

Abstract
Background
CYP1B1 variants and deletions are the most common cause of primary congenital glaucoma (PCG).
Methods
We investigated an individual with PCG from the Australian and New Zealand Registry of Advanced Glaucoma. We performed sequencing of the CYP1B1 gene, followed by Multiplex Ligation‐dependent Probe Amplification and SNP array.
Results
We identified a homozygous deletion of the CYP1B1 gene by Multiplex Ligation‐dependent Probe Amplification and confirmed that the father was heterozygous for a CYP1B1 deletion but the mother had normal gene copy number. SNP array identified paternal uniparental isodisomy of the entire chromosome 2.
Conclusions
This study is the first report of a homozygous CYP1B1 whole gene deletion due to paternal uniparental isodisomy of chromosome 2 as a cause of PCG. These results illustrate the importance of genetic testing in providing appropriate genetic counseling regarding the risks of recurrence.
Keywords: childhood glaucoma, chromosome 2, CYP1B1, gene deletion, primary congenital glaucoma, uniparental disomy
1. INTRODUCTION
Primary congenital glaucoma (PCG, MIM 231300) is a significant cause of irreversible blindness in children and results from developmental defects of the aqueous humor outflow pathway structures. It is characterized by increased intraocular pressure (IOP), buphthalmos, optic disc cupping, corneal edema, enlarged corneal diameter, and/or Haab's striae (Thau et al., 2018). A high incidence has been found among some populations with founder effects or a high rate of consanguinity (Slovakian Roma 1/1,250 (Gencik, 1989), Saudi Arabian 1/2,500 (Bejjani et al., 2000)) but incidence is lower in Western countries, including Australia (1/30,000 births) (MacKinnon, Giubilato, Elder, Craig, & Mackey, 2004).
PCG is most commonly associated with autosomal recessive variants in CYP1B1 on chromosome 2p21 (MIM 601771) (Stoilov, Akarsu, & Sarfarazi, 1997). Mice deficient in CYP1B1 have developmental defects of the ocular drainage structures resembling those reported in individuals with PCG, supporting loss of function as a mechanism of CYP1B1 variants causing PCG (Libby et al., 2003). Over 200 pathogenic sequence variants have been reported in CYP1B1 (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=CYP1B1), however deletions of the gene have seldom been reported (Damjanovich, Baldwin, Lewis, & Bayrak‐Toydemir, 2013; Kelberman et al., 2011; Milla et al., 2013; Stoilov, et al., 1997). In this study, we report an individual with PCG and a homozygous CYP1B1 deletion arising from paternal uniparental isodisomy of chromosome 2.
2. CASE REPORT
The study was conducted in accordance with the revised Declaration of Helsinki and participants were recruited with the approval of the Southern Adelaide Clinical Human Research Ethics Committee. The proband and his family members were enrolled as part of The Australian and New Zealand Registry of Advanced Glaucoma and gave their informed consent (Souzeau et al., 2012). The proband was a 15‐year‐old boy and the second child of nonconsanguineous Indian parents. He presented with bilateral significant corneal clouding, enlarged cornea 12 mm horizontally and raised IOP at birth. Right trabeculectomies were performed at 4 weeks and 3 months of age, however this eye became densely amblyopic. His better left eye had a trabeculectomy with Mitomycin C antimetabolite at age 32 months. Sequential left then right Ahmed tube surgery took place at age 4 and 5 years. These surgeries all took place in India. His family moved to Australia when he was 11 years. At that time his right eye was blind with perception of light, he had IOP of 30 mmHg, band keratopathy, a small pupil and dense cataract limiting the view of his optic nerve (Figure 1). The better left eye has nystagmus, vision of 3/15, IOP in the mid‐teens on maximal topical medical therapy and since age 13 also acetazolamide 250 mg bd. His most recent field test had generalized reduced sensitivity, with a left nasal step. Both parents and his older sister were examined and were unaffected.
Figure 1.
