

Abstract
Background
The minimal critical region in 2q23.1 deletion syndrome comprises one gene only, that is, the methyl‐CpG‐binding domain protein 5 (MBD5) gene. Since the phenotypes of patients with deletions, duplications or pathogenic variants of MBD5 show considerable overlap, the term MBD5‐associated neurodevelopmental disorder (MAND) was proposed. These syndromes are characterized by intellectual disability, seizures of any kind and symptoms from the autism spectrum. In a very limited number of patients, MAND may be associated with regression starting either at early infancy or at midlife.
Methods
The present paper describes a severely intellectually disabled autistic female with therapy resistant complex partial epilepsy starting at her 16the with gradual cognitive and behavioral regression towards her sixth decade.
Results
Cognitive and behavioral regression occurred towards the patient's sixth decade. Exome sequencing disclosed a novel heterozygous pathogenic frameshift mutation of MBD5 that was considered to be causative for the combination of intellectual disability, treatment‐resistant epilepsy and autism.
Conclusion
The presented patient is the second with a pathogenic MBD5 mutation in whom the course of disease is suggestive of early onset dementia starting in her fifth decade. These findings stress the importance of exome sequencing, also in elderly intellectually disabled patients, particularly in those with autism.
Keywords: autism, epilepsy, intellectual disability, MAND, MBD5
1. INTRODUCTION
By using genome‐wide analysis, in 2003 a microdeletion in the 2q23.1 region was for the first time identified by Vissers and co‐workers in a 12‐year old girl, out of a series of 20 patients with unexplained intellectual disability and additional malformations such as microcephaly and facial dysmorphisms. They suggested that one or more genes within the deleted region could be causative for the phenotype (Vissers et al., 2003). In subsequent years it became obvious that the minimal critical region in the 2q23.1 deletion in a group of 15 patients comprises one gene only, that is methyl‐CpG‐binding domain protein 5 (MBD5) [OMIM: *611472] which is a member of the methyl‐CpG‐binding domain protein family that also includes MECP2, the causative locus in Rett syndrome, and is demonstrated to be highly expressed in the brain, fetal testes and fetal ovaries (Van Bon et al., 2010; Laget et al., 2010). The major characteristics of patients with a deletion comprising MBD5 are moderate to severe intellectual disability, seizures, sleep disturbances, speech impairment, autistic‐like symptoms and behavioral problems. In addition, a broad range of dysmorphisms as well as somatic abnormalities such as constipation and hand/foot anomalies may be present (Van Bon et al., 2010; Bonnet et al., 2013; Talkowski et al., 2011; Williams et al., 2010). Interestingly, a great overlap has been observed in clinical characteristics between patients with a deletion or duplication of 2q23.1 and MBD5 point mutations, in that they all share developmental and motor delay, language impairment, behavioral problems and symptoms from the autism spectrum albeit that the 2q23.1 duplication syndrome seems to have a milder phenotype. In contrast to those with a mutation, MBD5‐specific deletion or 2q23.1 deletion, seizures are not common in patients with MBD5 duplications. These observations suggest that MBD5 can be considered as a dose‐sensitive gene for normal development and plays a pivotal role in the pathophysiology of autism spectrum disorder (Mullegama et al., 2014).
Since the phenotypes of patients with deletions, duplications or pathogenic variants of MBD5 show considerable overlap, the umbrella term MBD5‐associated neurodevelopmental disorder (MAND; OMIM: #156200) was proposed to describe a group of syndromes that is caused by haploinsufficiency of MBD5 (Mullegama & Elsea, 2016). These syndromes are characterized by intellectual disability with serious developmental delays, language impairment, symptoms from the autism spectrum, sleep disturbances, and seizures of any kind although most patients exhibit one seizure type only. It is assumed that the sleep disturbances originate from MBD5‐ associated dysregulation of the circadian rhythm pathway (Mullegama, Pugliesi, et al., 2015). Albeit that MAND is suggested to be consistent with normal lifespan, in some patients it may be associated with regression starting either at early infancy as described in two single case reports of female children (Chung et al., 2011; Noh & Graham, 2012) or at midlife as mentioned in the paper by Hodge et al. (2014). The latter concerned a 44‐year‐old male patient with early onset dementia, cataracts and behavioral regression with increase in autistic‐like symptoms without, however, a detailed description of the disease history.
