

Abstract
Background
Dilated cardiomyopathy (DCM) is the most common cardiomyopathy with a common presentation of heart failure. It has been reported that CASZ1 loss‐of‐function mutation contributes to familial DCM and congenital ventricular septal defect (VSD). To date, only two pathogenic variants in CASZ1 have been previously reported worldwide.
Methods
To identify the causative variant in an 11‐month‐old Chinese boy with DCM and left ventricular noncompaction cardiomyopathy (LVNC), trio‐whole‐exome sequencing was performed followed by mutational analysis and Sanger sequencing.
Results
An unreported de novo heterozygous frameshift variant (c.2443_2459delGTGGGCACCCCCAGCCT, p.Val815Profs*14) in CASZ1 was idenitified in the proband. The frameshift mutation in CASZ1 not only led to DCM but also presented an LVNC phenotype.
Conclusion
We have identified a novel CASZ1 variant in a patient with combined DCM and LVNC for the first time, thus broadening the phenotypic spectrum of CASZ1 variants. Furthermore, this study emphasized the usefulness of whole‐exome sequencing for genetic diagnosis of cardiomyopathy.
Keywords: CASZ1 variant, DCM, LVNC, whole‐exome sequencing
1. INTRODUCTION
Dilated cardiomyopathy (DCM) is the most common cardiomyopathy, characterized by dilatation and impaired contractility of the left or both ventricles, and its common presentation is heart failure (HF) (Bharucha et al., 2015; Richardson et al., 1996). Recently, extrapolation of epidemiological data for DCM and HF suggests a prevalence as high as 1:250 in the general population worldwide (Hershberger, Hedges, & Morales, 2013). Left ventricular noncompaction cardiomyopathy (LVNC) is an increasingly recognized type of cardiomyopathy, characterized by a 2‐layered myocardium, numerous prominent trabeculations, and deep intertrabecular recesses communicating with the left ventricular (LV) cavity (Jenni, Rojas, & Oechslin, 1999; Oechslin, Attenhofer Jost, Rojas, Kaufmann, & Jenni, 2000). The combined incidence of DCM and LVNC was 0.77 per 100,000 person years in Sweden (Fadl, Wahlander, Fall, Cao, & Sunnegardh, 2018).
Dilated cardiomyopathy frequently has a genetic etiology and approximately 40% patients with familial DCM have an identifiable genetic origin (Ganesh et al., 2013). DCM has been reported in association with mutations in >100 genes, which encode molecules that participate in sarcomere integrity, cytoskeletal and nuclear architecture, force generation, force transmission, electrolyte homeostasis, gene transcription and mitochondrial function (Burke, Cook, Seidman, & Seidman, 2016). There is evidence for genetic overlap among DCM and LVNC. To date, more than 40 genes have been implicated in LVNC (Finsterer, Stollberger, & Towbin, 2017).
CASZ1 is a para‐zinc‐finger transcription factor that is required for vertebrate heart development (Christine & Conlon, 2008). To date, only two pathogenic variants of CASZ1 have been reported. A CASZ1 missense mutation p.L38P has been identified in a family with congenital ventricular septal defect (VSD) (Huang et al., 2016), and a CASZ1 nonsense mutation p.K351X has been identified to be associated with DCM (Qiu et al., 2017). Here we present the clinical features of a patient with a de novo CASZ1 frameshift variant, c.2443_2459delGTGGGCACCCCCAGCCT (p.Val815Profs*14). The patient manifested DCM, LVNC and sudden cardiac arrest (SCA).
2. MATERIALS AND METHODS
2.1. Ethical compliance
The study was approved by the Ethics Committee of Beijing Children's Hospital, Capital Medical University.
2.2. Patient information
After obtaining informed consent, clinical data, pedigree information, blood samples were collected from the proband and his parents. All subjects underwent detailed physical evaluations by an experienced pediatrician, including blood examination, echocardiographic examination, electrocardiography (ECG), radiographs of chest, and other necessary tests.
2.3. Whole‐exome sequencing
DNA was extracted from peripheral blood samples of the proband and his parents using a QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) in accordance with the manufacturer's protocol. Trio‐whole‐exome sequencing was performed using the Agilent SureSelect Human All Exome V6 kit (Agilent Technologies Inc. Mississauga, ON, Canada) on an Illumina Hiseq X Analyzer (Illumina, San Diego, CA) with 150‐bp paired‐end runs.