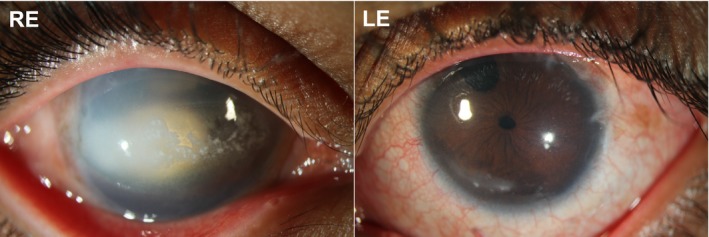
Slit lamp photographs of the proband. Right eye (RE) showing central calcified band keratopathy, unreactive pupil with surgical iridectomy occluded by cataract. Left eye (LE) showing iridectomy (iris resection in the superior part of the iris) from previous trabeculectomy, constriction of the pupil due to intraocular lowering topical therapy and limbal scars due to surgery (at the corner of the cornea and the sclera)
3. GENETIC ANALYSIS
Genomic DNA was extracted from venous blood of the proband, his parents, and his sister. CYP1B1 sequencing was performed on the proband's DNA through the NATA (National Association of Testing Authorities) accredited laboratories of SA Pathology at Flinders Medical Centre (Adelaide, Australia) as previously described (Souzeau, Hayes, Zhou, et al., 2015). Amplification of the two coding exons of CYP1B1 failed, suggesting a potential gene deletion (data not shown).
CYP1B1 was then analyzed for copy number variation by Multiplex Ligation‐dependent Probe Amplification (MLPA) using the SALSA P128 Cytochrome P450 probemix (MRC Holland, Amsterdam, The Netherlands) according to the manufacturers' protocol. MLPA analysis of CYP1B1 indicated an abnormal copy number in the proband's DNA, characteristic of a homozygous deletion of the entire gene. MLPA analysis of both parents was then performed, and revealed in the father the loss of a single allele of CYP1B1 but indicated a normal copy number for all exons of CYP1B1 in the mother (Figure 2).
Figure 2.
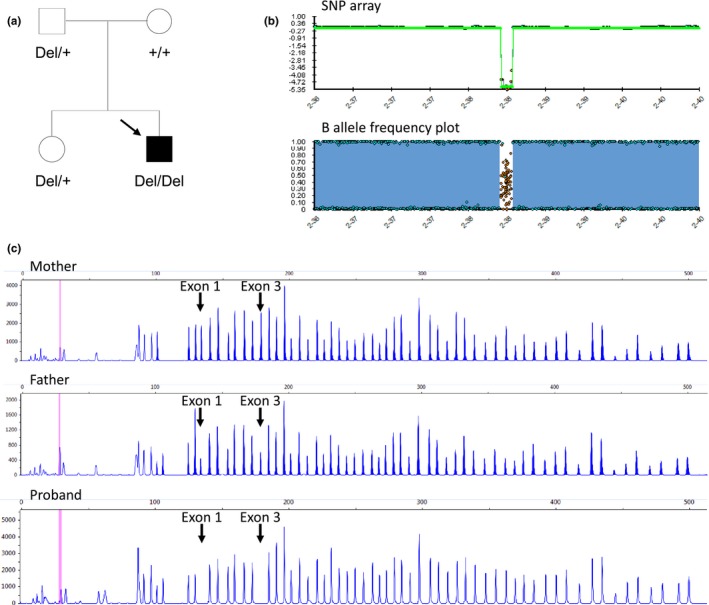
Pedigree and genetic analysis. (a) Pedigree of the family. The solid symbol indicates primary congenital glaucoma, the CYP1B1 deletion is represented by “Del” and the wild type allele by the “+’ sign. (b) Single nucleotide polymorphism (SNP) array confirming the homozygous CYP1B1 deletion (top panel) and B allele frequency plot showing complete homozygosity on chromosome 2, corresponding to uniparental isodisomy (bottom panel). The B allele frequency is a normalized measure of the allele frequency ratio of the two alleles: values of 0 or 1 indicate areas of homozygosity, whereas a value of 0.5 indicates areas of heterozygosity. (c) MLPA analysis showing normal CYP1B1 copy number in the proband's mother (upper panel), heterozygous CYP1B1 deletion in the proband's father (middle panel) and homozygous CYP1B1 deletion in the proband (bottom panel). The arrows show the two probes in exons 1 and 3, while the other peaks represent unrelated probes to other genes used as internal controls
To investigate the extent of the CYP1B1 gene deletion in the proband and the possibility of uniparental disomy (UPD), a SNP array (Illumina Infinium CytoSNP‐850K BeadChip) was conducted. Array result of the proband revealed an interstitial homozygous deletion of 119kb on chromosome 2q22.2 (chr2: 38 239 356–38 358 664, GRCh37/hg19 assembly) encompassing the entire CYP1B1 gene as well as the RMDN2 gene (MIM 611872). The RMDN2 gene has not been associated with any clinical phenotype. Paternal isodisomy for the entire chromosome 2 was identified, confirming that the homozygous CYP1B1 deletion in the child originated from a single parent (Figure 2). Chromosome 2 has not been assessed for the presence of other homozygous variants.
4. DISCUSSION
UPD is a rare event that arises when an individual with a diploid genome carries both homologs of a pair of chromosomes from one parent (uniparental heterodisomy) or both copies of a single parental chromosome (uniparental isodisomy) (Engel, 1980). UPD can result in clinically recognizable phenotypes through imprinting defects, mosaicism, or unmasking of homozygosity for pathogenic variants inherited from one parent in autosomal recessive genes. Paternal UPD2 is relatively rare and has generally been reported following genetic analysis of individuals homozygous for variants in autosomal recessive genes consistent with their phenotype (Baskin, Geraghty, & Ray, 2010; Chavez, Valdez, & Vilchis, 2000; Lopez‐Garrido, Campos‐Mollo, Harto, & Escribano, 2009; Shen et al., 2018; Thompson et al., 2002). One of these cases involved a homozygous CYP1B1 missense variant due to paternal UPD2 in a patient with PCG (Lopez‐Garrido et al., 2009).
We previously showed that deletion of the entire CYP1B1 gene is not a major contributor to PCG (Souzeau, Hayes, Ruddle, et al., 2015). CYP1B1 deletions have only been reported three times, with deletion sizes ranging from 146kb to 193kb (Figure 3) (Damjanovich et al., 2013; Kelberman et al., 2011; Milla et al., 2013). The deletion reported in this study is the smallest deletion encompassing the entire CYP1B1 gene, with a size of 119kb. All reported deletions included part of the RMDN2 gene that has not been associated with any clinical phenotype.
Figure 3.
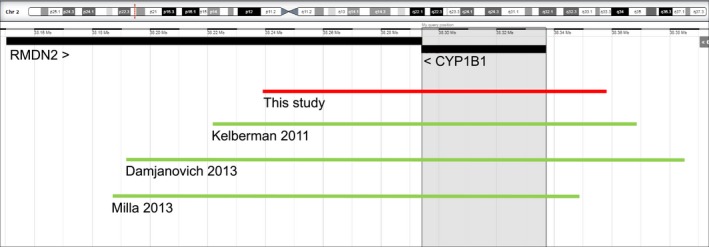
Summary of CYP1B1 entire gene deletions. Schematic drawing of the 2p22.2 region showing known human protein‐coding genes from the NCBI reference sequence (RefSeq) as seen in DECIPHER (http://decipher.sanger.ac.uk) using the GRCh37/hg19 assembly. The 119kb deletion reported in this study is shown in a solid red line (chr2: 38,239,356–38,358,664). Previously reported deletions are displayed in solid green lines (146kb, chr2: 38,222,086–38,368,231 (Kelberman, 2011); 193 kb, chr2: 38,191,823–38,385,253 (Damjanovich, 2013); 162kb, chr2: 38,187,289–38,349,505 (Milla, 2013))
The confirmation of UPD as the cause of variant homozygosity has important implications for the family in terms of genetic counseling. The parents can be counseled that the risk of recurrence for other children is low, as opposed to the 25% risk of recurrence usually reported for variants in autosomal recessive genes such as CYP1B1. Similarly, the occurrence of phenotypically normal individuals with paternal UPD2 does not support a significant role for paternally imprinted genes on chromosome 2 (Keller et al., 2009; Ou et al., 2013). This is supported by the absence of additional clinical features in the proband reported in this study.
In summary, this is the first report of a homozygous CYP1B1 whole gene deletion due to paternal UPD2 and associated with PCG. This study illustrates the importance of genetic testing in identifying the underlying molecular cause of the disease and in providing adequate genetic counseling regarding the possible recurrence risks.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
ACKNOWLEDGMENTS
This project was supported by the Australian National Health and Medical Research Council (NHMRC) Centres of Research Excellence Grant 1116360 (2017‐2021). Jamie E Craig is an NHMRC Practitioner Fellow (1154824).
Souzeau E, Dubowsky A, Ruddle JB, Craig JE. Primary congenital glaucoma due to paternal uniparental isodisomy of chromosome 2 and CYP1B1 deletion. Mol Genet Genomic Med. 2019;7:e774 10.1002/mgg3.774
REFERENCES
- Baskin, B. , Geraghty, M. , & Ray, P. N. (2010). Paternal isodisomy of chromosome 2 as a cause of long chain 3‐hydroxyacyl‐CoA dehydrogenase (LCHAD) deficiency. American Journal of Medical Genetics. Part A, 152A(7), 1808–1811. 10.1002/ajmg.a.33462 [DOI] [PubMed] [Google Scholar]
- Bejjani, B. A. , Stockton, D. W. , Lewis, R. A. , Tomey, K. F. , Dueker, D. K. , Jabak, M. , … Lupski, J. R. (2000). Multiple CYP1B1 mutations and incomplete penetrance in an inbred population segregating primary congenital glaucoma suggest frequent de novo events and a dominant modifier locus. Human Molecular Genetics, 9(3), 367–374. 10.1093/hmg/9.3.367 [DOI] [PubMed] [Google Scholar]
- Chavez, B. , Valdez, E. , & Vilchis, F. (2000). Uniparental disomy in steroid 5alpha‐reductase 2 deficiency. Journal of Clinical Endocrinology and Metabolism, 85(9), 3147–3150. 10.1210/jcem.85.9.6786 [DOI] [PubMed] [Google Scholar]
- Damjanovich, K. , Baldwin, E. E. , Lewis, T. , & Bayrak‐Toydemir, P. (2013). Novel homozygous CYP1B1 deletion in siblings with primary congenital glaucoma. Ophthalmic Genetics, 34(3), 180–181. 10.3109/13816810.2012.743571 [DOI] [PubMed] [Google Scholar]
- Engel, E. (1980). A new genetic concept: Uniparental disomy and its potential effect, isodisomy. American Journal of Medical Genetics, 6(2), 137–143. 10.1002/ajmg.1320060207 [DOI] [PubMed] [Google Scholar]
- Gencik, A. (1989). Epidemiology and genetics of primary congenital glaucoma in Slovakia. Description of a form of primary congenital glaucoma in gypsies with autosomal‐recessive inheritance and complete penetrance. Developments in Ophthalmology, 16, 76–115. [PubMed] [Google Scholar]
- Kelberman, D. , Islam, L. , Jacques, T. S. , Russell‐Eggitt, I. , Bitner‐Glindzicz, M. , Khaw, P. T. , … Sowden, J. C. (2011). CYP1B1‐related anterior segment developmental anomalies novel mutations for infantile glaucoma and von Hippel's ulcer revisited. Ophthalmology, 118(9), 1865–1873. 10.1016/j.ophtha.2011.01.044 [DOI] [PubMed] [Google Scholar]
- Keller, M. C. , McRae, A. F. , McGaughran, J. M. , Visscher, P. M. , Martin, N. G. , & Montgomery, G. W. (2009). Non‐pathological paternal isodisomy of chromosome 2 detected from a genome‐wide SNP scan. American Journal of Medical Genetics. Part A, 149A(8), 1823–1826. 10.1002/ajmg.a.32973 [DOI] [PubMed] [Google Scholar]
- Libby, R. T. , Smith, R. S. , Savinova, O. V. , Zabaleta, A. , Martin, J. E. , Gonzalez, F. J. , & John, S. W. (2003). Modification of ocular defects in mouse developmental glaucoma models by tyrosinase. Science, 299(5612), 1578–1581. 10.1126/science.1080095 [DOI] [PubMed] [Google Scholar]
- Lopez‐Garrido, M. P. , Campos‐Mollo, E. , Harto, M. A. , & Escribano, J. (2009). Primary congenital glaucoma caused by the homozygous F261L CYP1B1 mutation and paternal isodisomy of chromosome 2. Clinical Genetics, 76(6), 552–557. 10.1111/j.1399-0004.2009.01242.x [DOI] [PubMed] [Google Scholar]
- MacKinnon, J. R. , Giubilato, A. , Elder, J. E. , Craig, J. E. , & Mackey, D. A. (2004). Primary infantile glaucoma in an Australian population. Clinical and Experimental Ophthalmology, 32(1), 14–18. 10.1046/j.1442-9071.2004.00750.x. [DOI] [PubMed] [Google Scholar]
- Milla, E. , Mane, B. , Duch, S. , Hernan, I. , Borras, E. , Planas, E. , … Gamundi, M. J. (2013). Survey of familial glaucoma shows a high incidence of cytochrome P450, family 1, subfamily B, polypeptide 1 (CYP1B1) mutations in non‐consanguineous congenital forms in a Spanish population. Molecular Vision, 19, 1707–1722. [PMC free article] [PubMed] [Google Scholar]
- Ou, X. , Liu, C. , Chen, S. , Yu, J. , Zhang, Y. , Liu, S. , & Sun, H. (2013). Complete paternal uniparental isodisomy for Chromosome 2 revealed in a parentage testing case. Transfusion, 53(6), 1266–1269. 10.1111/j.1537-2995.2012.03863.x [DOI] [PubMed] [Google Scholar]
- Shen, W. , Young, B. A. , Bosworth, M. , Wright, K. E. , Lamb, A. N. , & Ji, Y. (2018). Prenatal detection of uniparental disomy of chromosome 2 carrying a CHRND pathogenic variant that causes lethal multiple pterygium syndrome. Clinical Genetics, 93(6), 1248–1249. 10.1111/cge.13164 [DOI] [PubMed] [Google Scholar]
- Stoilov, I. , Akarsu, A. N. , & Sarfarazi, M. (1997). Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Human Molecular Genetics, 6, 641–647. 10.1093/hmg/6.4.641. [DOI] [PubMed] [Google Scholar]
- Souzeau, E. , Goldberg, I. , Healey, P. R. , Mills, R. A. D. , Landers, J. , Graham, S. L. , … Craig, J. E. (2012). Australian and New Zealand Registry of Advanced Glaucoma: Methodology and recruitment. Clinical & Experimental Ophthalmology, 40(6), 569–575. 10.1111/j.1442-9071.2011.02742.x [DOI] [PubMed] [Google Scholar]
- Souzeau, E. , Hayes, M. , Ruddle, J. B. , Elder, J. E. , Staffieri, S. E. , Kearns, L. S. , … Craig, J. E. (2015). CYP1B1 copy number variation is not a major contributor to primary congenital glaucoma. Molecular Vision, 21, 160–164. [PMC free article] [PubMed] [Google Scholar]
- Souzeau, E. , Hayes, M. , Zhou, T. , Siggs, O. M. , Ridge, B. , Awadalla, M. S. , … Craig, J. E. (2015). Occurrence of CYP1B1 mutations in juvenile open‐angle glaucoma with advanced visual field loss. JAMA Ophthalmol, 133(7), 826–833. 10.1001/jamaophthalmol.2015.0980 [DOI] [PubMed] [Google Scholar]
- Thau, A. , Lloyd, M. , Freedman, S. , Beck, A. , Grajewski, A. , & Levin, A. V. (2018). New classification system for pediatric glaucoma: Implications for clinical care and a research registry. Current Opinion in Ophthalmology, 29(5), 385–394. 10.1097/icu.0000000000000516 [DOI] [PubMed] [Google Scholar]
- Thompson, D. A. , McHenry, C. L. , Li, Y. , Richards, J. E. , Othman, M. I. , Schwinger, E. , … Gal, A. (2002). Retinal dystrophy due to paternal isodisomy for chromosome 1 or chromosome 2, with homoallelism for mutations in RPE65 or MERTK, respectively. American Journal of Human Genetics, 70(1), 224–229. 10.1086/338455 [DOI] [PMC free article] [PubMed] [Google Scholar]