

Abstract
Recent proposals suggest that sleep may be a factor associated with accumulation of two core pathological features of Alzheimer's disease (AD): tau and β-amyloid (Aβ). Here we combined PET measures of Aβ and tau, electroencephalogram sleep recordings, and retrospective sleep evaluations to investigate the potential utility of sleep measures in predicting in vivo AD pathology in male and female older adults. Regression analyses revealed that the severity of impaired slow oscillation-sleep spindle coupling predicted greater medial temporal lobe tau burden. Aβ burden was not associated with coupling impairment but instead predicted the diminished amplitude of <1 Hz slow-wave-activity, results that were statistically dissociable from each other. Additionally, comparisons of AD pathology and retrospective, self-reported changes in sleep duration demonstrated that changes in sleep across the lifespan can predict late-life Aβ and tau burden. Thus, quantitative and qualitative features of human sleep represent potential noninvasive, cost-effective, and scalable biomarkers (current and future forecasting) of AD pathology, and carry both therapeutic and public health implications.
SIGNIFICANCE STATEMENT Several studies have linked sleep disruption to the progression of Alzheimer's disease (AD). Tau and β-amyloid (Aβ), the primary pathological features of AD, are associated with both objective and subjective changes in sleep. However, it remains unknown whether late life tau and Aβ burden are associated with distinct impairments in sleep physiology or changes in sleep across the lifespan. Using polysomnography, retrospective questionnaires, and tau- and Aβ-specific PET, the present study reveals human sleep signatures that dissociably predict levels of brain tau and Aβ in older adults. These results suggest that a night of polysomnography may aid in evaluating tau and Aβ burden, and that treating sleep deficiencies within decade-specific time windows may serve in delaying AD progression.
Keywords: aging, Alzheimer's disease, beta-amyloid, PET, sleep, tau
Introduction
Alzheimer's disease (AD) is an unquestioned public health crisis. A current pressing need is the development of biomarkers of underlying AD pathophysiology that are sensitive, noninvasive, cost-effective, and scalable, and that demonstrate independent sensitivity to each of the two hallmark features of AD: pathological tau and β-amyloid (Aβ) aggregates. A second issue concerns when, during the adult lifespan, changes in sleep quantity and/or quality signal a future forecasting sensitivity to AD pathology, measured in old age. Addressing this unresolved question would inform not only the timing of when such biomarkers should be measured, but also help define time-sensitive windows that could be used in earlier-life prevention approaches, rather than later-life treatment efforts.
Evidence suggests that sleep may represent one such potential biomarker of AD pathology, and one that is noninvasive, cost-effective, and potentially scalable, relative to current PET imaging options. For example, sleep quality and sleep-EEG alterations are recognized to co-occur with the transition into mild cognitive impairment and AD (Prinz et al., 1982; Hita-Yañez et al., 2013). In addition, subjective impairments in sleep (Spira et al., 2013, 2018; Sprecher et al., 2015; Carvalho et al., 2018) and the loss of <1 Hz non-rapid eye movement (NREM) slow-wave activity (SWA) (Mander et al., 2015) predict levels of Aβ aggregation as measured by PET.
However, these data leave at least two key questions currently unanswered. First, while tau and Aβ are related to NREM sleep disruption in early AD (Lucey et al., 2019), it remains unknown whether there are physiological sleep features that are uniquely associated with tau in the human brain, dissociable from those linked to Aβ. Data in animals support the proposition that tau accumulation is linked to disruption of sleep oscillations. Rodent models of tauopathy exhibit reductions in NREM sleep quality and related neural synchrony (Menkes-Caspi et al., 2015; Holth et al., 2017), and mice with aggregated tau in hippocampus show abnormal hippocampal sharp-wave ripple oscillations (Witton et al., 2016). These findings are of special relevance considering that ripples in the medial temporal lobe (MTL), a region of early tau accumulation (Braak and Braak, 1997), are causally linked with the coupled expression of NREM sleep spindles and slow oscillations (SOs) in the surface EEG (Clemens et al., 2007; Staresina et al., 2015). These data support an untested model whereby early tau accumulation in the MTL causally alters hippocampal generation of NREM sharp-wave ripples, with the measurable effect of impairing the expression and coupling of NREM SOs and spindles. If so, it may represent a candidate sleep physiological biomarker of tau accumulation in old age.
A second question is when in the time course of the human lifespan biomarkers of sleep disruption, specifically alterations in amount of sleep (duration), may be linked to abnormal Aβ and tau accumulation, resulting in elevated pathology in late stages of life. Understanding when in the lifespan AD pathophysiology may be particularly vulnerable to sleep disruption will be critical in determining when such biomarker screening should occur, and would also help in defining time-sensitive windows that could assist in developing earlier-life prevention approaches.
Building on this evidence, here we tested two interrelated hypotheses by combining overnight polysomnography recording, retrospective sleep evaluations, and PET measurements of tau and Aβ pathology in older adults: (1) micro-level changes in sleep physiology, specifically SO-spindle phase-amplitude coupling and <1 Hz SWA- predict MTL tau burden and cortical Aβ, respectively; and (2) macro-level changes (declines) in sleep quantity across specific windows of the adult lifespan forecast tau and amyloid pathological burden in old age.
Materials and Methods
Study participants.
The experimental sample comprised a total of 101 cognitively normal older adults from the Berkeley Aging Cohort Study, a longitudinal study of cognitive aging. Participants were enrolled in the present study if concurrently acquired PET and MRI data were available. The study was approved by the human studies committees at University of California, Berkeley and Lawrence Berkeley National Laboratories, with all participants providing written informed consent. Exclusion criteria included presence of neurologic or psychiatric disorders, and current use of antidepressant or hypnotic medications. Participants were free of depressive symptoms, scored ≥25 on the Mini Mental State Examination (Folstein et al., 1975), and displayed normal performance on neuropsychological testing (1.5 SDs within age, education, and sex adjusted means) (Schöll et al., 2016).
PET imaging analysis.
A total of n = 101 cognitively normal older adults (Table 1) received tau 18F-flortaucipir (FTP) and Aβ 11C-PiB (PiB) PET scans (Fig. 1); 1 × 1 × 1 mm resolution T1-weighted MPRAGE images were acquired for every subject on a 1.5T MRI scanner at Lawrence Berkeley National Laboratories (Schöll et al., 2016). MPRAGE scans were processed with FreeSurfer version 5.3.0 (http://surfer.nmr.mgh.harvard.edu/) to derive ROIs in each subject's native space using the Desikan-Killiany atlas. FreeSurfer ROIs were used to calculate regional 18F-FTP and 11C-PiB PET measures in native space for each subject (see below). MR images were also segmented into tissue types using SPM12 (Statistical Parametric Mapping; Wellcome Department of Cognitive Neurology, London). SPM-derived segments for noncerebral tissues were subsequently used for partial volume correction.
Table 1.
Demographics, PET, and sleep summary information (mean ± SD)a
| Variable | EEG subjects (n = 31) | Questionnaire subjects (n = 95) |
|---|---|---|
| Age (yr) | 75.7 ± 4.6 | 77.8 ± 6.7 |
| N (%) female | 24 (77%) | 56 (59%) |
| Education (yr) | 16.6 ± 1.8 | 17.0 ± 1.8 |
| Mini-Mental State Examination | 29.0 ± 1.0 | 28.7 ± 1.4 |
| PiB status (%) | PiB− 12 (39%); PiB+ 19 (61%); | PiB− 55 (56%); PiB+ 39 (41%) |
| FTP Braak staging (%) | Braak 0: 6 (19%); Braak I/II: 18 (61%); Braak III/IV: 7 (23%) | Braak 0: 22 (23%); Braak I/II: 60 (63%); Braak III/IV: 13 (14%) |
| Total sleep time (min) | 343 ± 61 | — |
| NREM S2 time (min) | 176 ± 57 | — |
| NREM SWS time (min) | 71 ± 49 | — |
| REM time (min) | 68 ± 24 | — |
| WASO (min) | 111 ± 74 | — |
aPiB status was determined based on a DVR threshold of 1.065 (Villeneuve et al., 2015). FTP Braak staging, indexing the progression of tau pathology, was assigned to 1 of 4 stages based on FTP uptake in Braak-based composite regions, as described previously (Maass et al., 2017).