2.4. Analysis and evaluation of variants
Sequence reads were mapped to the GRCh37/hg19 human reference genome. Variants were analyzed using four population databases (db‐SNP, genomAD browser, Exome Aggregation Consortium (ExAC), and 1000 Genomes Project Database) and three disease databases (OMIM, ClinVar, and HGMD). Variants were filtered if they had a minor allele frequency >0.5%, or >1% if they had been previously associated with disease (OMIM, ClinVar, and HGMD), or >2% if the variant was homozygous or if there was a second variant in the gene. Nonsynonymous variants and small indels in exons and splicing variants were selected to identify causal variants. Five applications (SIFT, Polyphen‐2, Mutation Taster, Mutation Assessor and CADD) were used to predict the potential impact of the identified mutations on protein function. We classified the variants in the cardiomyopathy‐associated gene according to the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015).
2.5. Sanger sequencing
DNA samples from the proband and his parents were amplified, and the variants detected by whole‐exome sequencing were verified using Sanger sequencing with a 3730xl DNA analyzer (Applied Biosystems, Foster City, CA).
3. RESULTS
3.1. Clinical findings
The patient was an 11‐month‐old boy. He was born at 39 of weeks gestation by vaginal delivery with birth weight 3.5 kg and 49 cm length. The boy was admitted to a local hospital because of fever and cough. After 5 days of oral treatment with cefixime, his fever continued with persistent cough and diarrhea. Then he was transferred to our hospital. Radiographs of chest displayed pneumonia and marked cardiomegaly with a cardiothoracic ratio of 0.69 (Figure 1a). Echocardiographic examination revealed a dilated left ventricle (left ventricular diastolic dimension of 51 mm) with prominent trabeculations, a depressed LV ejection fraction (LVEF) of 25%, and mild mitral regurgitation. The left atrial diameter was 20.3 mm. Right ventricular size, function, and wall structure were normal (Figure 1b). ECG displayed sinus rhythm with ST‐segment changes. Serum biochemistry tests showed high levels of N‐terminal pro‐brain natriuretic peptide (NT‐proBNP; 17,108 pg/ml; normal range <300 pg/ml) and D‐dimer (1.77 mg/L; normal range <0.5 mg/L). The patient had no extracardiac manifestations and was diagnosed with pneumonia, DCM and LVNC. He died on the second day of hospitalization due to ventricular fibrillation.
Figure 1.
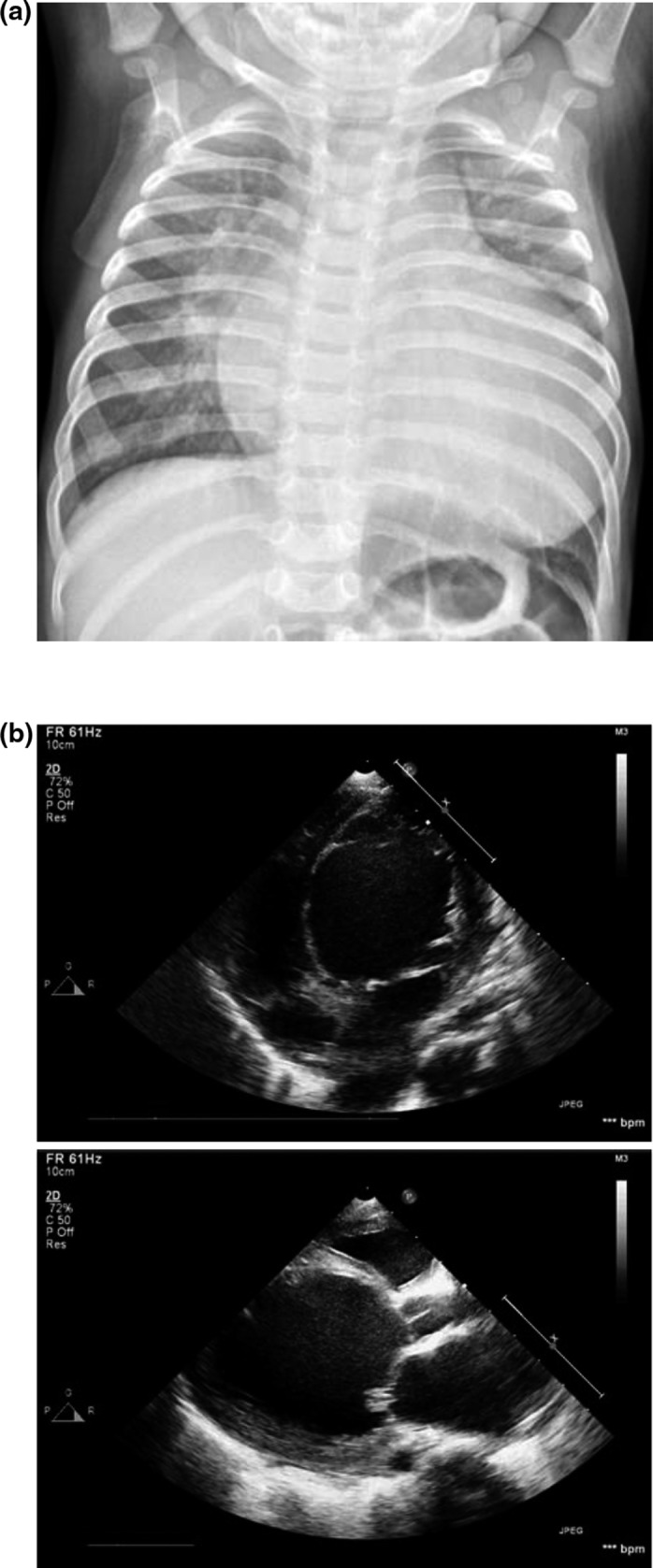
Radiographs of chest and echocardiographic examination of the proband. (a), the radiographs of chest displayed pneumonia and marked cardiomegaly with a cardiothoracic ratio of 0.69. (b), echocardiographic examination revealed a dilated left ventricle with prominent trabeculations, a depressed LV ejection fraction of 25%, and mild mitral regurgitation. The left atrial diameter was 20.3 mm. Right ventricular size, function, and wall structure were normal
The patient's parents are nonconsanguineous and there was no family history of cardiomyopathy and SCA. The mother had no complaints of abnormal manifestations during the perinatal period or history of reproductive system diseases.
3.2. Mutational analysis and Sanger sequencing
The mean coverage rate of target regions was 99.6% and on average 99.0% of the target bases were covered by >20× reads for each exome. The average sequencing depth of the exomes and their neighborhood was above 100×. A novel, de novo frameshift variant, c.2443_2459delGTGGGCACCCCCAGCCT (p.Val815Profs*14) of CASZ1 was identified in the patient and was confirmed using Sanger sequencing in the proband and his parents (Figure 2a).
Figure 2.
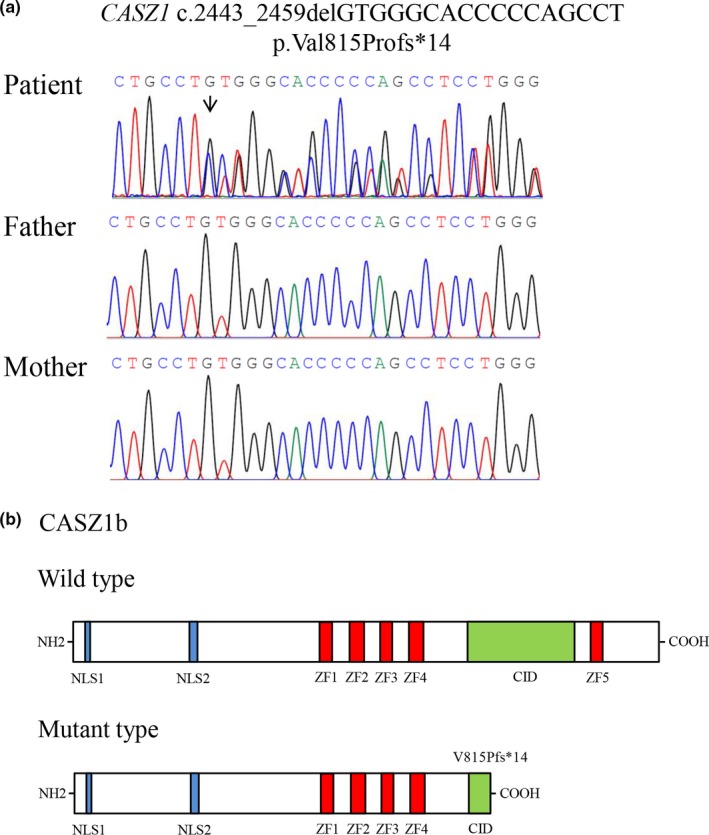
Sanger sequencing analysis and Schematic representations of protein structures. (a), Sanger sequencing analysis of CASZ1 in the patient and his parents. The arrows indicated the mutated nucleotides. The patient carries a de novo heterozygous frameshift mutation (c.2443_2459delGTGGGCACCCCCAGCCT) in CASZ1. (b), schematic of the full‐length CASZ1b protein and V815Pfs*14‐mutant CASZ1 protein structures. NH2 means amino terminus; CID, CHD5‐interacting domain; COOH, carboxyl terminus; NLS, nuclear location signal; ZF, zinc finger
3.3. Pathogenicity evaluation of the identified variant
Human CASZ1 is expressed in two major alternatively spliced isoforms, CASZ1a and CASZ1b. CASZ1b is the more evolutionarily conserved isoform and most functional characterizations of mutant CASZ1 proteins were focused on the evolutionarily conserved CASZ1b isoform (Qiu et al., 2017). The frameshift mutation (c.2443_2459delGTGGGCACCCCCAGCCT, p.Val815Profs*14) of CASZ1 in exon 11 is predicted to result in premature truncation of the protein, substituting valine at amino acid position 815 for proline. The severe truncation leads to a loss of 339 amino acids of CASZ1b, including zinc finger 5 and congenital heart disease 5 protein (CHD5)‐interacting domain (CID) of the C terminus of CASZ1 (Sojka et al., 2014; Figure 2b). The probability of loss‐of‐function intolerance (pLI; Lek et al., 2016) for CASZ1 in the ExAC Browser was 1.0, strongly supporting the fact that this variant is pathogenic. Neither the population databases nor the disease databases contain this variant. In line with the ACMG guidelines, the frameshift mutation c.2443_2459delGTGGGCACCCCCAGCCT of CASZ1 can be classified as pathogenic.
4. DISCUSSION
The patient reported here is a carrier of a de novo frameshift mutation in CASZ1. He presented at the age of 11 months with DCM and LVNC. CASZ1 contributes to VSD (Huang et al., 2016) and DCM (Qiu et al., 2017) in humans. However, none of the previously described patients carrying mutations in CASZ1 has been reported to display LVNC as well.
CASZ1 is a para‐zinc‐finger transcription factor and localizes to the nucleus. CASZ1 is expressed in cardiomyocytes during the earliest stages of mammalian heart development and is essential for the proliferation of cardiomyocytes in the first and second heart fields (Dorr et al., 2015). CASZ1 has been shown to be important in cardiac morphogenisis and development in Xenopus (Christine & Conlon, 2008) and mouse (Liu et al., 2014). In Xenopus, failure of a subset of progenitor cells to differentiate into cardiomyocytes and aberrant cardiac morphogenesis was identified in CASZ1 depleted embryos (Christine & Conlon, 2008). Consistent with this, cardiac noncompaction and VSD were identified in CASZ1 knockout mice (Liu et al., 2014). Abnormal expression of some CASZ1 targeted genes, such as muscle contraction genes (TNNI2, TNNT1, and CKM), contractile fiber gene (ACTA1), and cardiac arrhythmia associated ion channel coding genes (ABCC9 and CACNA1D), were identified in CASZ1 depleted embryonic hearts in mice, which might contribute to the heart defect (Liu et al., 2014). Furthermore, CASZ1 played an essential role in mammalian cardiomyocyte cell cycle progression (Dorr et al., 2015). Loss of CASZ1 leads to a marked reduction in DNA synthesis, an increase in phospho‐RB, and a decrease in the cardiac mitotic index due to a prolonged or arrested S phase (Dorr et al., 2015).
Recently, a CASZ1 missense mutation p.L38P was identified in a congenital VSD family (Huang et al., 2016). However, a CASZ1 nonsense mutation, p.K351X, was identified in a DCM family (Qiu et al., 2017). Functional studies revealed that both the L38P‐mutant and K351X‐mutant CASZ1 protein lost transcriptional activity (Huang et al., 2016; Qiu et al., 2017).
CASZ1 interacts with CHD5 and TBX20. CID of the C terminus of CASZ1 (amino acids 785–998) is necessary and sufficient to interact with CHD5. CHD5‐CASZ1 interaction is responsible for cardiac morphogenesis (Sojka et al., 2014). CASZ1 physically interacts with TBX20, and the four most N‐terminal zinc finger domains of CASZ1 are necessary for the CASZ1‐TBX20 interaction. Reduction or loss of physical interaction between TBX20 and CASZ1 leads to DCM (Kennedy et al., 2017). The frameshift mutation (c.2443_2459delGTGGGCACCCCCAGCCT, p.Val815Profs*14) was predicted to produce a truncated protein lacking the fifth zinc finger domain and the C terminus of CASZ1, including the CHD5‐interacting domain, but not the TBX20‐interacting domain. Nevertheless, the premature truncation of CASZ1 likely mediates mRNA decay to disrupt the whole function of CASZ1. Furthermore, the ExAC pLI of 1.0 for CASZ1 demonstrated that this gene is extremely intolerant of loss‐of‐function variation.