In this paper we describe a severely intellectually disabled autistic female with therapy resistant complex partial epilepsy starting at the age of 16, who developed cognitive and behavioral regression in her fifth decade and in whom ultimately exome sequencing disclosed a novel heterozygous pathogenic frameshift mutation in MBD5.
2. CASE REPORT
The patient was a 60‐year‐old intellectually disabled female born from nonconsanguineous parents. Because of imminent miscarriage, her mother was treated with diethylstilbestrol during pregnancy. She was born after a pregnancy of 6,5 months with a birth weight of 1,500 grams and has one younger and one older healthy brother. Early development was characterized by global delay especially concerning psychomotor skills. Due to a dysfunctional pedagogic climate with poor mother‐child attachment and alcohol abuse of both parents, already in her first years, severe oppositional behavioral problems became prominent that necessitated institutionalization at the age of four.
During subsequent years, the patient stayed in various institutes for intellectually disabled persons and sheltered home facilities but returned several times to her parental home because of lack of social interactions with her peers. At age 16, complex partial epileptic seizures occurred in combination with sleep disturbances and affective flattening for which she was admitted to a specialized hospital for epilepsy. Treatment with antiepileptics was started. She followed special education until her twenties and was employed on a part‐time basis for about five years in a sheltered work setting. Over the years, she alternately stayed at her parent's home until her mother became seriously ill and deceased, aged 65, from lung carcinoma and liver cirrhosis. At that time, the patient was 34 years. During that time, her younger brother developed a psychotic disorder for which he had to be admitted to a psychiatric hospital, was treated with depot antipsychotics and stayed thereafter for many years under outpatient control. In the meantime, the alcohol abuse of her father became increasingly problematic. Because of these circumstances, permanent institutionalization became necessary with, however, frequent changes in daycare settings because of mismatch between her needs and behavioral repertoire. Also, in this year, her father died at the age of 76 from acute cardiac failure.
In the following years the patient developed diabetes mellitus type 2, psoriasis vulgaris, hypertension and constipation for which adequate medications were prescribed. In addition, bilateral mild cataract was diagnosed. During this period, she was hospitalized several times because of her complex partial epilepsy that, however, persisted despite treatment with various combinations of antiepileptics, including valproic acid, levetiracetam, carbamazepine, clobazam and vigabatrine. At the age of 54, EEG‐recording showed multifocal epileptic features with spike‐wave configurations predominantly in the left frontal‐temporal region. MRI‐scanning of the brain disclosed no abnormalities apart from possibly rather pronounced folia of the cerebellar vermis. Testing of intelligence by WISC‐III administration resulted in a developmental age of about 9 years. Gradual decline of cognitive and behavioral functioning in the presence of enduring seizures and mood instability led, at age 60, to specialized neuropsychiatric consultation.
At referral, the patient was treated with the antiepileptics carbamazepine (600 mg; 7.6 mg/L), valproic acid (1,500 mg; 44 mg/L) and clobazam (10 mg; <50/520 µgr/L) whereas her somatic medications included metformine 500 mg daily, lisinopril 5 mg daily and macrogol in variable dose. In addition, for behavioral control and enduring sleep disturbances with frequent awakenings, 2 mg/day haloperidol and 100 mg/day sertraline were given that, however, were discontinued shortly after referral because of lack of effect and very low plasma concentrations due to interaction with carbamazepine.
