
- Hansen, M. F. , Johansen, J. , Sylvander, A. E. , Bjørnevoll, I. , Talseth‐Palmer, B. A. , Lavik, L. A. S. , … Sjursen, W. (2017). Use of multigene‐panel identifies pathogenic variants in several CRC‐predisposing genes in patients previously tested for Lynch Syndrome. Clinical Genetics, 92(4), 405–414. 10.1111/cge.12994 [DOI] [PubMed] [Google Scholar]
- Hass, C. S. , Gakhar, L. , & Wold, M. S. (2010). Functional characterization of a cancer causing mutation in human replication protein A. Molecular Cancer Research, 8(7), 1017–1026. 10.1158/1541-7786.MCR-10-0161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh, P. , & Yamane, K. (2008). DNA mismatch repair: Molecular mechanism, cancer, and ageing. Mechanisms of Ageing and Development, 129(7–8), 391–407. 10.1016/j.mad.2008.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X. , Gao, Y. , He, J. , Cai, J. , Ta, N. , Jiang, H. , … Zheng, J. (2016). The association between RFC1 G80A polymorphism and cancer susceptibility: Evidence from 33 studies. J Cancer, 7(2), 144–152. 10.7150/jca.13303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagmohan‐Changur, S. , Poikonen, T. , Vilkki, S. , Launonen, V. , Wikman, F. , Orntoft, T. F. , … Karhu, A. (2003). EXO1 variants occur commonly in normal population: Evidence against a role in hereditary nonpolyposis colorectal cancer. Cancer Research, 63(1), 154–158. [PubMed] [Google Scholar]
- Jokic, M. , Brcic‐Kostic, K. , Stefulj, J. , Catela Ivkovic, T. , Bozo, L. , Gamulin, M. , & Kapitanovic, S. (2011). Association of MTHFR, MTR, MTRR, RFC1, and DHFR gene polymorphisms with susceptibility to sporadic colon cancer. DNA and Cell Biology, 30(10), 771–776. 10.1089/dna.2010.1189 [DOI] [PubMed] [Google Scholar]
- Kanehisa, M. , Furumichi, M. , Tanabe, M. , Sato, Y. , & Morishima, K. (2017). KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Research, 45(D1), D353–D361. 10.1093/nar/gkw1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keijzers, G. , Liu, D. , & Rasmussen, L. J. (2016). Exonuclease 1 and its versatile roles in DNA repair. Critical Reviews in Biochemistry and Molecular Biology, 51(6), 440–451. 10.1080/10409238.2016.1215407 [DOI] [PubMed] [Google Scholar]
- Kuiper, R. P. , Vissers, L. E. L. M. , Venkatachalam, R. , Bodmer, D. , Hoenselaar, E. , Goossens, M. , … Ligtenberg, M. J. L. (2011). Recurrence and variability of germline EPCAM deletions in Lynch syndrome. Human Mutation, 32(4), 407–414. 10.1002/humu.21446 [DOI] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , … MacArthur, D. G. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , & Homer, N. , … 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16), 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindor, N. M. , Rabe, K. , Petersen, G. M. , Haile, R. , Casey, G. , Baron, J. , … Seminara, D. (2005). Lower cancer incidence in Amsterdam‐I criteria families without mismatch repair deficiency: Familial colorectal cancer type X. JAMA, 293(16), 1979–1985. 10.1001/jama.293.16.1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkin, S. M. , & Afrasiabi, K. (2007). Familial colorectal cancer syndrome X. Seminars in Oncology, 34(5), 425–427. 10.1053/j.seminoncol.2007.07.008 [DOI] [PubMed] [Google Scholar]
- Liu, H. X. , Zhou, X. L. , Liu, T. , Werelius, B. , Lindmark, G. , Dahl, N. , & Lindblom, A. (2003). The role of hMLH3 in familial colorectal cancer. Cancer Research, 63(8), 1894–1899. [PubMed] [Google Scholar]
- Lynch, H. T. , & de la Chapelle, A. (1999). Genetic susceptibility to non‐polyposis colorectal cancer. Journal of Medical Genetics, 36(11), 801–818. [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , … DePristo, M. A. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20(9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas, E. , Golemis, E. A. , & Arora, S. (2016). POLD1: Central mediator of DNA replication and repair, and implication in cancer and other pathologies. Gene, 590(1), 128–141. 10.1016/j.gene.2016.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palles, C. , Cazier, J.‐B. , Howarth, K. M. , Domingo, E. , Jones, A. M. , Broderick, P. , … Tomlinson, I. (2013). Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nature Genetics, 45(2), 136–144. 