In the DCM family with CASZ1 nonsense mutation p.K351X, the age at initial diagnosis of DCM was 25–45 years of age. However, our patient was diagnosed with DCM and HF at the age of 11 months, which was earlier than the patients reported. Furthermore, our patient displayed prominent trabeculations in the LV and was diagnosed with LVNC. The etiology of LVNC is uncertain, but it is speculated that it results from a disturbed compaction process during early development of the LV myocardium (Sedmera, Pexieder, Vuillemin, Thompson, & Anderson, 2000). CASZ1 depletion in the murine has been shown to be implicated in the development of LV compaction during embryogenesis (Liu et al., 2014). Our study reveals CASZ1 as a new causative gene responsible for human LVNC. It has been reported that subjects with LVNC and a dilated phenotype had higher rates of death or transplantation than matched subjects with DCM (Shi et al., 2018). The combination phenotype of DCM and LVNC may be explained the bad prognosis, including ventricular fibrillation and SCA of this patient.
In conclusion, we have identified a novel CASZ1 variant in a patient with combined DCM and LVNC for the first time, thus broadening the phenotypic spectrum of CASZ1 variants. Furthermore, this study emphasized the usefulness of whole‐exome sequencing for genetic diagnosis of cardiomyopathy.
CONFLICT OF INTEREST
None declared.
ACKNOWLEDGMENT
This work was supported by the National Natural Science Foundation of China (31830054; 91539204), and the Ministry of Science and Technology of China (2016YFC1000306).
Guo J, Li Z, Hao C, et al. A novel de novo CASZ1 heterozygous frameshift variant causes dilated cardiomyopathy and left ventricular noncompaction cardiomyopathy. Mol Genet Genomic Med. 2019;7:e828 10.1002/mgg3.828
Jun Guo and Zheng Li contributed equally to this work.
Contributor Information
Hengmiao Gao, Email: hengm_gao@163.com.
Wei Li, Email: liwei@bch.com.cn.
REFERENCES
- Bharucha, T. , Lee, K. J. , Daubeney, P. E. F. , Nugent, A. W. , Turner, C. , Sholler, G. F. , … Davis, A. M. ; Investigators, N . (2015). Sudden death in childhood cardiomyopathy: Results from a long‐term national population‐based study. Journal of the American College of Cardiology, 65(21), 2302–2310. 10.1016/j.jacc.2015.03.552 [DOI] [PubMed] [Google Scholar]
- Burke, M. A. , Cook, S. A. , Seidman, J. G. , & Seidman, C. E. (2016). Clinical and mechanistic insights into the genetics of cardiomyopathy. Journal of the American College of Cardiology, 68(25), 2871–2886. 10.1016/j.jacc.2016.08.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christine, K. S. , & Conlon, F. L. (2008). Vertebrate CASTOR is required for differentiation of cardiac precursor cells at the ventral midline. Developmental Cell, 14(4), 616–623. 10.1016/j.devcel.2008.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorr, K. M. , Amin, N. M. , Kuchenbrod, L. M. , Labiner, H. , Charpentier, M. S. , Pevny, L. H. , … Conlon, F. L. (2015). Casz1 is required for cardiomyocyte G1‐to‐S phase progression during mammalian cardiac development. Development, 142(11), 2037–2047. 10.1242/dev.119107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadl, S. , Wahlander, H. , Fall, K. , Cao, Y. , & Sunnegardh, J. (2018). The highest mortality rates in childhood dilated cardiomyopathy occur during the first year after diagnosis. Acta Paediatrica, 107(4), 672–677. 10.1111/apa.14183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterer, J. , Stollberger, C. , & Towbin, J. A. (2017). Left ventricular noncompaction cardiomyopathy: Cardiac, neuromuscular, and genetic factors. Nature Reviews Cardiology, 14(4), 224–237. 10.1038/nrcardio.2016.207 [DOI] [PubMed] [Google Scholar]