Somatic investigation disclosed a full nasal tip but no other dysmorphisms (Figure 1). Height, weight and head circumference were 157 cm (−2,3 SD), 53 kg (BMI: 21,5) and 53 cm (−1,3 SD), respectively. Neuropsychiatric examination revealed autistic behaviors with echolalia and perseverations as well as lack of initiative and low responsiveness. No neurological abnormalities could be detected although absences were regularly observed. No reliable WISC‐V scores could be obtained because of floor effects for most subtests. As assessed with the Vineland Adaptive Behavior Scale (De Bildt & Kraijer, 2003), for the factors communication, daily performance and socialization, a developmental age of 5;3, 3;8 and 3;9 (years;months), respectively, was established that were all clearly lower as compared to the results at the age of 56 (6;11, 6;11 and 5;1 year;months, respectively) confirming the observed decline in general functioning over the past years. By using the Dutch Scale for Emotional Development in people with intellectual disability (SEO‐R; Claes & Verduyn, 2012), her emotional developmental age was established to be maximally 18–24 months. With the AVZ‐R (Kraijer, 2004), a validated Dutch rating scale covering the autism parameters social, communicative and stereotyped behavior, a total score of 7 was established (range 0–19; scores 0–6 absent, 7–9 possible, 10–19 autism/pdd), indicative for possible autism. With the Autism Diagnostic Observation Schedule (ADOS‐2; De Bildt, Jonge, & Greaves‐Lord, 2013), a total score of 21 (cut‐off score = 10) corroborated a diagnosis of autism. Hematological and biochemical parameters were all within normal range. Earlier performed SNP array analysis did not show any abnormalities.
Figure 1.
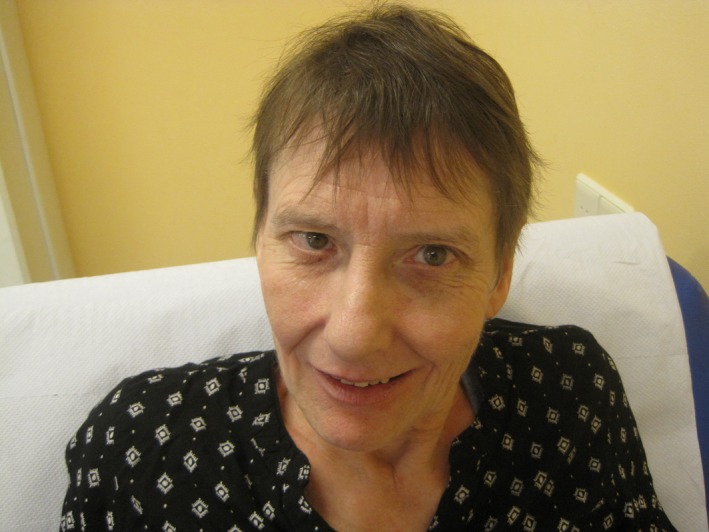
Picture of the face of the patient without remarkable dysmorphisms, apart from slight microcephaly
For reevaluation of her epilepsy, possible adjustment of antiepileptic medication and high‐resolution MRI‐scanning, the patient was referred to the Academic Centre for Epileptology Kempenhaeghe. As depicted in Figure 2, 24‐hr video‐EEG recording demonstrated sporadic left frontotemporal spike‐wave configurations (both awake and during relaxation and falling asleep). No changes in the combination of antiepileptics were judged necessary. MRI‐scanning of the brain under general anesthesia disclosed, comparable to the aforementioned MRI, no abnormalities (Figure 3). For further etiological analysis, whole exome sequencing was performed and subsequent analysis of variants within a panel of known intellectual disability disease genes resulted in the detection of a novel heterozygous pathogenic frameshift mutation in exon 9 of MBD5 (NG_017003.2) that leads to a premature stop codon within that same exon. The frameshift mutation concerns: Chr2(GRCh37):g.149227685del; NM_018328.4:c.2173del (p.[Ala725fs]). This finding was considered to be pathogenic and causative for the combination of intellectual disability, autism and treatment‐resistant epilepsy.
Figure 2.
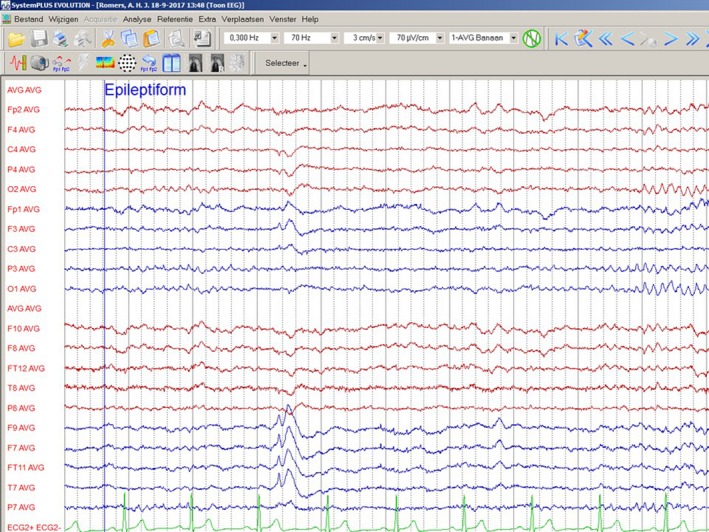
EEG‐recording bipolar lead. Spike visible in the left temporal region (F9, F7, FT11, T7). [3 cm/s, 70 µV/cm; high and low frequency filter characteristics mentioned in the figure]
Figure 3.
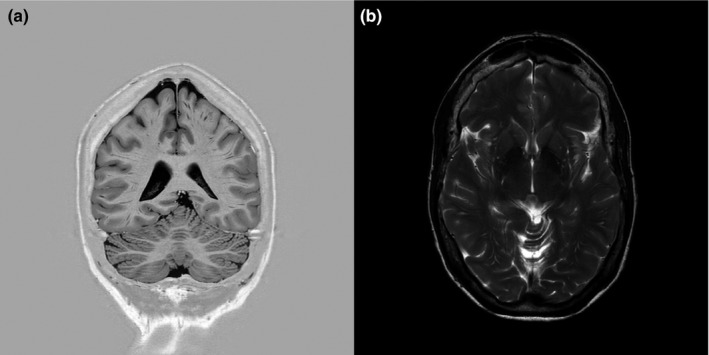
Brain MRI without abnormalities. (a) Coronal inversion recovery T2 image showing the cerebrum and cerebellum. (b) Axial T2 weighted image focused on the basal ganglia
3. DISCUSSION
In this paper, a detailed description of an older intellectually disabled autistic female with MBD5‐associated neurodevelopmental disorder caused by a not previously described heterozygous frameshift mutation in MBD5 is given. Her phenotypical characteristics closely match the previously described behavioral, somatic and neurological MAND‐profile (Mullegama & Elsea, 2016; Talkowski et al., 2011; Williams et al., 2010). Interestingly, this is the first patient in whom, apart from therapy resistant complex partial epilepsy, gradual regression occurred towards her sixth decade that could be objectivated with psychological and behavioral assessments.
With respect to brain imaging, no abnormalities could be detected either aged 54 or six years later. As to intellectual and cognitive function, however, six years before referral, the level of developmental was established to be about nine years whereas at referral she was no longer eligible for testing. This apparent deterioration was corroborated by several parameters as measured by the Vineland scales, that is, a significant decrease of especially daily performance skills and socialization. These converging observations are suggestive of early onset dementia.
The clinical features of MBD5 disruption with associated neurodevelopmental disorder and autism show similarities with other genetic syndromes such as Smith–Magenis, Rubinstein–Taybi, Angelman, Prader–Willi, Kleefstra and Pitt–Hopkins (Hodge et al., 2014; Mullegama, Alaimo, Chen, & Elsea, 2015). Indeed, network pathway analysis of nine ASD‐associated neurodevelopmental disorders disclosed that MBD5 regulates the expression of several autism related genes (UBE3A, TCF4, MEF2C, EHMT1, and RAI1) and that these and other genes (e.g., MECP2 and KMT2D) may combine into common or overlapping network pathways with phenotypical similarity (Mullegama, Alaimo, et al., 2015). This kind of functional convergence may be a key contributor to the identification of candidate genes and specific pharmacological interventions (Gigek et al., 2015; Iwase et al., 2017; Kleefstra et al., 2012; Koemans et al., 2017).
Summarizing, in the presented patient a diagnosis of MAND was established caused by a novel pathogenic mutation in MBD5. She is the second in whom the course of disease is suggestive of early onset dementia developing in the fifth decade. These findings stress both the clinical and scientific importance of exome sequencing techniques in patients with intellectual disability and psychopathology, particularly syndromes from the autism spectrum.
ACKNOWLEDGMENTS
This study is part of a collaborative project of the research group “Psychopathology and Genetics” of the Vincent van Gogh Centre of Excellence for Neuropsychiatry, Venray, the Donders Institute for Brain, Cognition and Behaviour, Nijmegen, and the Radboudumc Department of Human Genetics, Nijmegen. None of the authors has any conflict of interest. The first and second authors declare that they have had full access to all data and take responsibility for the integrity of the data and accuracy of the data analysis. The patient was referred to the first author by the Centre for Consultation and Expertise, West‐Gouda, the Netherlands. The authors are indebted to the neurological and neuroradiological staff members of the Academic Institute for Epileptology Kempenhaeghe, Heeze, the Netherlands, for their skillful examination of the patient including EEG‐recording and brain MRI scanning. MRIs of the brain were reevaluated by Dr. A. Laurent‐ de Gast, radiologist at the VieCuri Medical Centre in Venlo/Venray, the Netherlands, who also selected the most illustrative MRI brain images. Written informed consent was obtained from the legal representative of the patient for including a picture of the patient as well as images of MRI brain and EEG recording.
Verhoeven W, Egger J, Kipp J, et al. A novel MBD5 mutation in an intellectually disabled adult female patient with epilepsy: Suggestive of early onset dementia? Mol Genet Genomic Med. 2019;7:e849 10.1002/mgg3.849
REFERENCES
- Bonnet, C. , Ali Khan, A. , Bresso, E. , Vogouroux, C. , Béri, M. , Lejczak, S. , … Jonveaux, P. (2013). Extended spectrum of MBD5 mutations in neurodevelopmental disorders. European Journal of Human Genetics, 21, 1457–1461. 10.1038/ejhg.2013.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, B. H. Y. , Stavropoulos, J. , Marshall, C. R. , Weksberg, R. , Scherer, S. W. , & Yoon, G. (2011). 2q23.1 De novo microdeletion involving the MBD5 gene in a patient with developmental delay, postnatal microcephaly and distinct facial features. American Journal Medical Genetics, 155A, 424–429. 10.1002/ajmg.a.33821 [DOI] [PubMed] [Google Scholar]
- Claes, L. , & Verduyn, A. (2012). Scale for Emotional Development in People with Intellectual Disability [SEO‐R: Schaal voor Emotionele ontwikkeling bij mensen met een verstandelijke beperking – Revised]. Antwerpen‐Apeldoorn: Garant. [Google Scholar]
- De Bildt, A. , De Jonge, M. , & Greaves‐Lord, K. (2013). ADOS‐2 autism diagnostic observation schedule (Dutch version). Amsterdam, the Netherlands: Hogrefe. [Google Scholar]
- De Bildt, A. , & Kraijer, D. (2003). Dutch adaptation of the Vineland adaptive behavior scale of Sparrow, Balla, and Cicchetti. Leiden, the Netherlands: PITS. [Google Scholar]
- Gigek, C. , Chen, E. S. , Ota, V. K. , Maussion, B. , Peng, H. , Vaillancourt, K. , … Ernst, C. (2015). A Molecular model for neurodevelopmental disorders. Translational Psychiatry, 5, e565 10.1038/tp.2015.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge, J. C. , Mitchell, E. , Pillalamarri, V. , Toler, T. L. , Bartel, F. , Kearney, H. M. , … Talkowski, M. E. (2014). Disruption of MBD5 contributes to a spectrum of psychopathology and neurodevelopmental abnormalities. Molecular Psychiatry, 19, 368–379. 10.1038/mp.2013.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase, S. , Bérubé, N. G. , Zhou, Z. , Kasri, N. N. , Battagliolo, E. , Scandaglia, M. , & Barco, A. . (2017). Epigenetic etiology of intellectual disability. Journal of Neuroscience, 37, 10773–10782. 10.1523/JNEUROSCI.1840-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleefstra, T. , Kramer, J. M. , Neveling, K. , Willemsen, M. H. , Koemans, T. S. , Vissers, L. E. L. M. , … van Bokhoven, H. (2012). Disruption of an EHMT1‐associated chromatin‐modification module causes intellectual disability. American Journal Human Genetics, 91, 73–82. 10.1007/s00439-012-1216-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koemans, T. S. , Kleefstra, T. , Chubak, M. C. , Stone, M. H. , Reijnders, M. R. F. , de Munnik, S. , … Kramer, J. M. (2017). Functional convergence of histone methyltransferase EHMT1 and KMT2C involved in intellectual disability and autism spectrum disorder. Plos Genetics, 13, e1006864 10.1371/journal.pgen.1006864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraijer, D. (2004). AVZ‐R Manual, 4th ed. Amsterdam, the Netherlands: Harcourt Test Publishers. [Google Scholar]
- Laget, S. , Joulie, M. , Le Masson, F. , Sasai, N. , Christians, E. , Pradhan, S. , … Defossez, P. A. (2010). The human proteins MBD5 and MBD6 associate with heterochromatin but they do not bind methylated DNA. PLoS ONE, 5, e11982 10.1371/journal.pone.0011982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullegama, S. V. , Alaimo, J. T. , Chen, L. , & Elsea, S. H. . (2015). Phenotypic and molecular convergence of 2q23.1 deletion syndrome with other neurodevelopmental syndromes associated with autism spectrum disorder. International Journal Molecular Sciences, 16(12), 7627–7643. 10.3390/ijms16047627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullegama, S. V. , & Elsea, S. H. (2016). Clinical and molecular aspects of MBD5‐associated neurodevelopmental disorder (MAND). European Journal Medical Genetics, 24, 1235–1243. 10.1038/ejhg.2016.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullegama, S. V. , Pugliesi, L. , Burns, B. , Shah, A. , Tahir, R. , Gu, Y. , … Elsea, S. H. (2015). MBD5 haploinsufficiency is associated with sleep disturbance and disrupts circadian pathways common to Smith‐Magenis and fragile X syndromes. European Journal Medical Genetics, 23, 781–789. 10.1038/ejhg.2014.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullegama, S. V. , Rosenfeld, J. A. , Orellana, C. , van Bon, B. W. M. , Halbach, S. , Repnikova, E. A. , … Elsea, S. H. (2014). Reciprocal deletion and duplication at 2q23.1 indicates a role for MBD5 in autism spectrum disorder. European Journal Human Genetics, 22, 57–63. 10.1038/ejhg.2013.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh, G. J. , & Graham, J. M. Jr (2012). 2q23.1 microdeletion of the MBD5 gene in a female with seizures, developmental delay and distinct dysmorphic features. European Journal Medical Genetics, 55, 354–357. 10.1016/j.ejmg.2011.10.001 [DOI] [PubMed] [Google Scholar]
- Talkowski, M. E. , Mullegama, S. V. , Rosenfeld, J. A. , van Bon, B. W. M. , Shen, Y. , Repnikova, E. A. , … Elsea, S. H. (2011). Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. American Journal Human Genetics, 89, 551–563. 10.1016/j.ajhg.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bon, B. W. M. , Koolen, D. A. , Brueton, L. , McMullen, D. , Lihtenbelt, K. D. , Adès, L. C. , …, de Vries, B. B. A. (2010). The 2q23.1 microdeletion syndrome: clinical and behavioural phenotype. European Journal of Human Genetics, 18(2), 163–170. 10.1038/ejhg.2009.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers, L. E. L. M. , de Vries, B. A. , Osoegawa, K. , Janssen, I. M. , Feuth, T. , On Choy, C. , … Veltman, J. A. (2003). Array‐based genomic hybridization for the genome wide detection of submicroscopic chromosomal abnormalities. American Journal Human Genetics, 73, 1261–1270. 10.1086/379977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, S. R. , Mullegama, S. V. , Rosenfeld, J. A. , Dagli, A. I. , Hatchwell, E. , Allen, W. P. , … Elsea, S. H. (2010). Haploinsufficiency of MBD5 associated with a syndrome involving microcephaly, intellectual disabilities, severe speech impairment, and seizures. European Journal Human Genetics, 18, 436–441. 10.1038/ejhg.2009.199 [DOI] [PMC free article] [PubMed] [Google Scholar]