10.1038/ng.2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo, P. , Maia, S. , Pinto, C. , Pinto, P. , Monteiro, A. , Peixoto, A. , & Teixeira, M. R. (2018). Targeted next generation sequencing identifies functionally deleterious germline mutations in novel genes in early‐onset/familial prostate cancer. PLoS Genetics, 14(4), e1007355 10.1371/journal.pgen.1007355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Bigas, M. A. , Boland, C. R. , Hamilton, S. R. , Henson, D. E. , Srivastava, S. , Jass, J. R. , … Sobin, L. (1997). A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: Meeting highlights and Bethesda guidelines. Journal of the National Cancer Institute, 89(23), 1758–1762. 10.1093/jnci/89.23.1758 [DOI] [PubMed] [Google Scholar]
- Samadder, N. J. , Smith, K. R. , Wong, J. , Thomas, A. , Hanson, H. , Boucher, K. , … Curtin, K. (2017). Cancer Risk in Families Fulfilling the Amsterdam Criteria for Lynch Syndrome. JAMA Oncol, 3(12), 1697–1701. 10.1001/jamaoncol.2017.0769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinke, V. , Holzapfel, S. , Loeffler, M. , Holinski‐Feder, E. , Morak, M. , Schackert, H. K. , … Engel, C. (2014). Evaluating the performance of clinical criteria for predicting mismatch repair gene mutations in Lynch syndrome: A comprehensive analysis of 3,671 families. International Journal of Cancer, 135(1), 69–77. 10.1002/ijc.28650 [DOI] [PubMed] [Google Scholar]
- Talseth‐Palmer, B. A. , Bauer, D. C. , Sjursen, W. , Evans, T. J. , McPhillips, M. , Proietto, A. , … Scott, R. J. (2016). Targeted next‐generation sequencing of 22 mismatch repair genes identifies Lynch syndrome families. Cancer Medicine, 5(5), 929–941. 10.1002/cam4.628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umar, A. , Boland, C. R. , Terdiman, J. P. , Syngal, S. , Chapelle, A. D. L. , Ruschoff, J. , … Srivastava, S. (2004). Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. Journal of the National Cancer Institute, 96(4), 261–268. 10.1093/jnci/djh034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera, G. A. , Carneiro, M. O. , Hartl, C. , Poplin, R. , Del Angel, G. , Levy‐Moonshine, A. , DePristo, M. A. . (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics, 43, 11.10.1–11.10.33. 10.1002/0471250953.bi1110s43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasen, H. F. , Mecklin, J. P. , Khan, P. M. , & Lynch, H. T. (1991). The International Collaborative Group on Hereditary Non‐Polyposis Colorectal Cancer (ICG‐HNPCC). Diseases of the Colon and Rectum, 34(5), 424–425. 10.1007/BF02053699 [DOI] [PubMed] [Google Scholar]
- Vasen, H. F. , Watson, P. , Mecklin, J. P. , & Lynch, H. T. (1999). New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology, 116(6), 1453–1456. 10.1016/S0016-5085(99)70510-X [DOI] [PubMed] [Google Scholar]
- Vigeland, M. D. , Gjotterud, K. S. , & Selmer, K. K. (2016). FILTUS: A desktop GUI for fast and efficient detection of disease‐causing variants, including a novel autozygosity detector. Bioinformatics, 32(10), 1592–1594. 10.1093/bioinformatics/btw046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. , Li, M. , & Hakonarson, H. (2010). ANNOVAR: Functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Research, 38(16), e164 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Cortez, D. , Yazdi, P. , Neff, N. , Elledge, S. J. , & Qin, J. (2000). BASC, a super complex of BRCA1‐associated proteins involved in the recognition and repair of aberrant DNA structures. Genes & Development, 14(8), 927–939. [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Putnam, C. D. , Kane, M. F. , Zhang, W. , Edelmann, L. , Russell, R. , … Edelmann, W. (2005). Mutation in Rpa1 results in defective DNA double‐strand break repair, chromosomal instability and cancer in mice. Nature Genetics, 37(7), 750–755. 10.1038/ng1587 [DOI] [PubMed] [Google Scholar]
- Watson, P. , & Riley, B. (2005). The tumor spectrum in the Lynch syndrome. Familial Cancer, 4(3), 245–248. 10.1007/s10689-004-7994-z [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network , Weinstein, J. N. , Collisson, E. A. , Mills, G. B. , Shaw, K. R. , & Ozenberger, B. A. , … Stuart, J. M. (2013). The Cancer Genome Atlas Pan‐Cancer analysis project. Nature Genetics, 45(10), 1113–1120. 10.1038/ng.2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials