

Abstract
Background
Retinoblastoma (Rb) is the most prevalent intraocular pediatric malignancy of the retina. Significant genetic factors are known to have a role in the development of Rb.
Methods
Here, we report the mutation status of 4813 clinically significant genes in six patients with noncarrier of RB1 gene mutation and having normal RB1 promoter methylation from three families having higher risk for developing Rb in the study.
Results
A total of 27 variants were detected in the study. Heterozygous missense variants c.1162G > A (p.Gly388Arg) in the FGFR4 gene; c.559C > T (p.Pro187Ser) in the NQO1 gene were identified. The family based evaluation of the variants showed that the variant, c.714T > G (p.Tyr238Ter), in the CLEC7A gene in first family; the variant, c.55C > T (p.Arg19Ter), in the APOC3 gene and the variant, c.1171C > T (p.Gln391Ter), in the MUTYH gene in second family; and the variant, c.211G > A (p.Gly71Arg), in the UGT1A1 gene in the third family, were found statistically significant (p < 0.05).
Conclusion
This study might be an important report on emphazing the mutational status of other genes in patients without RB1 gene mutations and having high risk for developing Rb. The study also indicates the interaction between the retinoic acid pathway and Rb oncogenesis for the first time.
Keywords: mutation, next‐generation sequencing, RB1 gene, retinoblastoma, retinoic acid pathway
1. INTRODUCTION
The effects of tumor supressor genes in cancer were first identified in retinoblastoma (Rb) which is a rare pediatric cancer (Knudson, 1971). Therefore, retinoblastoma was described as a model system for the better understanding of the tumor suppressor genes. Rb is the most prevalent intraocular pediatric malignancy of the retina (Jagadeesan, Khetan, & Mallipatna, 2016). Rb is usually reported as two different forms; hereditary in 25%–35%, and nonhereditary in 65%–75%. Eighty‐five per cent of hereditary tumors are detected in the early age (Murphree, Samuel, Harbour, & Mansfield, 2006). Children with bilateral Rb account for approximately 40% of the patients (Draper, Sanders, Brownbill, & Hawkins, 1992). Aproximately, 20% of children diagnosed with bilateral Rb have a family history (Chintagumpala, Chevez‐Barrios, Paysse, Plon, & Hurwitz, 2007). All bilateral tumors are hereditary, some of the unilateral may be hereditary as well. Patients with hereditary Rb have a risk for developing secondary malignancies such as osteosarcoma, soft tissue sarcomas and melanomas (Wong et al., 1997).
The incidence of Rb is higher in developing countries (Pandey, 2014). The cause of this high incidence rate is unknown. Significant genetic factors are known to have a role in the development of Rb. The disease is known to be initiated by the mutations in the retinoblastoma gene (RB1) in accordance with the current literature. The RB1 gene (Gene ID: 5925, OMIM 614041) produces a nuclear protein called pRB weighing 105 kD. This protein functions as a tumor suppressor, and is involved in the cell regulation, proliferation, and prevents rapid or uncontrolled division of cells (Chaussade et al., 2018).
RB1 gene includes a wide variety of mutations, including single nucleotide variations, small insertions and deletions (INDELs), and large deletions or duplications. The genetic tests include the screening of genome of 27 exons of RB1 gene, and close intronic areas by Sanger sequencing, and detection of large rearrangements (large deletions, and duplications) by MLPA analysis. However, these methods are time and money consuming. The next‐generation sequencing (NGS) technology is an important research tool which is an effective and high throughput. However, it is unclear how the disease develops in patients who are noncarriers for the mutations of RB1 gene including large rearrangements after the mutation screening by Sanger method and MLPA analysis. The stuctural alterations of the other genes would be suggested to be responsible in the development of the disease detected in childhood. According to the recent literature, although many gene expression profiles (Chakraborty et al., 2007; Ganguly & Shields, 2010), and methylation levels (Indovina et al., 2010; Livide et al., 2012) were investigated in Rb disease, no information on the structural alterations of different genes were reported. Therefore, the role of the mutations in other genes that may be responsible for the disease occurrence is still unclear in Rb pathogenesis. The aim of this study was to investigate possible candidate genes associated with Rb oncogenesis in retinoblastoma patients without RB1 gene mutations including INDELS and large rearrangements and having normal RB1 promoter methylation and having a heavy family history by using NGS‐based technology.
2. MATERIALS AND METHODS
2.1. Editorial policies and ethical considerations
The study was approved by the Local and Clinical Research Ethics Committee of Istanbul University (Number of ethical approvall: 2016‐360); according to the tenets of the Declaration of Helsinki (JAMA 1997; 277:925‐926). Written informed consent was obtained from all participants or parents of children under 18 years of age. This work was supported by Scientific Research Projects Coordination Unit of Istanbul University (Project number: 21460).
2.2. Clinical diagnosis and patients
Five patients diagnosed with Rb and one patient with the retinoma, all diagnosed and treated in the Istanbul University, Oncology Institute, Division of Pediatric Hematology‐Oncology and in the Istanbul University, Istanbul Medical Faculty, Department of Ophtalmology between 2011 and 2016 were enrolled in the study. The blood specimens were collected from the patients without RB1 mutation including large rearrangements and without RB1 promoter methylation from three families. In two members of each three families RB1(RefSeq NM_000321.2 and chromosome 13 co‐ordinates in hg19) gene mutation was initially screened for small INDEL mutation with Sanger Sequencing and for large rearrengements by MLPA analysis. In the first family; a unilateral Rb patient and his second degree relative with retinoma were tested for RB1 gene mutation. In the second family; again a unilateral Rb patient, who has fibrosarcoma, and his first degree cousin with retinoblastoma were investigated for RB1 gene mutation. In the third family; two siblings with bilateral Rb were tested for RB1 gene mutation. At least two members from the same family who had retinoblastoma or retinoma without RB1 gene mutations were selected. Thus six patients from three families with two members in each family were selected (Table 1).
Table 1.
The clinical features of the patients
| Family ID | Patient no.a | Diagnosis and laterality | Tumor site/stageb | Age of diagnosis (months)/gender | Leukocoria | Treatment | OS (year)/final situation | Consanguinity |
|---|---|---|---|---|---|---|---|---|
| Fm1 | 1/II‐7 | Unilateral retinoma | L | 18/M | − | − | 32 years/alive | Uncle |
| Fm1 | 1/III‐2 | Unilateral Rb | L/Group C | 8/M | + | CT; LOT | 6 years/alive | Nephew |
| Fm2 | 2/IV‐2 | Unilateral Rb | L/Group E | 8/M | + | CT; RT; Enucleation(L) | 11 years/dead due to fibrosarcoma | Cousin |
| Fm2 | 2/IV‐7 | Bilateral Rb |
L/Group A R/Group D |
1.5/F | + | CT; LOT | 8 years/alive | Cousin |
| Fm3 | 3/III‐1 | Bilateral Rb |
L/Group B R/Group D |
7/M | + | CT; LOT; IAC; Enucleation(R) | 3 years/alive | Brother |
| Fm3 | 3/III‐2 | Bilateral Rb |
L/Group B R/Group E |
7/F | − | CT; LOT; IAC; Enucleation(R) | 6 years/alive | Sister |
Abbreviations: Fm, family; M, male; F, female; L, left eye; R, right eye; RT, radiotherapy; CT, chemotherapy; IAC, intraarterial chemotherapy; LOT, local ophthalmic treatment; OS, Overall Survival; FS, Final Situation
Patient numbers are coded according to the order in the family pedigree
Staging according to IRBC.
2.3. DNA sample preparation
The peripheral blood samples were collected from available members of the three families. Genomic DNA of all six patients were searched TruSight One panel of 4813 genes associated with human disease by NGS‐based sequencing technology.