Figure 1.
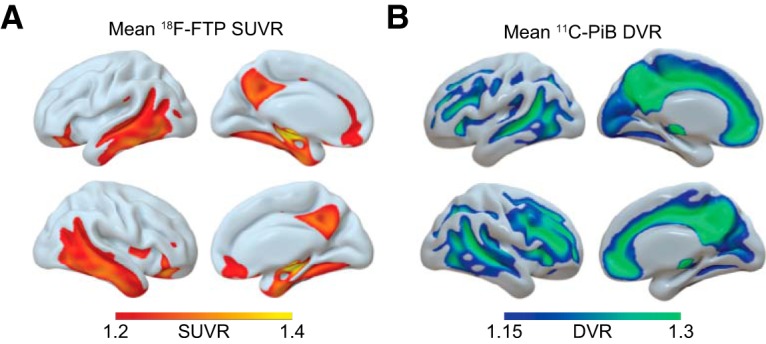
Mean PET binding demonstrates tau and Aβ aggregation. 18F-FTP tau and 11C-PiB Aβ PET show distinct binding patterns in healthy older adults. Using SPM12, PET images for all subjects were normalized to a common template; then a mean image was created for each radiotracer. A, Mean 18F-FTP SUVR for n = 101 PET scans, representing tau distribution in healthy older adults. B, Mean 11C-PiB DVR for n = 100 PET scans, representing Aβ distribution in healthy older adults.
A detailed description of 18F-FTP tau and 11C-PiB Aβ PET acquisition for Berkeley Aging Cohort Study has been published previously (Schöll et al., 2016). 18F-FTP standard uptake value ratio (SUVR) images were created based on mean uptake over 80–100 min after injection normalized by mean inferior cerebellar gray matter uptake. SUVR images were coregistered and resliced to structural MRI. To account for partial volume effects due to atrophy and spillover signal, the Geometric Transfer Matrix approach (Rousset et al., 1998) was used for partial volume correction based on FreeSurfer-derived ROIs, including corrections for extracerebral signal as previously described in detail (Baker et al., 2017).
Given our hypothesized associations between tau in the MTL and disruption of NREM sleep oscillations, sleep EEG and 18F-FTP tau PET analyses focused on an MTL ROI. Specifically, this ROI consisted of weighted mean 18F-FTP SUVR within bilateral MTL FreeSurfer regions (entorhinal cortex, hippocampus, amygdala, and parahippocampal gyrus), all of which were partial volume corrected (Baker et al., 2017; Maass et al., 2017). Given the experimental hypothesis that decreasing sleep duration would predict greater tau burden, analyses between questionnaires and PET focused on 18F-FTP SUVR within a composite ROI (“meta ROI”) considered to represent pathological severity of tau accumulation in the context of AD (Jack et al., 2017; Maass et al., 2017). This ROI was selected a priori because it was hypothesized that lifespan sleep change would impact overall tau burden, rather than tau aggregation within any specific brain region. Specifically, this ROI consisted of weighted mean 18F-FTP SUVR within bilateral entorhinal, amygdala, parahippocampal, fusiform, inferior temporal, and middle temporal FreeSurfer regions, all of which were partial volume corrected (Baker et al., 2017; Maass et al., 2017).
11C-PiB frames were coregistered and resliced to structural MRI. Distribution volume ratios (DVRs) for 11C-PiB images were generated with Logan graphical analysis on 11C-PiB frames corresponding to 35–90 min after injection using a cerebellar gray matter reference region (Logan et al., 1996; Price et al., 2005). One participant (with questionnaire data and not polysomnography (PSG) data) was excluded because full dynamic 11C-PiB data were not available. Cortical 11C-PiB DVR was calculated as a weighted mean across FreeSurfer-derived frontal, temporal, parietal, and posterior cingulate cortical regions. Participants were classified as Aβ-positive if their cortical 11C-PiB DVR was >1.065, a cutoff adapted from previous thresholds developed in our laboratory (Villeneuve et al., 2015).
Sleep EEG analysis.
A subset of n = 31 participants completed the sleep EEG assessment (Table 1). Data from 5 of these subjects were included in a previous publication (Mander et al., 2015). PSG data were acquired within 11.2 ± 6.8 months of the tau PET scan. All participants abstained from caffeine, alcohol, and daytime naps for the 48 h before and during the study. All participants completed the Berlin Questionnaire to assess sleep apnea risk (Netzer et al., 1999), which was used as a covariate in EEG biomarker analyses. Participants kept to their habitual sleep–wake rhythms and averaged 7–9 h of reported time in bed per night before study participation, verified by sleep logs. For 29 subjects, polysomnography was recorded using a Grass Technologies Comet XL system (Astro-Med), including 19-channel EEG placed using the 10–20 system. For 2 subjects, polysomnography was recorded using a Grass Technologies AURA PSG Ambulatory system (Astro-Med) using a 9-electrode EEG montage. All subjects had EOG recorded at the right and left outer canthi (right superior; left inferior) and EMG. Reference electrodes were recorded at both the left and right mastoid (A1, A2). The a priori electrode of interest for statistical tests (Fz, see below) was present in both montages. Data from the 2 subjects with a 9-channel montage were interpolated to match the 19-channel montage for the purpose of visualizing group-level results across the scalp.
Sleep was scored using standard criteria (Rechtschaffen and Kales, 1968) by a single trained scorer (B.A.M.). Sleep scoring was performed blinded to PET data. EEG data from the experimental night were imported into EEGLAB (http://sccn.ucsd.edu/eeglab/) and epoched into 5 s bins. Epochs containing artifacts were manually rejected, and the remaining epochs were filtered between 0.4 and 50 Hz. A fast Fourier transform was then applied to the filtered EEG signal at 5 s intervals with 50% overlap and using Hanning windowing. Slow-wave sleep was defined as NREM Stages 3–4, whereas NREM sleep encompassed Stages 2–4. Given that Stage 2 does not always exhibit pronounced SO activity (Helfrich et al., 2018), all correlational analyses focused on slow-wave sleep.
Event detection of SOs and spindles was performed for every channel separately based on previously established algorithms (Mölle et al., 2011; Staresina et al., 2015; Helfrich et al., 2018). (1) SOs: In brief, the continuous signal was first filtered between 0.16 and 1.25 Hz and detected zero crossings. Events were then selected based on time (0.8–2 s duration) and amplitude (75% percentile) criteria. Finally, artifact-free 5-s-long segments (±2.5 s around trough) were extracted from the raw signal. (2) Sleep spindles: the signal was filtered between 12 and 16 Hz, and the analytical amplitude was extracted after applying a Hilbert transform. The amplitude was smoothed with a 200 ms moving average and then thresholded at the 75% percentile (amplitude criterion). Only events that exceeded the threshold for 0.5–3 s (time criterion) were accepted. Artifact-free events were then defined as 5-s-long sleep-spindle epochs (±2.5 s), peak-locked. Events were normalized per subject by means of a z score before all subsequent analyses, alleviating power differences between subjects (Helfrich et al., 2018). The mean and SD were derived from the unfiltered event-locked average time course of either SO or spindle events in every participant.
Time-frequency representations for artifact-free normalized SO (Fig. 2B) were calculated after applying a 500 ms Hanning window. Spectral estimates (0.5–30 Hz; 0.5 Hz steps) were calculated between −2 and 2 s in steps of 50 ms and baseline-corrected by means of z score relative to a bootstrapped baseline distribution that was created from all trials (baseline epoch −2 to −1.5 s, 10,000 iterations) (Helfrich et al., 2018).
Figure 2.

Associations between SO-spindle coupling, tau, and Aβ. A, Peak-locked sleep spindle average across all detected events in NREM sleep (black). Low-pass filtered events (red) highlight that sleep spindles preferentially peaked before the SO “up-state.” Top right, Mean SO phase where sleep spindle power peaks. Red dots represent individual subjects. B, Baseline-corrected grand-average SO-locked time-frequency representation. Dashed white lines indicate the two largest SO peaks. Sleep spindle activity (12–16 Hz) is greatest before SO peak. C, Left, Negative association between SO-spindle coupling strength (resultant vector length) and MTL tau PET at Fz electrode. Right, Topography of correlation between SO-spindle coupling strength and MTL tau PET across all EEG electrodes. D, Left, No association between strength of SO-spindle coupling and cortical Aβ PET. Right, Topography of correlation across all EEG electrodes. E, Left, Bar plots represent mean vector length in high and low tau groups at electrode Fz. Error bars indicate SEM. Right, Topography of SO-spindle coupling strength in subjects with low versus high MTL tau burden.
For event-locked cross-frequency analyses (Dvorak and Fenton, 2014; Staresina et al., 2015; Helfrich et al., 2018), the normalized SO trough-locked data were first filtered into the SO component (0.1–1.25 Hz), and then the instantaneous phase angle was extracted after applying a Hilbert transform. Then the same trials were filtered between 12 and 16 Hz, and the instantaneous amplitude was extracted from the Hilbert transform. Only the time range from 2 to 2 s was considered, to avoid filter edge artifacts. For every subject, channel, and epoch, the maximal sleep spindle amplitude and corresponding SO phase angle were detected. The mean circular direction and resultant vector length across all NREM events were determined using the CircStat toolbox (Berens, 2009).
A summary proportional measure of SWA, a measure previously shown to be associated with Aβ accumulation in healthy older adults, was derived by dividing the spectral power between 0.6 and 1 Hz by the sum of spectral power between 0.6 and 4 μHz (Mander et al., 2015).
For all sleep EEG analyses, statistical tests were performed a priori at Fz (frontal midline) electrode derivation, based on previously demonstrated sensitivity to differences in both SO-spindle coupling and proportion 0.6–1 Hz SWA (Mander et al., 2015; Helfrich et al., 2018), our experimental hypothesis targets.
Sleep questionnaires analysis.
A subset of n = 95 subjects completed a retrospective questionnaire about lifespan sleep duration and sleep quality change. Questionnaire data were acquired within 8.6 ± 12.5 months of PET imaging. Subjects were asked to report their estimated retrospective average sleep duration in each decade across life. As in other retrospective life questionnaires in other fields aimed at improving veracity of recall (Friedenreich et al., 1998), subjects were encouraged to consider life circumstances during each decade that may have impacted their average sleep duration, such as education, births, moves, and occupation. For each decade, subjects chose one of six options: <5 h, 5–6, 6–7, 7–8, 8–9, and >9 h. These subject responses were then coded 4–9 for duration change analyses. Analyses were restricted to decade intervals from 40s to 70s. These data were used to generate two metrics: (1) percent sleep duration change by decade and (2) sleep duration slope. Percent sleep duration change by decade was calculated by dividing the reported average duration of sleep in one decade by the duration of the previous decade. For each decade bin, subjects with a negative percent sleep duration change were considered “sleep decrease” participants, and subjects with a positive percent sleep duration change were considered “sleep increase” participants. Sleep duration slope was calculated by fitting a linear regression to the decades by reported sleep duration for all reported decades from 40s to 70s, determined by the slope of the regression line.
Subjects also answered whether they felt their sleep had started to get worse as they got older. This yes or no response was used as a measure of subjective nonspecific sleep quality change.
Statistical analysis.
Associations between PET measures and sleep EEG measures were assessed using linear regression. The strength of correlations between sleep EEG measures and PET measures were compared using the Robust Correlation Toolbox (Pernet et al., 2012). Differences in sleep EEG measures between PET-defined groups were assessed using t tests.
Differences in PET between individuals on the basis of self-reported changes in sleep duration and quality were assessed using t tests, with false discovery rate (FDR) correction used to adjust for multiple decade comparisons. Associations between reported change in sleep duration and PET measures were examined using nonparametric Kendall's τ correlations. Nonparametric correlations were applied since they do not require the data to be normally distributed.
Results
Associations between NREM sleep oscillation coupling, tau, and Aβ
Focusing first on tau, we determined whether the disrupted coupling between SOs and sleep spindles predicted MTL tau burden. As previously reported in older adults, mean sleep spindle activity occurred during the rise of the SO, before the SO peak (Fig. 2A,B) (Helfrich et al., 2018). To quantitatively assess the coupling strength between spindles and SOs, we used a validated event-locked approach that extracted the mean resultant vector length across all SO-spindle events at every electrode for every participant (Helfrich et al., 2018), where greater vector length indicates greater coupling strength. Analyses focused a priori on the frontal midline electrode (Fz), since this location has been demonstrated to be sensitive to both SO-spindle coupling and SWA (Mander et al., 2015; Helfrich et al., 2018).
Supporting the experimental prediction, the measure of SO-spindle coupling strength at Fz negatively predicted the severity of MTL tau burden (Fig. 2C; r = −0.47, p = 0.01). Specifically, individuals with weaker SO-spindle coupling had greater accumulation of tau within MTL. This association remained significant in a linear regression adjusting for age, sex, sleep apnea risk, and time-interval differences across subjects between PSG and tau PET scan (r = −0.49, p = 0.02).
Next, we examined whether this SO-spindle association was unique to tau pathology, or instead, showed a similar relationship with Aβ accumulation. There was no significant relationship between SO-spindle coupling strength and the degree of cortical Aβ burden (Fig. 2D; r = −0.16, p = 0.39). Furthermore, these two associations (tau and Aβ) were significantly different from each other. Specifically, comparison of the two correlations (based on 1000 bootstrap samples of correlation coefficient pairs) revealed that the MTL tau association with coupling was significantly stronger than the cortical Aβ association (Δr = 0.31, p = 0.04). Therefore, SO-spindle coupling reflects an electrophysiological sleep measure that is associated with the degree of tau deposition within MTL, dissociable from a relationship with Aβ burden.
While these data provided an analytical approach based on continuous variables (i.e., correlational), the applicability for biomarker utility is frequently examined using a categorical approach (i.e., high/low, positive/negative). To address this question, we explored the relationship between tau burden and disrupted SO-spindle coupling, categorically separating participants into high and low tau groups, based on median FTP SUVR within MTL. Due to the limited number and cognitively healthy status of the subject sample, tau PET groups defined by in vivo Braak staging (Maass et al., 2017) are small and nonuniform (Table 1). A median split approach was therefore chosen since, unlike Aβ, there is no current consensus regarding an SUVR threshold determining “tau positive/negative” with a tau PET radiotracer (Jack et al., 2017). The categorical separation of individuals as Aβ-positive (high cortical Aβ) or Aβ-negative (low cortical Aβ) was defined by the PiB DVR threshold of 1.065.
SO-spindle coupling differentiated the two tau groups, such that individuals with categorically high MTL tau had significantly lower (i.e., weaker) SO-spindle coupling than individuals with categorically low MTL tau burden (Fig. 2E; at Fz, low MTL tau: 0.16 [0.01], high MTL tau: 0.10 [0.00], t = 2.30, p = 0.03). Conversely, SO-spindle coupling did not significantly differ between Aβ-positive and Aβ-negative groups (p = 0.53).
MTL tau and cortical Aβ were significantly correlated across participants (r = 0.45, p = 0.01). This relationship is in line with the amyloid cascade hypothesis of AD (Jack et al., 2013), which posits that tau accumulation typically only accelerates in the presence of cortical amyloid. Hence, these associations between tau and SO-sleep spindle coupling are likely AD-relevant, beyond normative aging-related changes.
Therefore, both categorical and correlational analysis approaches confirmed the experimental hypothesis that the disruption of NREM SO-spindle coupling, specifically the progressive loss of precise coupling strength, provides an electrophysiological sleep signature that is predictive of (relatively and categorically) individuals with high relative to low MTL tau burden.
Associations between NREM SWA, tau, and Aβ
Having established that the measure of SO-spindle coupling strength represents a sleep physiological feature that is associated with magnitude of MTL tau burden, we next examined whether the relationship between the proportion of SWA between 0.6–1 Hz and Aβ burden is stronger for Aβ, or instead, predicts both Aβ as well as tau.
Replicating previous findings (Mander et al., 2015), the proportion 0.6–1 Hz SWA predicted cortical Aβ, such that lower proportion 0.6–1 Hz SWA was related to higher cortical Aβ burden (Fig. 3A; r = −0.36, p = 0.04). This association remained significant in a linear regression adjusting for age, sex, sleep apnea risk, and interval between PSG and tau PET scan (r = −0.43, p = 0.04).
Figure 3.

Associations between NREM SWA, tau, and Aβ. A, Left, Negative association between proportion of 0.6–1 Hz SWA and cortical Aβ PET at electrode Fz. Right, Topography of correlation across all EEG electrodes. B, Left, No association between proportion of 0.6–1 Hz SWA and MTL tau PET. Right, Topography of correlation between proportion of 0.6–1 Hz SWA and MTL tau PET across all EEG electrodes. C, Top, Topography of 0.6–1 Hz SWA in Aβ-negative and Aβ-positive subjects. Bottom, Bar plots represent proportion of 0.6–1 Hz SWA in Aβ-negative and Aβ-positive subjects at electrode Fz. Error bars indicate SEM.
In contrast, this same measure of proportion 0.6–1 Hz SWA demonstrated no predictive relationship with the degree of MTL tau burden (Fig. 3B; r = 0.05, p = 0.80). Additionally, comparison of the two correlations (based on 1000 bootstrap samples of correlation coefficient pairs) revealed that this SWA disruption measure and Aβ correlation was stronger than the correlation between SWA and MTL tau (Δr = 0.41, p = 0.04). These findings support the hypothesis that the sleep marker of 0.6–1 Hz SWA is significantly sensitive to Aβ burden, and not tau burden, which added to the results described earlier, establishing a double dissociation (SWA/SO-spindle coupling, Aβ/tau).
We additionally explored whether specific slow-wave frequency ranges (<4 Hz) were associated with MTL tau burden. There was no association between MTL tau and SWA frequencies (0.6–1 Hz, r = −0.04, p = 0.84; 1–2 Hz, r = −0.14, p = 0.44; 2–3 Hz, r = 0.27, p = 0.15; 3–4 Hz, r = −.08, p = 0.67).
Finally, we examined whether the measure of 0.6–1 Hz SWA was able to differentiate Aβ-positive and Aβ-negative categorical groups significantly, and we furthermore examined whether this was also evident for low and high MTL tau groups.
The proportion 0.6–1 Hz SWA differentiated Aβ-positive and Aβ-negative groups, with significantly lower proportion 0.6–1 Hz SWA in Aβ-positive individuals, relative to Aβ-negative individuals (Fig. 3C; PiB−, n = 12, 0.42 [0.09]; PiB+, n = 19, 0.35 [0.08]; t = 2.24, p = 0.03). Conversely, the proportion 0.6–1 Hz SWA did not differ between the low tau and high tau groups (p = 0.43), further highlighting dissociable sensitivity to these two pathological entities.
Together, the results suggest a double dissociation in the predictive associations between two unique features of sleep (SO-spindle coupling, NREM SWA) and the two prototypical neuropathological features of AD: tau and Aβ. Significant continuous and categorical associations demonstrated that SO-spindle coupling disruption was unique to MTL tau burden. Serving as a complementary pathological biomarker, proportion 0.6–1 Hz SWA was uniquely associated with cortical Aβ. Associations between sleep metrics and tau (FTP SUVR) and Aβ (PiB DVR) within brain regions beyond our a priori targets of interest are reported for in Table 2 for completeness, and without further discussion.
Table 2.
Correlations between regional PET uptake and sleep EEG variables of interesta
| Correlation with coupling strength (r) | Correlation with proportion 0.6–1 Hz SWA (r) | |
|---|---|---|
| PiB DVR ROI | ||
| mPFC | −0.161 | −0.372* |
| dlPFC | −0.144 | −0.431* |
| Parietal | −0.131 | −0.360* |
| Temporal | −0.157 | −0.377* |
| FTP SUVR ROI | ||
| Entorhinal cortex | −0.338 | 0.081 |
| Braak I/II regions | −0.454* | 0.056 |
| Braak III/IV regions | −0.362* | −0.066 |
| Braak V/VI regions | −0.401* | −0.136 |
| Meta ROI | −0.327 | −0.048 |
| Global | −0.404* | −0.110 |
| Parahippocampal cortex | −0.492* | −0.040 |
| Orbitofrontal cortex | −0.532* | −0.315 |
| Precuneus | −0.443* | −0.026 |
| Inferior parietal cortex | −0.290 | −0.110 |
| Inferior temporal cortex | −0.298 | −0.080 |
aRows represent each ROI. Columns represent Pearson's correlations between EEG metric and PET uptake (see Materials and Methods). PiB DVR ROIs are comprised of weighted PiB DVR means within FreeSurfer regions, as described by Mander et al. (2015). FTP SUVR ROIs are comprised of weighted FTP SUVR means within FreeSurfer regions after PVC, as described by Maass et al. (2017).
*Significant r value (p < 0.05).
Associations between retrospective change in sleep and Aβ
Having demonstrated that changes in “micro-level” sleep oscillation features, SO-spindle phase-amplitude coupling and <1 Hz SWA, dissociably predicted MTL tau and cortical Aβ, respectively, we next sought to determine whether macro-level changes in sleep across the lifespan predicted later-life pathological burden of both Aβ and tau. Specifically, we tested the hypothesis that self-reported sleep reduction in mid- and later-life decade windows, those that overlap with current Alzheimer's pathological models of Aβ and tau escalation, predicts later-life Aβ and tau burden.
First, we tested the prediction that decade-by-decade changes in sleep duration predict later-life pathological burden, starting with Aβ. Consistent with this hypothesis, subjects who reported a decrease in sleep duration in their 50s (relative to the previous decade) had significantly higher Aβ in late-stage life compared with subjects whose sleep duration increased (Fig. 4A; sleep decrease: n = 6, PiB DVR 1.25 [0.34]; sleep increase: n = 12, PiB DVR 1.04 [0.05]; t = 2.19, p = 0.04). That is, a relative reduction in sleep amount in one's 50s was associated with having greater global Aβ burden in late life.
Figure 4.

Associations between retrospective change in sleep duration, tau, and Aβ. A, Bar plots represent mean late-life Aβ burden (cortical PiB DVR) for subjects with self-reported decreasing sleep duration versus subjects with increasing sleep duration in each decade from age 40 to 70. Aβ burden was significantly higher in subjects whose sleep duration decreased both for the 50s and 70s decades (p < 0.05), with a trend in the same direction for the 60s (p < 0.1). B, Left, Bar plot represents that subjects with a negative sleep duration slope, indicating their sleep duration decreased over their lifespan, had significantly greater Aβ burden in late life compared with those with positive slope (p < 0.05). Right, Negative association between slope of sleep duration change and late-life Aβ burden, demonstrating that greater loss of sleep duration was predictive of greater Aβ. C, Mean late-life tau burden (meta ROI FTP SUVR) for subjects with self-reported decreasing sleep duration versus subjects with increasing sleep duration for each decade. Late-life tau burden was significantly higher for individuals with decreasing sleep duration in their 60s (p < 0.05). D, Left, Tau burden is not significantly different in subjects with negative sleep duration slope relative to positive slope subjects. Right, No association between slope of sleep duration change and late-life tau burden. *Significant t value (p < 0.05). +Trending t value (p < 0.10). Error bars indicate SEM.
A similar relationship was also observed for decreasing sleep times in an individual's 70s, wherein a decline in sleep was again associated with greater Aβ burden later in life, relative to those whose sleep duration increased in their 70s (Fig. 4A; sleep decrease: n = 12, PiB DVR 1.28 [0.31]; sleep increase: n = 19, PiB DVR 1.06 [0.10]; t = 2.82, p = 0.01). Nonparametric correlations, which included subjects whose sleep duration did not change, showed a negative association between percent change in reported sleep duration and Aβ within only the 70s decade (n = 84, τ = −0.17, p = 0.04). This association indicates that the magnitude of sleep change in the 70s decade, beyond the categorical decrease or increase of sleep duration, was predictive of Aβ burden.
Changes in duration of sleep in 60s decade were not significantly predictive of later-life Aβ burden (sleep decrease: n = 11, PiB DVR 1.20 [0.26]; sleep increase: n = 19, PiB DVR 1.07 [0.12]; t = 2.05, p = 0.07), unlike the 50s and 70s age windows. However, it should be noted that the association in the 60s trended in a similar direction. After FDR correction for multiple comparisons, the 70s window association remained significant. Together, these findings suggest that decreases in sleep duration starting with the 50s, a prototypical time period of escalating Aβ accumulation (Jack et al., 2013), predicts greater Aβ burden late in life.
As an alternative analytical test of the hypothesis, we examined the rate of decline in sleep duration across the surveyed lifespan, indexed by the slope of change in sleep duration across all ages assessed (40s-70s). Subjects with a negative slope of sleep duration across this time period (i.e., a decline in sleep duration across the assessed lifespan window as a whole) exhibited significantly greater Aβ burden in later life, relative to those with an increase in sleep duration across the lifespan (Fig. 4B; negative slope: n = 22, PiB DVR 1.24 [0.29]; positive slope: n = 41, PiB DVR 1.07 [0.11]; t = 3.41, p = 0.001). This association was also revealed when using nonparametric correlation between sleep duration slope value and Aβ across all subjects, including those with no change in sleep duration (n = 94, τ = −0.15, p = 0.04).
Thus, the association between lifespan changes in sleep and late-life Aβ burden was not only observable on a decade-by-decade basis, but also using the overall change in individual's adult-span sleep duration. This finding may suggest that decreasing sleep duration in mid to late life is significantly associated with an increased risk of late-life Aβ burden, and that a profile of maintained (or even subtle increase) in sleep duration throughout this time period is statistically associated with a reduced predicted risk of Aβ accumulation in late life.
Associations between retrospective change in sleep and tau
We next examined whether the same predictive associations were observable for tau burden. Suggesting a time sensitivity distinct from that identified for Aβ, subjects who reported a decrease in sleep duration in their 60s had significantly greater tau in later life, relative to subjects whose sleep duration did not decline (Fig. 4C; sleep decrease: n = 11, 1.29 [0.09]; sleep increase n = 20, 1.19 [0.10]; t = 2.70, p = 0.01). This association remained significant after FDR correction for multiple comparisons. To test this association in all subjects, we used a nonparametric correlation that included subjects whose sleep duration did not change during this interval. This correlation was significant (n = 94, τ = −0.17, p = 0.03), further demonstrating that declining sleep duration in the 60s decade was predictive of greater late-life tau burden. No other decade showed such sleep-duration sensitivity to tau (all p values >0.05).
In addition to examining whether decade-by-decade changes in sleep duration predicted tau burden, we also examined the association between tau and the change in sleep duration across the entire time interval, as indexed by sleep duration slope; a similar analysis to that above for Aβ. Unlike the lifespan-predictive trend identified for Aβ, there was no predictive lifespan association between the decline in adult lifespan sleep duration (slope) and late-life tau burden (Fig. 4D; negative slope: n = 22, FTP SUVR 1.27 [0.14]; positive slope: n = 42, 1.24 [0.13]; t = 0.80, p = 0.58). This was also true when using a nonparametric correlation approach (n = 95, τ = −0.08, p = 0.27).
These associations between sleep duration change and pathology suggest that, while reductions in sleep duration from 40s onward appear to be associated with greater Aβ burden, sensitivity for tau burden is not generalized to sleep change across the adult lifespan. Rather, a predictive association for tau burden was specific to changes in 60s decade interval, suggesting a possible model in which only a select period of the lifespan may hold sensitivity in forecasting the extent of later-life tau pathology. Retrospective sleep change versus tau (FTP SUVR) and Aβ (PiB DVR) statistics for other brain areas outside of our a priori ROIs are reported in Table 3 for completeness only.
Table 3.
Regional PET uptake related to retrospective change in sleep durationa
|
t value |
Kendall's τ slope (corr) | ||||
|---|---|---|---|---|---|
| 40s–50s | 50s–60s | 60s–70s | Slope (t test) | ||
| PiB DVR ROI | |||||
| mPFC | 2.17* | 1.76 | 3.00* | 3.56* | −0.17* |
| dlPFC | 2.27* | 1.68 | 2.81* | 3.41* | −0.16* |
| Parietal | 2.42* | 1.85 | 2.75* | 3.29* | −0.15* |
| Temporal | 2.03 | 2.18* | 2.59* | 3.41* | −0.15 |
| FTP SUVR ROI | |||||
| Entorhinal cortex | −0.21 | 1.27 | 1.69 | 0.83 | −0.06 |
| Braak I/II regions | −0.80 | 1.14 | 1.03 | 0.30 | −0.07 |
| Braak III/IV regions | −1.29 | 2.56* | 0.48 | 0.70 | −0.08 |
| Braak V/VI regions | −1.11 | 1.73 | 1.73 | 1.23 | −0.10 |
| MTL | −0.80 | 1.49 | 0.86 | 0.40 | −0.05 |
| Global | −1.19 | 1.97 | 1.32 | 1.09 | −0.09 |
aRows represent each ROI. Columns represent statistical tests (see Materials and Methods). PiB DVR ROIs are comprised of weighted PiB DVR means within FreeSurfer regions, as described by Mander et al. (2015). FTP SUVR ROIs are comprised of weighted FTP SUVR means within FreeSurfer regions after PVC, as described by Maass et al. (2017).
*p < 0.05.
Separate from sleep duration, we finally examined whether subjective changes in sleep quality across the lifespan, defined by subjects on the questionnaire as worsening sleep quality, were associated with late-life Aβ and tau burden. Unlike the sensitivity observed for sleep duration, individuals who reported that their subjective sense of sleep quality had worsened with age (n = 57; 60%) did not show significantly greater (or lesser) Aβ or tau burden, compared with those who reported maintained (or improved) subjective sleep quality (n = 38; 40%, all p values > 0.05).
Discussion
Together, these findings help to establish that changes in sleep quantity and electrophysiological quality are associated with in vivo human tau pathology, relative to Aβ pathology. Moreover, these predictive relationships were observed in two temporally unique ways: (1) current moment, where sleep EEG features were uniquely predictive of tau and Aβ in older adults; and (2) as a future forecast, such that sleep changes in earlier mid- to later-life phases were associated with measures of tau and Aβ burden in late life.
Impaired NREM SO-spindle coupling predicted greater tau accumulation in MTL. Importantly, this association with impaired SO-spindle coupling was unique and specific to tau accumulation, with no association for Aβ burden. Instead, a dissociable NREM-EEG signature, specifically impairments in 0.6–1 Hz SWA (Mander et al., 2015), predicted cortical Aβ. Conversely, 0.6–1 Hz SWA was itself not associated with tau burden in the MTL, nor any other cortical region (Table 2). These findings imply a double dissociation wherein sleep-EEG measures of SO-spindle coupling and SWA are separately and uniquely associated with tau and Aβ burden.
The observed impairments in sleep EEG features offer mechanistic insights within the context of AD. In early AD, both tau and Aβ have been shown to be related to the disruption of NREM SWA (Lucey et al., 2019). Since tau and Aβ pathology are both thought to begin accumulating before the onset of AD-related cognitive impairment (Braak and Braak, 1997; Jack et al., 2013), biomarkers should be sensitive to early pathology in healthy adults who may be years away from signs of cognitive impairment. While the current research definition of AD requires the presence of both pathologies (Jack et al., 2018), tau and Aβ can occur independently of one another (Arriagada et al., 1992; Giannakopoulos et al., 2003), demanding the use of dissociable biomarkers that are specific to each pathological protein.
Intracranial EEG recordings in the human MTL have demonstrated that hippocampal sharp-wave ripples, spindles, and SOs interact in a phase-locked coordinated manner. Specifically, hippocampal SOs orchestrate spindle bursts, with peak ripple activity occurring at the spindle trough (Clemens et al., 2007; Staresina et al., 2015). This fine-tuned interaction of the three NREM oscillations indicates that the hippocampus can play a functional role in facilitating SO-spindle coupling, which can be measured on the scalp. Building on these data, our findings suggest that early tauopathy in the human MTL, potentially through tau-related disruption of either hippocampal-network integrity or individual neuronal transmission (Palop and Mucke, 2016), significantly impairs the coordinated interplay between sharp-wave ripples, spindles, and SOs, impairing the precision of phase-locked oscillation timing. Supporting this proposal, mouse models of tauopathy have demonstrated that MTL tau disposition impairs the generation of hippocampal sharp-wave ripple events (Witton et al., 2016). Moreover, dysfunctional oscillatory coupling between cortical networks has been observed in mouse models of tau overexpression (Ahnaou et al., 2017), although SO-spindle coupling has yet to been studied in rodent models of AD.
Independent of sleep, studies using tau PET have recently shown that early tau pathology is associated with impaired hippocampal-related memory performance (Schöll et al., 2016; Maass et al., 2018). This is germane considering that coupling of SO-spindles is proposed to serve a functional role in sleep-dependent hippocampal memory processing (Diekelmann and Born, 2010) yet is significantly impaired in older adults before mild cognitive impairment (Helfrich et al., 2018). Combined with the findings reported here, a testable hypothesis emerges for future examination: disrupted SO-spindle coupling represents a novel mediating pathway through which MTL tau pathology disrupts long-term hippocampal memory, thereby contributing to AD-related cognitive decline. Falsification/affirmation of this proposal will require longitudinal studies combining tau and Aβ PET imaging, sleep EEG assessment, and hippocampal-memory measures.
In the current study of cognitively healthy older adults, there was no association between tau and deficits in SWA reported in mouse models of tauopathy (Menkes-Caspi et al., 2015), and a human study that included cognitively impaired individuals (Lucey et al., 2019). However, supporting the tau sensitivity of these regions reported by Lucey et al. (2019), exploratory analyses of these ROIs in our dataset revealed similar associations with impaired SO-spindle coupling our older adult sample (Table 2), showing congruency between the studies. One explanation of these differences is that the association between tau and disruption of SWA only emerges in later stages of tau progression, when individuals transition into cognitive decline. This hypothesis is tenable given that tau has differential effects on human cortical network activity at different stages of its spread throughout cortex (Adams et al., 2018). Since SOs have principally been considered a cortical phenomenon (Murphy et al., 2009), the above reasoning would make temporal sense as tau aggregation moves out of MTL into limbic cortical regions wherein slow-wave source generation centers have been identified (Braak and Braak, 1997; Murphy et al., 2009).
Due to small group sizes, EEG biomarker analyses were not separated based on Aβ-positivity status. Importantly, Aβ and tau were significantly correlated in this group of subjects. This association is to be expected in accordance with the amyloid cascade hypothesis of AD (Jack et al., 2013), as well as considerable data (Jagust, 2018), which posits that tau accumulation typically accelerates in the presence of cortical amyloid. Supporting this widely accepted hypothesis, recent papers have shown that that high levels of in vivo tau are rare in amyloid-negative individuals (Johnson et al., 2016; Jack et al., 2018; Lucey et al., 2019; Soldan et al., 2019). Therefore, deficits in SO-sleep spindle coupling appear to be sensitive to, and linked with, biological features of AD pathogenesis that extend beyond normal brain aging.
Beyond testing present-moment associations between sleep EEG, tau, and Aβ discussed above, our second complementary hypothesis sought to determine whether changes in earlier mid- and later-life sleep prospectively predicted late-life tau and Aβ. Retrospective questionnaire tools have been used to measure other age-related changes outside of sleep, such as physical and cognitive activities (Friedenreich et al., 1998; Landau et al., 2012; Nucci et al., 2012), and memory function (Perrotin et al., 2012), with similar measures being validated by longitudinal prospective studies (Sobell et al., 1989; Schmidt et al., 2006). Here, we extended this line of inquiry to changes in sleep across the lifespan. It is, however, important to consider the limitations of these data, since retrospective self-reports are fallible to subjective biases across individuals. Specifically, this questionnaire will need to be validated for test-retest reliability and compared with longitudinal studies of sleep over the lifespan.
Another caveat in interpreting these results is that the two sleep-duration subgroups (increasers and decreasers) within each decade interval were comprised of a relatively small number of individuals. This necessarily limits the generalizability of the findings, which must now be replicated in large longitudinal cohorts. Nevertheless, these exploratory data are unique in comparing AD pathology with retrospective changes in sleep quality. Moreover, such preliminary data can further serve to inform larger studies to examine these associations with guided a priori hypothesis and targets.
Individuals reporting a decrease in sleep duration during their 50s and 70s exhibited greater late life Aβ burden. Additionally, declining sleep over the adult lifespan, as measured by the slope of duration in each decade, further predicted greater Aβ burden in late life. In contrast, late-life tau burden was associated with a reported decrease in sleep only during an individual's 60s. These results illustrate that the sleep history of an individual carries statistically significant future forecast of late-life Aβ and tau accumulation.
Seminal cross-sectional studies have shown that reported short sleep duration is linked to greater Aβ measured by PET (Spira et al., 2013; Sprecher et al., 2015). We extend these findings, demonstrating that Aβ burden was associated with declining sleep duration across the surveyed interval beginning in individuals' 40s. That is, decreasing sleep time across the adult lifespan predicts later-life Aβ burden. However, since pathology may begin to accumulate within the surveyed interval (Braak and Braak, 1997), these data cannot determine the directionality of this association. Indeed, the presence of Aβ is associated with disrupted sleep physiology both in mouse models (Roh et al., 2012) and humans (Ju et al., 2013; Mander et al., 2015; Lucey et al., 2019), suggesting a bidirectional relationship (Mander et al., 2016). Experimental disruption of sleep has been linked to accelerated accumulation of Aβ in mouse models of AD (Kang et al., 2009). Moreover, recent longitudinal studies in humans have demonstrated that self-reported sleepiness predicts Aβ accumulation over time (Carvalho et al., 2018; Spira et al., 2018). Thus, while the presence of early Aβ during the surveyed interval may be one factor influencing these findings, evidence also supports a parsimonious (and nonmutually exclusive) hypothesis that decreasing sleep duration accelerates the aggregation of Aβ, resulting in greater late-life Aβ burden observed in the present study.
In contrast to Aβ, late-life tau burden was not associated with declining sleep duration across the surveyed interval. Chronic sleep restriction has been shown to accelerate tau accumulation and spread in mouse models of tauopathy (Zhu et al., 2018; Holth et al., 2019), and a night of sleep deprivation in healthy middle-aged human adults leads to increases in CSF Aβ and tau (Holth et al., 2019). Should changes in sleep duration affect tau in addition to Aβ, this link may only be present in individuals at later stages of AD progression that were not included in the present study. Importantly, current models of AD pathological progression propose that tau deposition is consequent to Aβ (Jack et al., 2013). The retrospective questionnaire revealed that 50s sleep duration was linked to late-life Aβ, and 60s to late-life tau. This timeline, whereby changes in sleep predict subsequent pathology, supports the hypothesized order of pathological events.
In conclusion, the current data support the position that quantitative and qualitative markers of human sleep are sensitive to early tau and Aβ pathology. Regarding the identified physiological sleep oscillation changes, EEG, especially a single channel of high sensitivity, may represent an affordable, safe, noninvasive, and scalable tool for measuring AD pathophysiology. Sleep assessment may therefore allow for quantification of AD disease status, disease progression, as well as measurement of therapeutic treatment efficacy. While remaining preliminary, and thus speculative, our macro-sleep questionnaire findings may signify lifespan windows of sensitivity where targeted sleep treatment could be most effective. Reporting race was optional in the study, preventing sufficient subject responses to covary for race in analyses, representing a limitation of the current report. If validated in larger longitudinal studies, these sleep-sensitive windows would have the potential to be included in public health recommendations with the goal shifting from a model of late-stage AD treatment to earlier-life AD prevention.
Footnotes
This work was supported by the National Institutes of Health (R01AG031164, RF1AG054019, and RF1AG054106 to M.P.W.; R01AG034570 to W.J.J.; R37NS21135 and 1U19NS107609–01 to R.T.K.; F32AG057107 to T.M.H.). Avid Radiopharmaceuticals enabled the use of the 18F-flortaucipir radiotracer but did not provide direct funding and were not involved in data analysis or interpretation. We thank Rachel Bell, Laura Fenton, Samika Kumar, Benjamin Miller Mills, and Shai Porat for assistance.
The authors declare no competing financial interests.
References
- Adams JN, Lockhart SN, Li L, Jagust WJ (2018) Relationships between tau and glucose metabolism reflect Alzheimer's disease pathology in cognitively normal older adults. Cereb Cortex 29:1997–2009. 10.1093/cercor/bhy078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahnaou A, Moechars D, Raeymaekers L, Biermans R, Manyakov NV, Bottelbergs A, Wintmolders C, Van Kolen K, Van De Casteele T, Kemp JA, Drinkenburg WH (2017) Emergence of early alterations in network oscillations and functional connectivity in a tau seeding mouse model of Alzheimer's disease pathology. Sci Rep 7:14189. 10.1038/s41598-017-13839-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 42:631–639. 10.1212/WNL.42.3.631 [DOI] [PubMed] [Google Scholar]
- Baker SL, Maass A, Jagust WJ (2017) Considerations and code for partial volume correcting [18F]-AV-1451 tau PET data. Data Brief 15:648–657. 10.1016/j.dib.2017.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berens P. (2009) CircStat: a MATLAB toolbox for circular statistics. J Stat Softw 31:1–21. [Google Scholar]
- Braak H, Braak E (1997) Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 18:351–357. 10.1016/S0197-4580(97)00056-0 [DOI] [PubMed] [Google Scholar]
- Carvalho DZ, St Louis EK, Knopman DS, Boeve BF, Lowe VJ, Roberts RO, Mielke MM, Przybelski SA, Machulda MM, Petersen RC, Jack CR Jr, Vemuri P (2018) Association of excessive daytime sleepiness with longitudinal β-amyloid accumulation in elderly persons without dementia. JAMA Neurol 75:672–680. 10.1001/jamaneurol.2018.0049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens Z, Mölle M, Eross L, Barsi P, Halász P, Born J (2007) Temporal coupling of parahippocampal ripples, sleep spindles and slow oscillations in humans. Brain 130:2868–2878. 10.1093/brain/awm146 [DOI] [PubMed] [Google Scholar]
- Diekelmann S, Born J (2010) The memory function of sleep. Nat Rev Neurosci 11:114–126. 10.1038/nrn2762 [DOI] [PubMed] [Google Scholar]
- Dvorak D, Fenton AA (2014) Toward a proper estimation of phase-amplitude coupling in neural oscillations. J Neurosci Methods 225:42–56. 10.1016/j.jneumeth.2014.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR (1975) “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12:189–198. 10.1016/0022-3956(75)90026-6 [DOI] [PubMed] [Google Scholar]
- Friedenreich CM, Courneya KS, Bryant HE (1998) The Lifetime Total Physical Activity Questionnaire: development and reliability. Med Sci Sports Exerc 30:266–274. 10.1097/00005768-199805001-01514 [DOI] [PubMed] [Google Scholar]
- Giannakopoulos P, Herrmann FR, Bussière T, Bouras C, Kövari E, Perl DP, Morrison JH, Gold G, Hof PR (2003) Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology 60:1495–1500. 10.1212/01.WNL.0000063311.58879.01 [DOI] [PubMed] [Google Scholar]
- Helfrich RF, Mander BA, Jagust WJ, Knight RT, Walker MP (2018) Old brains come uncoupled in sleep: slow wave-spindle synchrony, brain atrophy, and forgetting. Neuron 97:221–230.e4. 10.1016/j.neuron.2017.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hita-Yañez E, Atienza M, Cantero JL (2013) Polysomnographic and subjective sleep markers of mild cognitive impairment. Sleep 36:1327–1334. 10.5665/sleep.2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holth JK, Mahan TE, Robinson GO, Rocha A, Holtzman DM (2017) Altered sleep and EEG power in the P301S Tau transgenic mouse model. Ann Clin Transl Neurol 4:180–190. 10.1002/acn3.390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holth JK, Fritschi SK, Wang C, Pedersen NP, Cirrito JR, Mahan TE, Finn MB, Manis M, Geerling JC, Fuller PM, Lucey BP, Holtzman DM (2019) The sleep–wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 363:880–884. 10.1126/science.aav2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Pankratz VS, Donohue MC, Trojanowski JQ (2013) Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 12:207–216. 10.1016/S1474-4422(12)70291-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Wiste HJ, Weigand SD, Therneau TM, Lowe VJ, Knopman DS, Gunter JL, Senjem ML, Jones DT, Kantarci K, Machulda MM, Mielke MM, Roberts RO, Vemuri P, Reyes DA, Petersen RC (2017) Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimers Dement 13:205–216. 10.1016/j.jalz.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R (2018) NIA-AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement 14:535–562. 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust W. (2018) Imaging the evolution and pathophysiology of Alzheimer disease. Nat Rev Neurosci 19:687–700. 10.1038/s41583-018-0067-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Schultz A, Betensky RA, Becker JA, Sepulcre J, Rentz D, Mormino E, Chhatwal J, Amariglio R, Papp K, Marshall G, Albers M, Mauro S, Pepin L, Alverio J, Judge K, Philiossaint M, Shoup T, Yokell D, Dickerson B, et al. (2016) Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol 79:110–119. 10.1002/ana.24546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YE, McLeland JS, Toedebusch CD, Xiong C, Fagan AM, Duntley SP, Morris JC, Holtzman DM (2013) Sleep quality and preclinical Alzheimer disease. JAMA Neurol 70:587–593. 10.1001/jamaneurol.2013.2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM (2009) Amyloid-β dynamics are regulated by orexin and the sleep–wake cycle. Science 326:1005–1007. 10.1126/science.1180962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau SM, Marks SM, Mormino EC, Rabinovici GD, Oh H, O'Neil JP, Wilson RS, Jagust WJ (2012) Association of lifetime cognitive engagement and low β-amyloid deposition. Arch Neurol 69:623–629. 10.1001/archneurol.2011.2748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL (1996) Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab 16:834–840. 10.1097/00004647-199609000-00008 [DOI] [PubMed] [Google Scholar]
- Lucey BP, McCullough A, Landsness EC, Toedebusch CD, McLeland JS, Zaza AM, Fagan AM, McCue L, Xiong C, Morris JC, Benzinger TL, Holtzman DM (2019) Reduced non-rapid eye movement sleep is associated with tau pathology in early Alzheimer's disease. Sci Transl Med 11:eaau6550. 10.1126/scitranslmed.aau6550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass A, Landau S, Baker SL, Horng A, Lockhart SN, La Joie R, Rabinovici GD, Jagust WJ (2017) Comparison of multiple tau-PET measures as biomarkers in aging and Alzheimer's disease. Neuroimage 157:448–463. 10.1016/j.neuroimage.2017.05.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass A, Lockhart SN, Harrison TM, Bell RK, Mellinger T, Swinnerton K, Baker SL, Rabinovici GD, Jagust WJ (2018) Entorhinal tau pathology, episodic memory decline and neurodegeneration in aging. J Neurosci 38:530–543. 10.1523/JNEUROSCI.2028-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mander BA, Marks SM, Vogel JW, Rao V, Lu B, Saletin JM, Ancoli-Israel S, Jagust WJ, Walker MP (2015) β-Amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat Neurosci 18:1051–1057. 10.1038/nn.4035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mander BA, Winer JR, Jagust WJ, Walker MP (2016) Sleep: a novel mechanistic pathway, biomarker, and treatment target in the pathology of Alzheimer's disease? Trends Neurosci 39:552–566. 10.1016/j.tins.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menkes-Caspi N, Yamin HG, Kellner V, Spires-Jones TL, Cohen D, Stern EA (2015) Pathological tau disrupts ongoing network activity. Neuron 85:959–966. 10.1016/j.neuron.2015.01.025 [DOI] [PubMed] [Google Scholar]
- Mölle M, Bergmann TO, Marshall L, Born J (2011) Fast and slow spindles during the sleep slow oscillation: disparate coalescence and engagement in memory processing. Sleep 34:1411–1421. 10.5665/SLEEP.1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M, Riedner BA, Huber R, Massimini M, Ferrarelli F, Tononi G (2009) Source modeling sleep slow waves. Proc Natl Acad Sci U S A 106:1608–1613. 10.1073/pnas.0807933106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netzer NC, Stoohs RA, Netzer CM, Clark K, Strohl KP (1999) Using the Berlin Questionnaire to identify patients at risk for the sleep apnea syndrome. Ann Intern Med 131:485–491. 10.7326/0003-4819-131-7-199910050-00002 [DOI] [PubMed] [Google Scholar]
- Nucci M, Mapelli D, Mondini S (2012) Cognitive Reserve Index questionnaire (CRIq): a new instrument for measuring cognitive reserve. Aging Clin Exp Res 24:218–226. 10.3275/7800 [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L (2016) Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat Rev Neurosci 17:777–792. 10.1038/nrn.2016.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernet CR, Wilcox R, Rousselet GA (2012) Robust correlation analyses: false positive and power validation using a new open source Matlab toolbox. Front Psychol 3:606. 10.3389/fpsyg.2012.00606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrotin A, Mormino EC, Madison CM, Hayenga AO, Jagust WJ (2012) Subjective cognition and amyloid deposition imaging: a Pittsburgh compound B positron emission tomography study in normal elderly individuals. Arch Neurol 69:223–229. 10.1001/archneurol.2011.666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JC, Klunk WE, Lopresti BJ, Lu X, Hoge JA, Ziolko SK, Holt DP, Meltzer CC, DeKosky ST, Mathis CA (2005) Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh compound-B. J Cereb Blood Flow Metab 25:1528–1547. 10.1038/sj.jcbfm.9600146 [DOI] [PubMed] [Google Scholar]
- Prinz PN, Vitaliano PP, Vitiello MV, Bokan J, Raskind M, Peskind E, Gerber C (1982) Sleep, EEG and mental function changes in senile dementia of the Alzheimer's type. Neurobiol Aging 3:361–370. 10.1016/0197-4580(82)90024-0 [DOI] [PubMed] [Google Scholar]
- Rechtschaffen A, Kales A (1968) A manual standardized terminology, techniques and scoring system for sleep stages of human subjects. Bethesda, MD: Department of Health. [DOI] [PubMed] [Google Scholar]
- Roh JH, Huang Y, Bero AW, Kasten T, Stewart FR, Bateman RJ, Holtzman DM (2012) Disruption of the sleep–wake cycle and diurnal fluctuation of beta-amyloid in mice with Alzheimer's disease pathology. Sci Transl Med 4:150ra122. 10.1126/scitranslmed.3004291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset OG, Ma Y, Evans AC (1998) Correction for partial volume effects in PET: principle and validation. J Nucl Med 39:904–911. [PubMed] [Google Scholar]
- Schmidt ME, Slanger T, Chang-Claude J, Wahrendorf J, Steindorf K (2006) Evaluation of a short retrospective questionnaire for physical activity in women. Eur J Epidemiol 21:575–585. 10.1007/s10654-006-9042-9 [DOI] [PubMed] [Google Scholar]
- Schöll M, Lockhart SN, Schonhaut DR, O'Neil JP, Janabi M, Ossenkoppele R, Baker SL, Vogel JW, Faria J, Schwimmer HD, Rabinovici GD, Jagust WJ (2016) PET imaging of tau deposition in the aging human brain. Neuron 89:971–982. 10.1016/j.neuron.2016.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobell J, Block G, Koslowe P, Tobin J, Andres R (1989) Validation of a retrospective questionnaire assessing diet 10–15 years ago. Am J Epidemiol 130:173–187. 10.1093/oxfordjournals.aje.a115310 [DOI] [PubMed] [Google Scholar]
- Soldan A, Pettigrew C, Fagan AM, Schindler SE, Moghekar A, Fowler C, Li QX, Collins SJ, Carlsson C, Asthana S, Masters CL, Johnson S, Morris JC, Albert M, Gross AL (2019) ATN profiles among cognitively normal individuals and longitudinal cognitive outcomes. Neurology 92:e1567–e1579. 10.1212/WNL.0000000000007248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spira AP, Gamaldo AA, An Y, Wu MN, Simonsick EM, Bilgel M, Zhou Y, Wong DF, Ferrucci L, Resnick SM (2013) Self-reported sleep and beta-amyloid deposition in community-dwelling older adults. JAMA Neurol 70:1537–1543. 10.1001/jamaneurol.2013.4258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spira AP, An Y, Wu MN, Owusu JT, Simonsick EM, Bilgel M, Ferrucci L, Wong DF, Resnick SM (2018) Excessive daytime sleepiness and napping in cognitively normal adults: associations with subsequent amyloid deposition measured by PiB PET. Sleep 41:zsy152. 10.1093/sleep/zsy152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprecher KE, Bendlin BB, Racine AM, Okonkwo OC, Christian BT, Koscik RL, Sager MA, Asthana S, Johnson SC, Benca RM (2015) Amyloid burden is associated with self-reported sleep in nondemented late middle-aged adults. Neurobiol Aging 36:2568–2576. 10.1016/j.neurobiolaging.2015.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staresina BP, Bergmann TO, Bonnefond M, van der Meij R, Jensen O, Deuker L, Elger CE, Axmacher N, Fell J (2015) Hierarchical nesting of slow oscillations, spindles and ripples in the human hippocampus during sleep. Nat Neurosci 18:1679–1686. 10.1038/nn.4119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve S, Rabinovici GD, Cohn-Sheehy BI, Madison C, Ayakta N, Ghosh PM, La Joie R, Arthur-Bentil SK, Vogel JW, Marks SM, Lehmann M, Rosen HJ, Reed B, Olichney J, Boxer AL, Miller BL, Borys E, Jin LW, Huang EJ, Grinberg LT, et al. (2015) Existing Pittsburgh compound-B positron emission tomography thresholds are too high: statistical and pathological evaluation. Brain 138:2020–2033. 10.1093/brain/awv112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witton J, Staniaszek LE, Bartsch U, Randall AD, Jones MW, Brown JT (2016) Disrupted hippocampal sharp-wave ripple-associated spike dynamics in a transgenic mouse model of dementia. J Physiol 594:4615–4630. 10.1113/jphysiol.2014.282889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Zhan G, Fenik P, Brandes M, Bell P, Francois N, Shulman K, Veasey S (2018) Chronic sleep disruption advances the temporal progression of tauopathy in P301S mutant mice. J Neurosci 38:10255–10270. 10.1523/JNEUROSCI.0275-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]