- Ganesh, S. K. , Arnett, D. K. , Assimes, T. L. , Basson, C. T. , Chakravarti, A. , Ellinor, P. T. , … Waldman, S. A. ; American Heart Association Stroke, C . (2013). Genetics and genomics for the prevention and treatment of cardiovascular disease: Update: A scientific statement from the American Heart Association. Circulation, 128(25), 2813–2851. 10.1161/01.cir.0000437913.98912.1d [DOI] [PubMed] [Google Scholar]
- Hershberger, R. E. , Hedges, D. J. , & Morales, A. (2013). Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nature Reviews Cardiology, 10(9), 531–547. 10.1038/nrcardio.2013.105 [DOI] [PubMed] [Google Scholar]
- Huang, R.‐T. , Xue, S. , Wang, J. , Gu, J.‐Y. , Xu, J.‐H. , Li, Y.‐J. , … Yang, Y.‐Q. (2016). CASZ1 loss‐of‐function mutation associated with congenital heart disease. Gene, 595(1), 62–68. 10.1016/j.gene.2016.09.044 [DOI] [PubMed] [Google Scholar]
- Jenni, R. , Rojas, J. , & Oechslin, E. (1999). Isolated noncompaction of the myocardium. New England Journal of Medicine, 340(12), 966–967. 10.1056/NEJM199903253401215 [DOI] [PubMed] [Google Scholar]
- Kennedy, L. , Kaltenbrun, E. , Greco, T. M. , Temple, B. , Herring, L. E. , Cristea, I. M. , & Conlon, F. L. (2017). Formation of a TBX20‐CASZ1 protein complex is protective against dilated cardiomyopathy and critical for cardiac homeostasis. PLoS Genetics, 13(9), e1007011 10.1371/journal.pgen.1007011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , … MacArthur, D. G. ; Exome Aggregation Consortium . (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Li, W. , Ma, X. , Ding, N. , Spallotta, F. , Southon, E. , … Thiele, C. J. (2014). Essential role of the zinc finger transcription factor Casz1 for mammalian cardiac morphogenesis and development. Journal of Biological Chemistry, 289(43), 29801–29816. 10.1074/jbc.M114.570416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oechslin, E. N. , Attenhofer Jost, C. H. , Rojas, J. R. , Kaufmann, P. A. , & Jenni, R. (2000). Long‐term follow‐up of 34 adults with isolated left ventricular noncompaction: A distinct cardiomyopathy with poor prognosis. Journal of the American College of Cardiology, 36(2), 493–500. 10.1016/S0735-1097(00)00755-5 [DOI] [PubMed] [Google Scholar]
- Qiu, X.‐B. , Qu, X.‐K. , Li, R.‐G. , Liu, H. , Xu, Y.‐J. , Zhang, M. , … Yang, Y.‐Q. (2017). CASZ1 loss‐of‐function mutation contributes to familial dilated cardiomyopathy. Clinical Chemistry and Laboratory Medicine, 55(9), 1417–1425. 10.1515/cclm-2016-0612 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. ; ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson, P. , McKenna, W. , Bristow, M. , Maisch, B. , Mautner, B. , O'Connell, J. , … Nordet, P. (1996). Report of the 1995 world health organization/international society and federation of cardiology task force on the definition and classification of cardiomyopathies. Circulation, 93(5), 841–842. [DOI] [PubMed] [Google Scholar]
- Sedmera, D. , Pexieder, T. , Vuillemin, M. , Thompson, R. P. , & Anderson, R. H. (2000). Developmental patterning of the myocardium. Anatomical Record, 258(4), 319–337. [DOI] [PubMed] [Google Scholar]
- Shi, W. Y. , Moreno‐Betancur, M. , Nugent, A. W. , Cheung, M. , Colan, S. , Turner, C. , … Weintraub, R. G. ; National Australian Childhood Cardiomyopathy, S . (2018). Long‐term outcomes of childhood left ventricular non‐compaction cardiomyopathy: Results from a national population‐based study. Circulation, 138(4), 367–376. 10.1161/CIRCULATIONAHA.117.032262 [DOI] [PubMed] [Google Scholar]
- Sojka, S. , Amin, N. M. , Gibbs, D. , Christine, K. S. , Charpentier, M. S. , & Conlon, F. L. (2014). Congenital heart disease protein 5 associates with CASZ1 to maintain myocardial tissue integrity. Development, 141(15), 3040–3049. 10.1242/dev.106518 [DOI] [PMC free article] [PubMed] [Google Scholar]