First, lymphocyte isolation was performed from the whole blood samples using the Ficoll (Sigma‐Aldrich, Darmstadt, Germany) separation method. The DNA isolation was performed from the pellets of lymphocytes using the QIAamp DNA mini kit (Qiagen, 40724 Hilden, Germany) in accordance with the manufacturer's instruction. Quantification of genomic DNAs was measured by Qubit fluorimeter (ThermoFisher Scientific, Paisley PA4 9RF, UK) and then the concentration of DNAs was adjusted to 10 ng/µl using 10 mM pH 8.5 Tris‐HCl. The fluorometric measurement was repeated, and the concentration was adjusted to 5 ng/µl with the same buffer solution, and 50 ng was prepared for use.
2.4. Library generation and next‐generation sequencing
The TruSight One “clinical exome” panel kit (Illumina, San Diego, CA) was used for sequencing the whole gene regions of 4813 genes associated with human disease in the study. In accordance with the kit protocol; genomic DNA tagmentation, cleaning up of the tagmented DNA, cleaning up of the accumulated DNA, hybridization of the probes, catching the hybridized probes, second hybridization, second catch, cleaning up of the catched library, accumulation of the enriched library, cleaning up of the accumulated enriched library, and bioanalyser device (Agilent, Santa Clara, CA) were performed. The generated library was sequenced on the Illumina NextSeq 500 device (Illumina, San Diego, CA) in accordance with the manufacturer's instructions. The 27 pathogenic variants were identified in selected six patients from three families are indicated position according to reference transcript ACADS (NM_000017.3); APOC3 (NM_000040.2); ATP6V0A4 (NM_020632.2); C2 (NM_000063.4); CFB (NM_001710.5); CLEC7A (NM_197947.2); CX3CR1 (NM_001171174.1); DSPP (NM_014208.3); FGFR4 (NM_002011.4); FUT6 (NM_000150.2); GBE1 (NM_000158.3); GHRL (NM_001134944.1); GNPAT (NM_014236.3); HBD (NM_000519.3); HFE (NM_000410.3); KRT85 (NM_002283.3); MBL2 (NM_000242.2); MCCC2 (NM_022132.4); MUTYH (NM_001128425.1); NQO1 (NM_000903.2); RHAG (NM_000324.2); RPGRIP1 (NM_020366.3); SERPINA1 (NM_001002235.2); SLC34A1 (NM_003052.4); TYR (NM_000372.4); UGT1A1 (NM_000463.2).
2.5. Data analysis and interpretation of the results
The Variant Studio v3.0 (Illumina) software was used for the analysis of data. The data obtained after sequencing from the Illumına NextSeq 500 device were first converted into VCF file format, and the files were uploaded to the software program using the Illumina VariantStudio desktop receiver. The data were annoted in the Illumina VariantStudio program. The comprehensive database of this software catches the explanations in variant, gene, and transcript levels. The variant effect predictor is a central resource for the annotation of the transcript results (McLaren et al., 2016), which is a variant program that uses the databases such as NCBI Reference sequence database (RefSeq) (O'Leary et al., 2016), and the in silico algorithms such as Polymorphism Phenotyping (PolyPhen) (Adzhubei et al., 2010), and SIFT (Kumar, Henikoff, & Ng, 2009). The information about the association with the disease was obtained through the Catalogue of Somatic Mutations in Cancer (COSMIC) (Forbes et al., 2017), from ClinVar database (Landrum et al., 2018), and from the catalogue of the Online Mendelian Inheritance in Man (OMIM) (McKusick, 2007). The resources, dbSNP (Sherry et al., 2001), Exome Aggregation Consortium (ExAC) and Genome Aggregation Database (gnomAD) (Lek et al., 2016), and Ensembl 1000 Genomes Project (Genomes Project et al., 2015) provide information about the occurrence, and frequencies of the variants in a population. The obtained variants were evaluated considering the >Q30 reading quality, and >50 confidence score. All the data about the variants, and information on the algorithms were evaluated in all the related databases. Various filtering options were used for identification of the phenotypes of the variants that were performed annotation procedure. In the study, the variants with particularly pathogenic according to ClinVar records were investigated in details. The variants have not been previously reported in the literature or databases with Rb were identified as candidate variants. The defined variants were labeled in accordance with the recommendation standards of the American College of Medical Genetics, and Genomics (ACMG) (Richards et al., 2008). In order to confirm the all pathogenic variants identified by NGS, PCR amplification, and bidirectional Sanger sequencing was performed using standard reagents and conditions, and oligonucleotide primers flanking the variants.
2.6. The analysis of the functional association between genes
The database for annotation, visualization and integrated discovery (DAVID) v.6.8 [Laboratory of Immunopathogenesis and Bioinformatics (LIB)], and STRING Functional Protein Association Network v.10.5 were used for the interpretation of the functional association between the genes that were known to have pathogenic variants after the analysis (Huang da, Sherman, & Lempicki, 2009a, 2009b; Szklarczyk et al., 2017).
2.7. Statistical analysis
All clinical and genetic data were evaluated using the IBM Statistical Package for the Social Sciences (SPSS) Statistics v.20 (SPSS Inc., Chicago, IL) program. The Chi‐square test was used to compare the results of VariantStudio analysis for both based on patient, and between the patients for the clinical, and genetic data. The results with a p < 0.05 were accepted as statistically significant.
3. RESULTS
3.1. The clinical and genetic information of the patients
Six patients from three families with two members in each family who were noncarriers of RB1 gene mutations and normal RB1 promoter methylation were selected. The cases in the first family consisted of an uncle and a nephew, in the second family consisted of two of five first‐degree cousins, and in the third family consisted of two siblings. The uncle in the first family was diagnosed with unilateral retinoma and has been under follow‐up. Two patients (33.3%) had unilateral Rb, three patients (50%) had bilateral Rb and one (16.7%) had unilateral retinoma. Four patients (66.7%) were male and two were female (33.3%). The median age of the patients was 7.5 months with arange of 1.5–18 months at diagnosis. Four patients (66.7%) had presented with leukocoria and esotropia; one patient (16.7%) had exotropia at diagnosis. One of the patients (2/IV‐2) developed fibrosarcoma as a second malignancy 104/12 years after the diagnosis of retinoblastoma at the radiation site and died due to progressive disease.
Four patients, patients 1/III‐2, 2/IV‐7, 3/III‐1, 3/III‐2, recieved systemic chemotherapy (CT) for chemoreduction and local ophthalmic treatment (LOT) (laser, cryotherapy); one patient, 2/IV‐2, had CT and radiotherapy (RT) and underwent enucleation due to relapse. Two patients, 3/III‐1 and 3/III‐2, recieved CT and LOT, on follow‐up developed new lesions, they received intraarterial chemotherapy, due to further progression underwent enucleation. Only one patient, 2/IV‐2, developed fibrosarcoma, 11 years after primary treatment in the irradiated site. The clinical characteristics, treatment and outcome of the six patients are given in Table 1. The pedigrees of families who were included in the study are given in Figures 1, 2, 3.
Figure 1.
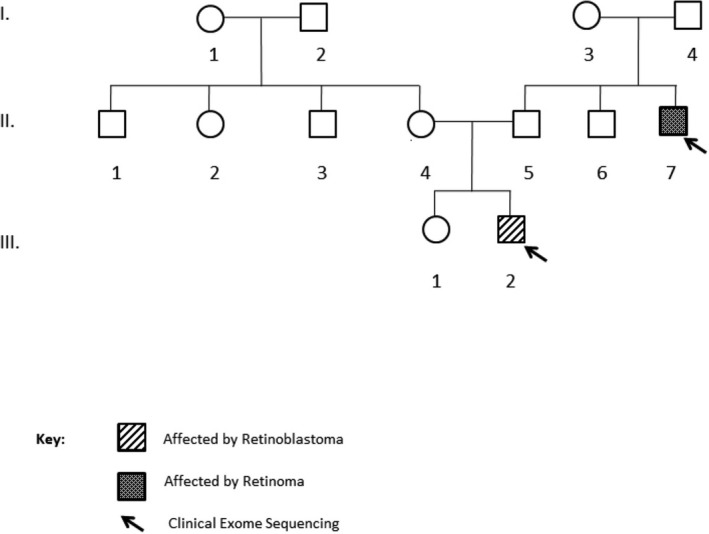
The pedigree of first family. The affected individuals were illustrated with filled box. Two family members marked by arrows were chosen for DNA sequencing, 4813 genes which are clinically important were sequenced by NGS. The sequenced genes were named as a Clinical Exome Sequencing
Figure 2.
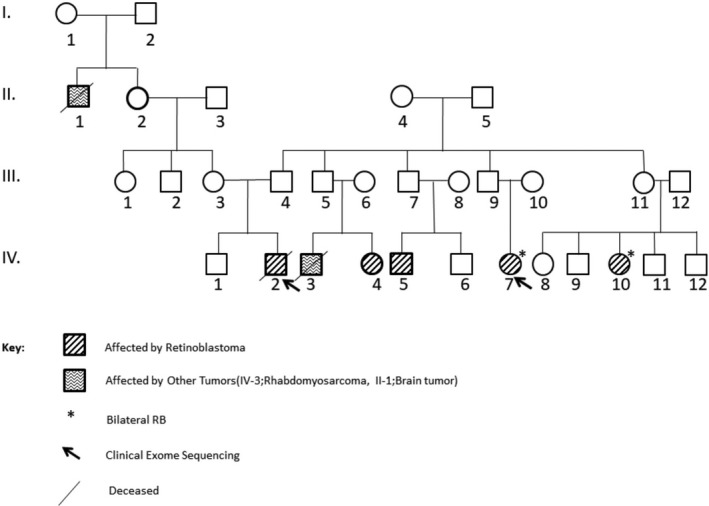
The pedigree of second family. The affected individuals were illustrated with filled box. Two family members marked by arrows were chosen for DNA sequencing, 4813 genes which are clinically important were sequenced by NGS. The sequenced genes were named as a Clinical Exome Sequencing
Figure 3.
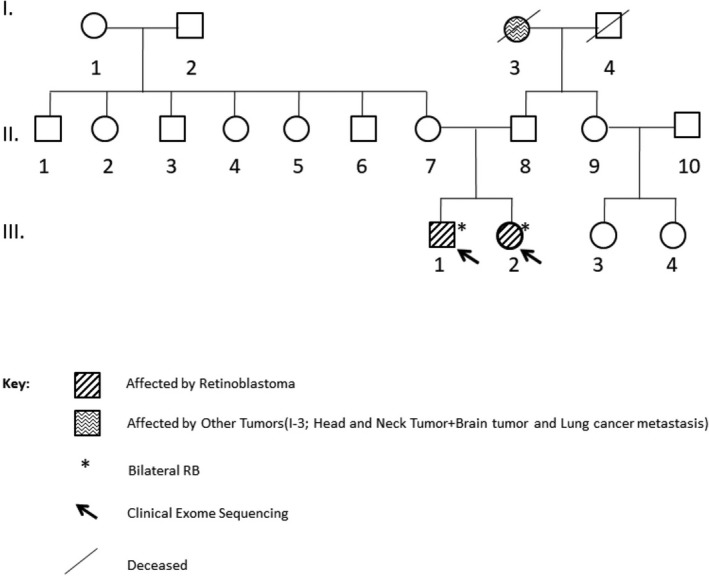
The pedigree of third family. The affected individuals were illustrated with filled box. Two family members marked by arrows were chosen for DNA sequencing, 4813 genes which are clinically important were sequenced by NGS. The sequenced genes were named as a Clinical Exome Sequencing
3.2. Evaluation of the analysis results
The mutation status of 4813 clinically significant genes were screened using the TruSight One “clinical exome” panel by NGS in RB1‐negative six patients from three families. The number of variants for each patient before, and after the annotation and filtration process are shown in Table 2.
Table 2.
The numbers of variants found in patients after the process of annotation and filtering
| Family ID | Patient noa | Number of variants after annotation | Number of variants after filtering | Number of deleterious variants | Number of genes having deleterious variant |
|---|---|---|---|---|---|
| Fm1 | 1/II‐7 | 121.757 | 17 | 15 | 15 |
| Fm1 | 1/III‐2 | 107.327 | 9 | 7 | 7 |
| Fm2 | 2/IV‐2 | 60.491 | 8 | 8 | 8 |
| Fm2 | 2/IV‐7 | 99.490 | 13 | 10 | 10 |
| Fm3 | 3/III‐1 | 108.627 | 9 | 5 | 5 |
| Fm3 | 3/III‐2 | 110.976 | 7 | 4 | 4 |
Abbreviation: Fm, family.
Patient numbers are coded according to the order in the family pedigree.
A total of 608.668 variants were found in the evaluation of the sequenced data of all patients (1/II‐7, 1/III‐2, 2/IV‐2, 2/IV‐7, 3/III‐1, 3/III‐2). However, the number of these variants decreased to 63 when the variants were filtered according to ClinVar pathogenic records about frameshift, stop gained, stop lost, initiator codon, inframe insertion, inframe deletion, and splice region mutations and according to Polyphen for “damaging” and to SIFT for “deleterious” about missense. Then, 27 pathogenic variants were detected after scanning on ALAMUT, HGMD and dbSNP databases. The information of the variants is shown in Table 3.
Table 3.
The list of 27 pathogenic mutations
| Patient No. | Genes (Reference transcript according to HGVS) | Mutations | dbSNP number | Type of Mutations | Primary Region of Effected in COSMIC | Cited cancer in COSMIC | MAF | SIFTc | PolyPhend | ClinVar |
|---|---|---|---|---|---|---|---|---|---|---|
| 1/II‐7; 1/III‐2; 3/III‐2 |
ACADS
(NM_000017.3) |
c.625G > A (p.Gly209Ser) | rs1799958 | missense_variant | Liver; soft tissue; breast | Carcinoma; rhabdomyosarcoma; carcinoma | 0.2586 | Deleterious (0.01) | Benign (0.342) | Pathogenic: benign |
| 2/IV‐2; 2/IV‐7 |
APOC3a
(NM_000040.2) |
c.55C > T (p.Arg19Ter) | rs76353203 | nonsense_variant | na | na | 0.0006032 | na | na | Pathogenic |
| 1/II‐7 |
ATP6V0A4
(NM_020632.2) |
c.1739T > C (p.Met580Thr) | rs3807153 | missense_variant | Skin; soft tissue | Malign melanoma; rhabdomyosarcoma | 0.06794 | Deleterious (0.03) | Benign (0.392) | Pathogenic |
| 1/II‐7 |
C2
(NM_000063.4) |
c.954G > C (p.Glu318Asp) | rs9332739 | missense_variant | Central nervous system; soft tissue | Primitive neuroectodermal tumor‐medulloblastoma; rhabdomyosarcoma | 0.03853 | Tolerated (0.23) | Probably damaging (0.933) | Pathogenic |
| 1/II‐7 |
CFB
(NM_001710.5) |
c.26T > A (p.Leu9His) | rs4151667 | missense_variant | Soft tissue | Rhabdomyosarcoma | 0.03865 | Tolerated (score: 0.3) | Probably damaging (0.999) | Pathogenic |
|
1/II‐7; 1/III‐2 |
CLEC7Aa
(NM_197947.2) |
c.714T > G (p.TYR238Ter) | rs16910526 | nonsense_variant | Soft tissue | Rhabdomyosarcoma | 0.06091 | na | na | Pathogenic |
|
1/II‐7; 1/III‐2; 2/IV‐7 |
CX3CR1
(NM_001171174.1) |
c.935C > T (p.Thr312Met) | rs3732378 | missense_variant | Pancreas; soft tissue | Carcinoma; rhabdomyosarcoma | 0.1376 | Deleterious (0.03) | Benign (0.333) | Pathogenic |
| 2/IV‐2 |
DSPPb
(NM_014208.3) |
c.202A > T (p.Arg68Trp) | rs36094464 | missense_variant | soft tissue | Rhabdomyosarcoma | 0.09294 | na | Probably damaging (0.992) | Pathogenic |
| 1/II‐7; 2/IV‐2; 2/IV‐7; 3/III‐1; 3/III‐2 |
FGFR4
(NM_002011.4) |
c.1162G > A (p.Gly388Arg) | rs351855 | missense_variant | Thyroid; soft tissue; soft tissue | Other; rhabdomyosarcoma; hemangioblastoma | 0.3209 | Tolerated (0.2) | Possibly damaging (0.742) | Pathogenic |
| 2/IV‐7 |
FUT6
(NM_000150.2) |
c.739G > A (p.Glu247Lys) | rs17855739 | missense_variant | Soft tissue; hematopoietic and lymphatic tissue | Rhabdomyosarcoma; hematologic tumors | 0.08068 | Deleterious (0) | Probably damaging (0.917) | Pathogenic |
|
2/IV‐7 3/III‐1; 3/III‐2 |
GBE1
(NM_000158.3) |
c.986A > G (p.TYR329Cys) | rs80338671 | missense_variant | na | na | 0.0004343 | Deleterious (0) | Probably damaging (0.999) | Pathogenic |
| 2/IV‐7 |
GHRL
(NM_001134944.1) |
c.178C > A (p.Leu60Met) | rs696217 | missense_variant | Soft tissue | Rhabdomyosarcoma | 0.08584 | Deleterious (score: 0.04) | Probably damaging (1.000) | Pathogenic |
| 1/II‐7 |
GNPAT
(NM_014236.3) |
c.1556A > G (p.Asp519Gly) | rs11558492 | missense_variant | Hematopoietic and lymphatic tissue | Hematologic tumors | 0.1608 | Deleterious (0.03) | Benign (0.097) | Pathogenic |
| 3/III‐1 |
HBD
(NM_000519.3) |
c.82G > T (p.Ala28Ser) | rs35152987 | missense_variant | na | na | 0.002054 | Tolerated (0.11) | Possibly damaging (0.68) | Pathogenic |
| 1/II‐7 |
HFE
(NM_000410.3) |
c.187C > G (p.His63Asp) | rs1799945 | missense_variant | Pancreas; soft tissue | Carcinoma;rhabdomyosarcoma | 0.1083 | Tolerated (0.74) | Probably damaging (0.974) | Pathogenic |
| 1/II‐7; 1/III‐2; 2/IV‐7 |
KRT85
(NM_002283.3) |
c.233G > A (p.Arg78His) | rs61630004 | missense_variant | Thyroid | Other | 0.03779 | Tolerated (0.38) | Probably damaging (0.991) | Pathogenic |
| 2/IV‐2 |
MBL2b
(NM_000242.2) |
c.161G > A (p.Gly54Asp) | rs1800450 | missense_variant | Skin; soft tissue | Malign melanoma; rhabdomyosarcoma | 0.1378 | Deleterious (0) | Probably damaging (0.999) | Pathogenic |
| 1/III‐2 |
MBL2
(NM_000242.2) |
c.154C > T (p.Arg52Cys) | rs5030737 | missense_variant | na | na | 0.05500 | Deleterious (0) | Probably damaging (0.988) | Pathogenic |
| 3/III‐1 |
MCCC2
(NM_022132.4) |
c.1015G > A (p.Val339Met) | rs150591260 | missense_variant | na | na | 0.0007506 | Deleterious ‐ low confidence (0.01) | Probably damaging (0.952) | Pathogenic |
| 2/IV‐2; 2/IV‐7 |
MUTYHa
(NM_001128425.1) |
c.1171C > T (p.Gln391Ter) | rs587783057 | missense_variant | Colon | Carcinoma | 0.00001629 | na | na | Pathogenic |
|
1/II‐7; 1/III‐2; 2/IV‐2; 2/IV‐7 |
NQO1
(NM_000903.2) |
c.559C > T (p.Pro187Ser) | rs1800566 | missense_variant | Large_intestine;biliary_tract;prostate;stomach;soft_tissue | Colon; bile tract; prostate; stomach; soft tissue | 0.2469 | Deleterious (0) | Probably damaging (0.999) | Pathogenic:drug response |
| 1/II‐7 |
RHAG
(NM_000324.2) |
c.808G > A (p.Val270Ile) | rs16879498 | missense_variant | na | na | 0.04170 | Deleterious (0) | Possibly damaging (0.519) | Pathogenic |
| 1/II‐7; 2/IV‐2 |
RPGRIP1
(NM_020366.3) |
c.1639G > T (p.Ala547Ser) | rs10151259 | missense_variant | na | na | 0.2041 | Deleterious (0.04) | Benign (0.259) | Pathogenic:benign |
| 1/III‐2 |
SERPINA1
(NM_001002235.2) |
c.1177C > T (p.Pro393Ser) | rs61761869 | missense_variant | na | na | 0.0002741 | Deleterious (0) | Probably damaging (0.988) | Pathogenic |
| 1/II‐7 |
SLC34A1
(NM_003052.4) |
c.272_292del21 (p.Val91_Ala97del) | rs199844043 | inframe_ deletion | na | na | 0 | na | na | Pathogenic |
| 1/II‐7; 2/IV‐2; 2/IV‐7 |
TYR
(NM_000372.4) |
c.1205G > A (p.Arg402Gln) | rs1126809 | missense_variant | Skin; esophagus; cervix | Malign melanoma; carcinoma; carcinoma | 0.1764 | Deleterious (0.03) | Probably damaging (0.941) | Pathogenic |
| 3/III‐1; 3/III‐2 |
UGT1A1a
(NM_000463.2) |
c.211G > A (p.Gly71Arg) | rs4148323 | missense_variant | Soft tissue; hematopoietic and lymphatic tissue | Rhabdomyosarcoma; hematologic tumors | 0.02130 | Tolerated (score: 0.42) | Probably damaging (0.982) | Pathogenic:likely benign:likely pathogenic:drug response |
Abbreviations: COSMIC, The Catalogue of Somatic Mutations in Cancer; MAF; minor allele frequency from the Exome Aggregation Consortium (ExAC) and Genome Aggregation Database (gnomAD) datasets; na, not available.
Family‐specific pathogenic variants.
Patient 2/IV‐2 specific pathogenic variants in terms of prognosis and survival.
SIFT value predication ranges from 0 to 1. Prediction of damaging or tolerated if the score shows ≤ 0.05 or > 0.05, respectively.
Polyphen value predication ranges from 0 to 1. A variant is appraised qualitatively, as benign (0.00–0.15), possibly damaging (0.16–0.85), or probably damaging (0.86–1.00).
The heterozygous variant in FGFR4 gene (GRCh37 Chr5:176520243, NM_002011.4:c.1162G > A p.Gly388Arg) commonly detected in five out of six patients (83.3%), was striking. Four patients (66.7%) had a pathogenic variant in NQO1 gene (GRCh37 Chr16:69745145, NM_000903.2:c.559C > T p.Pro187Ser). Commonly observed variants in three of the six patients (50%) were ACADS gene (GRCh37 Chr12:121176083, NM_000017.3:c.625G > A p.Gly209Ser), CX3CR1 gene (GRCh37 Chr3:39307162, NM_001171174.1:c.935C > T p.Thr312Met), GBE1 gene (GRCh37 Chr3:81691938, NM_000158.3:c.986A > G p.Tyr329Cys), KRT85 gene (GRCh37 Chr12:52760957, NM_002283.3:c.233G > A p.Arg78His), and TYR gene (GRCh37 Chr11:89017961, NM_000372.4:c.1205G > A p.Arg402Gln). The presence of mutations in the determined genes in the majority of patients indicates a statistically significant relationship between these genes, and Rb (p < 0.05).
The family‐based evaluation of the analysis results showed a variant in CLEC7A gene (GRCh37 Chr12:10271087, NM_197947.2:c.714T > G p.Tyr238Ter) in the first family; a variant in APOC3 gene (GRCh37 Chr11:116701353, NM_000040.2:c.55C > T p.Arg19Ter), and a variant in MUTYH gene (GRCh37 Chr1:45797348, NM_001128425.1:c.1171C > T p.Gln391Ter) in the second family, and a variant in UGT1A1 gene (GRCh37 Chr2:234669144, NM_000463.2:c.211G > A p.Gly71Arg) in the third family were found to be statistically significant (p < 0,05). Family‐specific pathogenic variants were shown in Table 3.
The evaluation of the patients in terms of prognosis, and survival showed that the patient 2/IV‐2 was diagnosed with a secondary tumor and died. The comparison of 2/IV‐2, with other patients, showed that there was a pathogenic variant in the DSPP gene (GRCh37 Chr4:88533540, NM_014208.3: c.202A > T p.Arg68Trp); and a variant in the MBL2 gene (GRCh37 Chr10:54531242, NM_000242.2:c.161G > A p.Gly54Asp). Patient 2/IV‐2 specific pathogenic variants in terms of prognosis and survival were shown in Table 3.
3.3. The associations of the genes with the metabolic pathways
The data consisting of 27 genes belonging to six patients in the study were uploaded into DAVID, and STRING databases were found associated with the Kyoto Encyclopedia of Genes and Genomes (KEGG), and REACTOME pathways (Table 4).
Table 4.
The gene sets associated with metabolic pathways
| Pathway | Effects of genes on metabolic pathways in cells or org anisms | Associated genes |
|---|---|---|
| KEGG Pathway | Complement and coagulation cascade | C2; SERPINA1; MBL2; CFB |
| KEGG Pathway | Fagosome | ATP6V0A4; CLEC7A; MBL2 |
| KEGG Pathway | Staphylococcus Aureus Infection | C2; MBL2; CFB |
| REACTOME Pathway | Catalysis | C2; MBL2 |
| KEGG Pathway | Tuberculosis | ATP6V0A4; CLEC7A |
| REACTOME Pathway | Regulation of the complement cascade | C2; CFB |
| KEGG Pathway | Valine, Leucine, and isoleucine catabolism | ACADS; MCCC2 |
| REACTOME Pathway | Spontaneous Separation of the C3 converters | C2; CFB |
| REACTOME Pathway | Catalysis | C2; CFB |
| KEGG Pathway | Starch and sucrose metabolism | UGT1A1; GBE1 |
Abbreviation: KEGG, Kyoto Encyclopedia of Genes and Genomes.
Three particular significant metabolic pathways were detected in DAVID database in the study. Four genes, C2, CFB, MBL2, and SERPINA1 (p: 0.00055) were found effective in complement and coagulation cascade on the immune system, three genes, C2, CFB, MBL2 (p: 0.008) were found effective in staphylococcus aureus infection and three genes, ATP6V0A4; CLEC7A, MBL2 (p: 0.05) were found effective in the occurrence of cellular phagocytosis and there was an association between mutations and Rb in the study.
3.4. The analysis of protein–protein interactions
The interactions between proteins in the STRING database were analyzed, and shown in Figure 4. Accordingly, a total of 27 nodes (circles), and 12 edges were identified. The results of the evaluation of this database are intended to be specific and meaningful, that is the proteins contribute to a common function, but this does not mean that they are physically linked to each other. This protein network obtained after the analysis showed a more significant level of protein interactions than expected (p: 0.0000097). This fact means that the protein set obtained in the study has more interactivity than would be expected from a random set of proteins at the same size in the genome. This indicates that the protein group is at least partly biologically involved or associated with each other.
Figure 4.
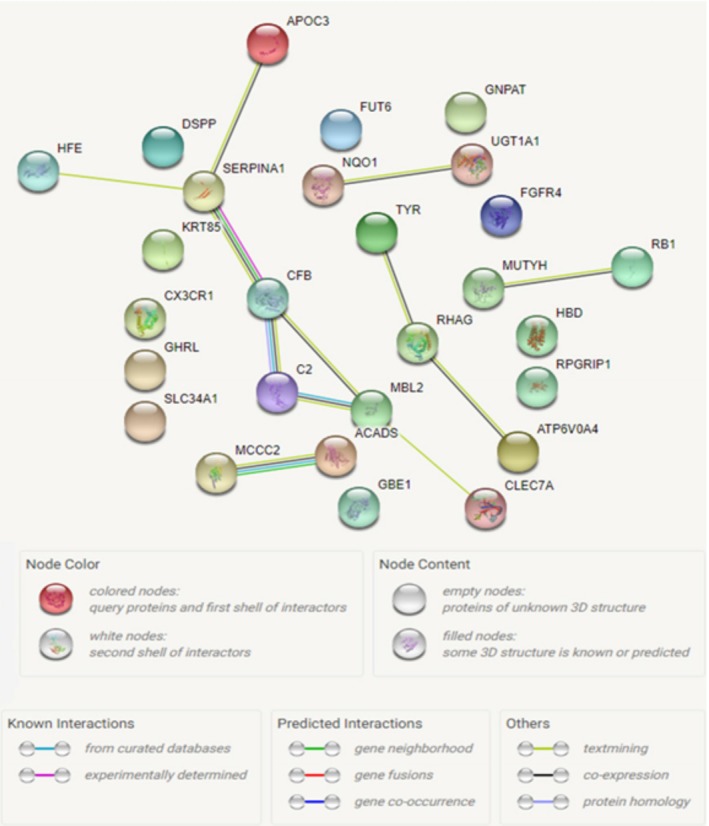
Protein–protein interactions of the genes mutated in the study. Each node represents all proteins produced by a single protein‐encoding gene locus. The edges that link between the proteins, which are determined to be related to each other, showed the protein‐protein interactions
4. DISCUSSION
Significant genetic factors are known to have a role in the development of Rb. Development of Rb is thought to be due to mutations in the RB1 gene. Genetic factors that are responsible for retinoblastoma are not all yet identified in patients who do not have the RB1 gene mutations. This is an important problem for Rb oncogenesis and need to be investigated. Changes in the number of the copies in the other genes in addition to RB1 are frequently detected in Rb. An acquisition ranging 4–10 copies in the MDM4, KIF14 (1q32), MYCN (2p24), DEK, and E2F3 (6p22) oncogenes, and a loss in the CDH11 (16q22‐24) tumor supressor gene has been reported (Corson & Gallie, 2007). The different expression profiles of some microRNAs on Rb have been suggested to be related to the let‐7b downregulation (Huang et al., 2007). Single nucleotide deletion, and insertions on the genes BCOR and CREBBP might be associated with Rb (Kooi et al., 2016). Zhang et al. reported that SYK protooncogene was shown to be overexpressed in Rb, and thus may have triggered the development of malignant cell. Furthermore, in the same study, after the full gene sequencing of 11 genes in patients diagnosed with Rb, the mutation merely reported was on the gene BCOR (Zhang et al., 2012). According to McEvoy et al., mutations in BCOR gene as well chromothripsis as a cause of retinoblastoma (McEvoy et al., 2014). In our study we detected 26 genes that had 27 pathogenic variants that may play a role in the pathogenesis of Rb.
This study differs from other studies in two points. First, none of the six patients had the RB1 mutation and abnormal RB1 promoter methylation. Second, all patients had a family history of retinoblastoma since there were two members with Rb in each family all of whom had consanguinity. The results of this study would provide significant clues about the Rb oncogenesis, and could identify new the pathway of Rb disease. The study also indicated the commonly detected genes in patients and the genes specified for the families were found remarkable and informative for Rb disease. Particularly the CLEC7A gene in the first family; APOC3 and MUTYH genes in the second family, and UGT1A1 gene in the third family may be new candidate and specific genes for these families that triggered the occurrence of Rb, since none had a RB1 gene mutation and abnormal RB1 promoter methylation. To understand effects of these genes on the heritage of disease based on families and roles in oncogenesis of retinoblastoma, it is recommended to investigate the patients throughout at least three generations in the future.
We detected the c.714T > G (p.Tyr238Ter) variant in the CLEC7A gene in the first family (1/II‐7,1/III‐2). CLEC7A is also known as the Dectin‐1. According to literature, an association between the Dectin‐1 immunodeficiency and mucocutaneous fungal infections have been detected in the eye (Klotz, Penn, Negvesky, & Butrus, 2000). Four women from the same family who were immunodeficient were reported to have the c.714T > G (p.Tyr238Ter) mutation in the CLEC7A gene and fungal infection (Ferwerda et al., 2009). This mutation was detected in patients 1/II‐7 and 1/III‐2 from the same family in our study. There was no significant history of immunodeficiency or infection in our patients. Moreover, two pathogen recognition receptors, Dectin‐1 and Toll‐like receptor 2 (TLR2) metabolizes Vitamin A, and transforms to retinoic acid in dendritic cells (DCs)(Manicassamy et al., 2009). CLEC7A gene has been demonstrated to be effective in the retinoic acid pathway. This gene might be a candidate gene in the pathogenesis of the retinoblastoma disease in the first family and also oncogenesis of retinoblastoma.
The pathogenic c.55C > T (p.Arg19Ter) variant was found in APOC3 gene in the second family (2/IV‐2, 2/IV‐7). APOC3 is a lipoprotein with a significantly low density. The increase in the level of APOC3 results in hypertriglyceridemia which is a metabolic complication of the retinoid therapy. Retinoids increase the APOC3 expression in transcriptional level through retinoid X receptor (RXR). The increase in APOC3 expression and its release by the retinoids in the liver demonstrating APOC3 might be a retinoid response gene (Vu‐Dac et al., 1998). The change in this gene which is known to have an association with the retinal pathway was suggested to be associated with Rb. However, pathogenic c.1171C > T (p.Gln391Ter) variant was detected in the MUTYH gene in the same family. MUTYH is known to have a role in the DNA damage repair. This gene cannot inhibit the accumulation and occurrence of mutation on DNA when it has a mutation. The mutations on the MUTYH gene have been associated with the autosomal recessive form of the syndrome of familial adenomatous polyposis (MYH associated polyposis) (Ali et al., 2008). The detection of a pathogenic variant on MUTYH gene in two patients, 2/IV‐2 and 2/IV‐7, in our study may suggest a risk for MYH‐associated polyposis, and colon cancer in the future. The patient 2/IV‐2 was diagnosed with unilateral Rb, and died of fibrosarcoma in the proceeding years of life; 2/IV‐7 was diagnosed with bilateral Rb, and three first‐degree cousins in the same family were diagnosed with unilateral Rb, and one cousin was diagnosed with rhabdomyosarcoma; which suggested that this variant might be associated with the Rb disease. The STRING protein‐protein analysis showed that MUTYH gene, and RB1 gene had a significant association. This association between MUTYH gene and RB1 gene may suggest the possibility that this variant might be responsible for the occurrence of Rb in this family. In addition, in families with MUTYH gene mutation exist a risk for a predisposition to juvenile colon cancer as others reported having. To clarify this association, this pathogenic variant must be investigated in future studies in the other individuals diagnosed with Rb in the family and also in large patients cohort and population‐based healthy controls.
The c.211G > A (p.Gly71Arg), pathogenic variant was detected in UGT1A1 gene in the third family (3/III‐1, and 3/III‐2). UGT1A1, performs a chemical reaction named as glucuronidation (Gong et al., 2001). An association was demonstrated on chemical reaction of UGT1A1 and 13‐cis retinoic acid in the literature. 13‐cis retinoic acid is known as the retinol derivative which organizes numerous biological procedures including embriyogenesis, growth, differentiation, vision, and reproduction (Evans & Kaye, 1999). Twenty‐one functional UGT isoforms, which catalyze the glucuronidation most of which consisting of various environmental carcinogens, nutritional chemopreventives, and anticancer agents in human, have been described (Nagar & Remmel, 2006). The detection of c.211G > A (p.Gly71Arg) pathogenic variant in UGT1A1 gene in patients 3/III‐1 and 3/III‐2 suggested that this mutation might have triggered the occurrence of cancer by affecting the retinoic acid metabolism in patients.
In addition, the FGFR4 and NQO1 genes detected in the majority of the patients might be thought to be effective candidate genes in the Rb etiology and pathogenesis. To understand the exact role of these genes in Rb etiology and pathogenesis, the alterations of these genes must be investigated in large patient groups with the familial segregation and compared with population‐based healthy controls. We detected the c.559C > T (p.Pro187Ser) pathogenic variant in the gene NQO1 in patients, 1/II‐7, 1/III‐2, 2/IV‐2, and 2/IV‐7. NQO1 gene is named as the anticancer enzyme because NQO1 gene protects the cells from oxidative damage. In addition to the protective role in the carcinogenesis, NQO1 gene functions as the drug metabolizing enzyme in the antitumor treatment. The mutations in this gene were associated with Tardive dyskinesia (TD), an increase in the risk of hematoxocity after exposure to benzene, and predisposition to various cancer types (Smith, 1999; Zai et al., 2010). The modified expression of this protein was detected in various tumors such as lung, bladder, breast, hepatocellular carcinoma, acute myeloid leukemia (AML), colorectal cancer, and gastrointestinal cancers, and in addition it was associated with the Alzheimer's disease (Chao, Zhang, Berthiller, Boffetta, & Hashibe, 2006; Chhetri, King, & Gueven, 2017; Valenzuela et al., 2014). The variant of c.559C > T (p.Pro187Ser) in NQO1 gene decreased the enzymatic activity and increased the risk of lung cancer. This variant caused the predisposition to bladder and colorectal cancer (Chao et al., 2006). Similarly, c.559C > T (p.Pro187Ser) variant detected in our patients 1/II‐7, 1/III‐2, 2/IV‐2, and 2/IV‐7 was suggested to increase the risk of lung, bladder, and colorectal cancers. The increase in the NQO1 target gene transcription affected the retinoic acid pathway, and prevent from cancer (Valenzuela et al., 2014). Therefore, the detection of the pathogenic variant of the gene NQO1 in four patients from two different families suggested that it might be associated with the pathogenesis of Rb. However, c.1162G > A (p.Gly388Arg) variant detected in FGFR4 gene in our patients 1/II‐7, 2/IV‐2, 2/IV‐7, 3/III‐1, and 3/III‐2. FGFR4 gene, is a member of fibroblast growth factor (FGF) family which has a role in various mechanisms such as cellular proliferation, differentiation, tissue repair, invasion, regulation of the lipid metabolism, bile acid biosynthesis, glucose intake, Vitamin D metabolism, and phosphate balance. The c.1162G > A (p.Gly388Arg) variant in FGFR4 gene, and the increase in the FGFR4 expression were associated with the development of breast, and colon cancer. In addition, it was reported to be statistically associated with the lymph node metastasis, and increased TNM stage, and demonstrated to trigger the cancer progression (Bange et al., 2002). The FGFR4 expression was associated with pancreatic cancers (Leung, Gullick, & Lemoine, 1994). Cancer progression and tumor cell motility were associated with the c.1162G > A (p.Gly388Arg) change in FGFR4 gene (Bange et al., 2002). The variant in the gene FGFR4 was effective in the initiation, and in the progression of prostate cancer (Wang, Stockton, & Ittmann, 2004). FGFR4 gene is also known with its oncogenic transformation activity which is required in the down‐regulation of the expression of the speed limiting enzyme of CYP7A1 in the synthesis of bile acid as a response to FGF19. Some fibroblast growth factors are known to have neuroprotective effects against the retinal photoreceptor degeneration. The expression of FGFR4 in the photoreceptors suggested a specific ligand of FGF‐19 might be beneficial. FGF‐19 is important for the development of the ocular tissue, and is a molecule expressed by the embryonic retina. Therefore, the potential role of FGF‐19 has been investigated in many studies in the literature. FGF‐19 had neuroprotective effects on mammalian photoreceptors (Siffroi‐Fernandez, Felder‐Schmittbuhl, Khanna, Swaroop, & Hicks, 2008). Photoreceptor degeneration develops as a pathologic response to numerous environmental and genetic disorders, and causes progressive vision loss and blindness. The hereditary retinal diseases such as retinitis pigmentosa and age associated macular degeneration (AMD) cause significant difficulties in the affected patients. FGF‐19 was expressed by the cells adjacent to photoreceptor layer, and FGF‐19 induced the dose and time‐dependent phosphorylation of FGFR4 in purified adult photoreceptor cultures, upregulated the expression of the specific transcription factors, and increased the survival (Siffroi‐Fernandez et al., 2008). Therefore, it was suggested to be a beneficial therapeutic approach in the treatment of retinal degeneration. In this regard, our results suggested the c.1162G > A (p.Gly388Arg) pathogenic variant commonly detected in FGFR4 gene that is known to have a role in cancer progression, and retinal development in patients 1/II‐7, 2/IV‐2, 2/IV‐7, 3/III‐1, and 3/III‐2, might be a candidate mechanism triggering the development of Rb. Furthermore, the common variant was found only in the FGFR4 gene among the 4813 genes and may be a biomarker of Rb disease. The presence of the gene variants should be investigated with larger patient groups and population‐based healthy controls in the future studies.
In conclusion, in this study we investigated candidate genes that may trigger Rb oncogenesis in six patients with retinoblastoma or retinoma within three families and who did not have a RB1 gene mutation and abnormal RB1 promoter methylation. This is the first study suggesting that these genes, FGFR4, NQO1, ACADS CX3CR1, GBE1, KRT85, and TYR genes, may play a role in the etiology of Rb. Although, in the literature database these genes were not reported to be involved in Rb promotion, they have found to be associated with the retinoic acid pathway; that has been suggesting to play a role in the Rb oncogenesis. It is recommended that these genes should be investigated in larger cohorts of patients and compared with population‐based healthy controls in the future.
CONFLICT OF INTEREST
No conflict of interest was declared by the authors.
ACKNOWLEDGMENTS
This work was supported by Scientific Research Projects Coordination Unit of Istanbul University (Project number: 21460).
Akdeniz D, Tuncer SB, Kebudi R, et al. Investigation of new candidate genes in retinoblastoma using the TruSight One “clinical exome” gene panel. Mol Genet Genomic Med. 2019;7:e785 10.1002/mgg3.785
REFERENCES
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , … Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali, M. , Kim, H. , Cleary, S. , Cupples, C. , Gallinger, S. , & Bristow, R. (2008). Characterization of mutant MUTYH proteins associated with familial colorectal cancer. Gastroenterology, 135(2), 499–507. 10.1053/j.gastro.2008.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project, C. , Auton, A. , Brooks, L. D. , Durbin, R. M. , Garrison, E. P. , Kang, H. M. , … Abecasis, G. R. (2015). A global reference for human genetic variation. Nature, 526(7571), 68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bange, J. , Prechtl, D. , Cheburkin, Y. , Specht, K. , Harbeck, N. , Schmitt, M. , … Ullrich, A. (2002). Cancer progression and tumor cell motility are associated with the FGFR4 Arg(388) allele. Cancer Research, 62(3), 840–847. [PubMed] [Google Scholar]
- Chakraborty, S. , Khare, S. , Dorairaj, S. K. , Prabhakaran, V. C. , Prakash, D. R. , & Kumar, A. (2007). Identification of genes associated with tumorigenesis of retinoblastoma by microarray analysis. Genomics, 90(3), 344–353. 10.1016/j.ygeno.2007.05.002 [DOI] [PubMed] [Google Scholar]
- Chao, C. , Zhang, Z. F. , Berthiller, J. , Boffetta, P. , & Hashibe, M. (2006). NAD(P)H:Quinone oxidoreductase 1 (NQO1) Pro187Ser polymorphism and the risk of lung, bladder, and colorectal cancers: A meta‐analysis. Cancer Epidemiology, Biomarkers & Prevention, 15(5), 979–987. 10.1158/1055-9965.EPI-05-0899 [DOI] [PubMed] [Google Scholar]
- Chaussade, A. , Millot, G. , Wells, C. , Brisse, H. , Lae, M. , Savignoni, A. , … Houdayer, C. (2018). Correlation between RB1germline mutations and second primary malignancies in hereditary retinoblastoma patients treated with external beam radiotherapy. European Journal of Medical Genetics, 62(3), 217 10.1016/j.ejmg.2018.07.017 [DOI] [PubMed] [Google Scholar]
- Chhetri, J. , King, A. E. , & Gueven, N. (2017). Alzheimer's disease and NQO1: Is there a link? Current Alzheimer Research, 10.2174/1567205014666170203095802 [DOI] [PubMed] [Google Scholar]
- Chintagumpala, M. , Chevez‐Barrios, P. , Paysse, E. A. , Plon, S. E. , & Hurwitz, R. (2007). Retinoblastoma: Review of current management. The Oncologist, 12(10), 1237–1246. 10.1634/theoncologist.12-10-1237 [DOI] [PubMed] [Google Scholar]
- Corson, T. W. , & Gallie, B. L. (2007). One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes, Chromosomes & Cancer, 46(7), 617–634. 10.1002/gcc.20457 [DOI] [PubMed] [Google Scholar]
- da Huang, W. , Sherman, B. T. , & Lempicki, R. A. (2009a). Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Research, 37(1), 1–13. 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Huang, W. , Sherman, B. T. , & Lempicki, R. A. (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols, 4(1), 44–57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- Draper, G. J. , Sanders, B. M. , Brownbill, P. A. , & Hawkins, M. M. (1992). Patterns of risk of hereditary retinoblastoma and applications to genetic counselling. British Journal of Cancer, 66(1), 211–219. 10.1038/bjc.1992.244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, T. R. , & Kaye, S. B. (1999). Retinoids: Present role and future potential. British Journal of Cancer, 80(1–2), 1–8. 10.1038/sj.bjc.6690312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferwerda, B. , Ferwerda, G. , Plantinga, T. S. , Willment, J. A. , van Spriel, A. B. , Venselaar, H. , … Netea, M. G. (2009). Human dectin‐1 deficiency and mucocutaneous fungal infections. New England Journal of Medicine, 361(18), 1760–1767. 10.1056/NEJMoa0901053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes, S. A. , Beare, D. , Boutselakis, H. , Bamford, S. , Bindal, N. , Tate, J. , … Campbell, P. J. (2017). COSMIC: Somatic cancer genetics at high‐resolution. Nucleic Acids Research, 45(D1), D777–D783. 10.1093/nar/gkw1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly, A. , & Shields, C. L. (2010). Differential gene expression profile of retinoblastoma compared to normal retina. Molecular Vision, 16, 1292–1303. [PMC free article] [PubMed] [Google Scholar]
- Gong, Q. H. , Cho, J. W. , Huang, T. , Potter, C. , Gholami, N. , Basu, N. K. , … Popescu, N. C. (2001). Thirteen UDPglucuronosyltransferase genes are encoded at the human UGT1 gene complex locus. Pharmacogenetics, 11(4), 357–368. 10.1097/00008571-200106000-00011 [DOI] [PubMed] [Google Scholar]
- Huang, J. C. , Babak, T. , Corson, T. W. , Chua, G. , Khan, S. , Gallie, B. L. , … Morris, Q. D. (2007). Using expression profiling data to identify human microRNA targets. Nature Methods, 4(12), 1045–1049. 10.1038/nmeth1130 [DOI] [PubMed] [Google Scholar]
- Indovina, P. , Acquaviva, A. , De Falco, G. , Rizzo, V. , Onnis, A. , Luzzi, A. , … Giordano, A. (2010). Downregulation and aberrant promoter methylation of p16INK4A: A possible novel heritable susceptibility marker to retinoblastoma. Journal of Cellular Physiology, 223(1), 143–150. 10.1002/jcp.22019 [DOI] [PubMed] [Google Scholar]
- Jagadeesan, M. , Khetan, V. , & Mallipatna, A. (2016). Genetic perspective of retinoblastoma: From present to future. Indian Journal of Ophthalmology, 64(5), 332–336. 10.4103/0301-4738.185585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz, S. A. , Penn, C. C. , Negvesky, G. J. , & Butrus, S. I. (2000). Fungal and parasitic infections of the eye. Clinical Microbiology Reviews, 13(4), 662–685. 10.1128/CMR.13.4.662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson, A. G. Jr (1971). Mutation and cancer: Statistical study of retinoblastoma. Proceedings of the National Academy of Sciences of the USA, 68(4), 820–823. 10.1073/pnas.68.4.820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooi, I. E. , Mol, B. M. , Massink, M. P. , Ameziane, N. , Meijers‐Heijboer, H. , Dommering, C. J. , … Dorsman, J. C. (2016). Somatic genomic alterations in retinoblastoma beyond RB1 are rare and limited to copy number changes. Scientific Reports, 6, 25264 10.1038/srep25264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, P. , Henikoff, S. , & Ng, P. C. (2009). Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nature Protocols, 4(7), 1073–1081. 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- Landrum, M. J. , Lee, J. M. , Benson, M. , Brown, G. R. , Chao, C. , Chitipiralla, S. , … Maglott, D. R. (2018). ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Research, 46(D1), D1062–D1067. 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , … Exome Aggregation, C. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung, H. Y. , Gullick, W. J. , & Lemoine, N. R. (1994). Expression and functional activity of fibroblast growth factors and their receptors in human pancreatic cancer. International Journal of Cancer, 59(5), 667–675. 10.1002/ijc.2910590515 [DOI] [PubMed] [Google Scholar]
- Livide, G. , Epistolato, M. C. , Amenduni, M. , Disciglio, V. , Marozza, A. , Mencarelli, M. A. , … Ariani, F. (2012). Epigenetic and copy number variation analysis in retinoblastoma by MS‐MLPA. Pathology & Oncology Research, 18(3), 703–712. 10.1007/s12253-012-9498-8 [DOI] [PubMed] [Google Scholar]
- Manicassamy, S. , Ravindran, R. , Deng, J. , Oluoch, H. , Denning, T. L. , Kasturi, S. P. , … Pulendran, B. (2009). Toll‐like receptor 2‐dependent induction of vitamin A‐metabolizing enzymes in dendritic cells promotes T regulatory responses and inhibits autoimmunity. Nature Medicine, 15(4), 401–409. 10.1038/nm.1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvoy, J. , Nagahawatte, P. , Finkelstein, D. , Richards‐Yutz, J. , Valentine, M. , Ma, J. , … Dyer, M. A. (2014). RB1 gene inactivation by chromothripsis in human retinoblastoma. Oncotarget, 5(2), 438–450. 10.18632/oncotarget.1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKusick, V. A. (2007). Mendelian Inheritance in Man and its online version, OMIM. The American Journal of Human Genetics, 80(4), 588–604. 10.1086/514346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren, W. , Gil, L. , Hunt, S. E. , Riat, H. S. , Ritchie, G. R. , Thormann, A. , … Cunningham, F. (2016). The ensembl variant effect predictor. Genome Biology, 17(1), 122 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphree, A. L. , Samuel, M. , Harbour, J. W. , & Mansfield, N. C. (2006). Retinoblastoma (4th ed.). Philadelphia, PA: Mosby Elsevier. [Google Scholar]
- Nagar, S. , & Remmel, R. P. (2006). Uridine diphosphoglucuronosyltransferase pharmacogenetics and cancer. Oncogene, 25(11), 1659–1672. 10.1038/sj.onc.1209375 [DOI] [PubMed] [Google Scholar]
- O'Leary, N. A. , Wright, M. W. , Brister, J. R. , Ciufo, S. , Haddad, D. , McVeigh, R. , … Pruitt, K. D. (2016). Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Research, 44(D1), D733–745. 10.1093/nar/gkv1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey, A. N. (2014). Retinoblastoma: An overview. Saudi Journal of Ophthalmology, 28(4), 310–315. 10.1016/j.sjopt.2013.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, C. S. , Bale, S. , Bellissimo, D. B. , Das, S. , Grody, W. W. , Hegde, M. R. , …Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee. (2008). ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genetics in Medicine, 10(4), 294–300. 10.1097/GIM.0b013e31816b5cae [DOI] [PubMed] [Google Scholar]
- Sherry, S. T. , Ward, M. H. , Kholodov, M. , Baker, J. , Phan, L. , Smigielski, E. M. , & Sirotkin, K. (2001). dbSNP: The NCBI database of genetic variation. Nucleic Acids Research, 29(1), 308–311. 10.1093/nar/29.1.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siffroi‐Fernandez, S. , Felder‐Schmittbuhl, M. P. , Khanna, H. , Swaroop, A. , & Hicks, D. (2008). FGF19 exhibits neuroprotective effects on adult mammalian photoreceptors in vitro. Investigative Ophthalmology & Visual Science, 49(4), 1696–1704. 10.1167/iovs.07-1272 [DOI] [PubMed] [Google Scholar]
- Smith, M. T. (1999). Benzene, NQO1, and genetic susceptibility to cancer. Proceedings of the National Academy of Sciences of the USA, 96(14), 7624–7626. 10.1073/pnas.96.14.7624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk, D. , Morris, J. H. , Cook, H. , Kuhn, M. , Wyder, S. , Simonovic, M. , … von Mering, C. (2017). The STRING database in 2017: Quality‐controlled protein‐protein association networks, made broadly accessible. Nucleic Acids Research, 45(D1), D362–D368. 10.1093/nar/gkw937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela, M. , Glorieux, C. , Stockis, J. , Sid, B. , Sandoval, J. M. , Felipe, K. B. , … Buc Calderon, P. (2014). Retinoic acid synergizes ATO‐mediated cytotoxicity by precluding Nrf2 activity in AML cells. British Journal of Cancer, 111(5), 874–882. 10.1038/bjc.2014.380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu‐Dac, N. , Gervois, P. , Torra, I. P. , Fruchart, J. C. , Kosykh, V. , Kooistra, T. , … Staels, B. (1998). Retinoids increase human apo C‐III expression at the transcriptional level via the retinoid X receptor. Contribution to the hypertriglyceridemic action of retinoids. Journal of Clinical Investigation, 102(3), 625–632. 10.1172/JCI1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Stockton, D. W. , & Ittmann, M. (2004). The fibroblast growth factor receptor‐4 Arg388 allele is associated with prostate cancer initiation and progression. Clinical Cancer Research, 10(18 Pt 1), 6169–6178. 10.1158/1078-0432.CCR-04-0408 [DOI] [PubMed] [Google Scholar]
- Wong, F. L. , Boice, J. D. Jr , Abramson, D. H. , Tarone, R. E. , Kleinerman, R. A. , Stovall, M. , … Li, F. P. (1997). Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA, 278(15), 1262–1267. 10.1001/jama.1997.03550150066037 [DOI] [PubMed] [Google Scholar]
- Zai, C. C. , Tiwari, A. K. , Basile, V. , de Luca, V. , Muller, D. J. , Voineskos, A. N. , … Kennedy, J. L. (2010). Oxidative stress in tardive dyskinesia: Genetic association study and meta‐analysis of NADPH quinine oxidoreductase 1 (NQO1) and Superoxide dismutase 2 (SOD2, MnSOD) genes. Progress in Neuro‐Psychopharmacology and Biological Psychiatry, 34(1), 50–56. 10.1016/j.pnpbp.2009.09.020 [DOI] [PubMed] [Google Scholar]
- Zhang, J. , Benavente, C. A. , McEvoy, J. , Flores‐Otero, J. , Ding, L. , Chen, X. , … Dyer, M. A. (2012). A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature, 481(7381), 329–334. 10.1038/nature10733 [DOI] [PMC free article] [PubMed] [Google Scholar]