

Abstract
Congenital muscular dystrophy with megaconial myopathy (MDCMC) is an autosomal recessive disorder characterized by progressive muscle weakness and wasting. The observation of megamitochondria in skeletal muscle biopsies is exclusive to this type of MD. The disease is caused by loss of function mutations in the choline kinase beta (CHKB) gene which results in dysfunction of the Kennedy pathway for the synthesis of phosphatidylcholine. We have previously reported a rostrocaudal MD (rmd) mouse with a deletion in the Chkb gene resulting in an MDCMC-like phenotype, and we used this mouse to test gene therapy strategies for the rescue and alleviation of the dystrophic phenotype. Introduction of a muscle-specific Chkb transgene completely rescues motor and behavioral function in the rmd mouse model, confirming the cell-autonomous nature of the disease. Intramuscular gene therapy post-disease onset using an adeno-associated viral 6 (AAV6) vector carrying a functional copy of Chkb is also capable of rescuing the dystrophy phenotype. In addition, we examined the ability of choline kinase alpha (Chka), a gene paralog of Chkb, to improve dystrophic phenotypes when upregulated in skeletal muscles of rmd mutant mice using a similar AAV6 vector. The sum of our results in a preclinical model of disease suggest that replacement of the Chkb gene or upregulation of endogenous Chka could serve as potential lines of therapy for MDCMC patients.
Introduction
Congenital muscular dystrophies (CMDs) are a group of autosomal recessive disorders exhibiting muscle weakness, wasting and hypotonia at or soon after birth with progressively increasing symptoms (1). There are currently at least 30 different types of CMD. In 2006, we reported a spontaneous mouse mutation displaying MD in a rostral to caudal gradient, with its hindlimbs being more affected than its forelimbs coupled with the presence of giant/megamitochondria, leading us to name it rostrocaudal MD (rmd) (2). The rmd mouse carries a 1.6 kb deletion in the choline kinase beta (Chkb) gene, resulting in complete loss of CHKB activity. Chkb is an important enzyme in the Kennedy pathway, required for de novo synthesis of phosphatidylcholine (PC). In mammals, choline kinase is the enzyme in the first step conversion of choline to phosphocholine which is ultimately converted to PC, one of the four major biolipids in all cellular membranes (3). In mammals, choline kinase activity is encoded by two separate paralogous genes, choline kinase alpha (Chka) and Chkb, which function either as homo- or hetero-dimers. Both of these genes have wide tissue expression profiles and both encoded enzymes phosphorylate choline to phosphocholine.
Following the publication of the rmd mouse model, a group of 15 patients with CMD and megamitochondria were sequenced and found to contain loss of function mutations in the CHKB gene. The disease was classified as CMD with megaconial myopathy (MDCMC), OMIM: 602541 (4). Both MDCMC patients and mice have loss of function mutations in the Chkb or Chkb gene and develop progressive MD indicates that the rmd mice have good face and construct validity as models of MDCMC. To date, there are at least 48 reported cases of MDCMC worldwide. These were children born of unaffected parents and having none or one affected sibling, indicating an autosomal recessive inheritance. In all reported cases, patients showed generalized muscle weakness starting at ~ 5 years of age. Some patients were reported as early as 22 months of age. These patients missed all major motor milestones and showed speech defects, with initial speech occurring at ~ 5 years of age and pronounced cognitive impairments at ~ 12 years of age. Many of these patients were first reported due to presentation of floppiness and increased tendencies to fall while walking or running and/or due to the presence of diffuse skin disorders like mild ichthyosis (5–14).
Here we test several methods of functional rescue of dystrophy using the rmd mouse model. These tests included examining the effects of dietary intervention with CDP-choline, a downstream compound of the Kennedy pathway, in an effort to circumvent the defects in the pathway. We also tested pre- and post-disease onset upregulation of CHKB in the rescue of MD phenotype in the rmd mutant mice. Pre-disease onset rescue of rmd was tested by overexpression of the Chkb gene in an engineered muscle-specific transgenic mouse where Chkb expression was driven by the Titin (Ttn) gene promoter in the skeletal and cardiac muscles. These transgenic rmd (Tg-rmd) mice also allowed us to examine the cell-type specificity required for rescue of MD. Post-disease onset rescue was tested by intramuscular injections of adeno-associated viral (AAV) vectors expressing either the Chka or Chkb genes in rmd mutant mice. Expression of the Chka gene normally shuts down during postnatal muscle differentiation (15), and our previous work suggested that residual CHKA activity in anterior muscle groups of the rmd mouse correlated with reduced severity of dystrophic symptoms (15). Thus, we sought to determine if CHKA could compensate for the lack of Chkb as an alternative rescue mechanism in rmd mice. We found that viral delivery of Chka was also efficacious with comparable potency to rescue with the Chkb gene.
Results
Muscle-specific expression of Chkb transgene prevents MD
We engineered a full-length cDNA of the mouse Chkb gene under the control of the muscle-specific Ttn promoter and created a line of Tg-rmd mice (Fig. 1A) (16). The Ttn-Chkb transgene was also carried on a wild-type (WT) background. The Tg-rmd and Tg-WT mice were physically indistinguishable from their WT littermates at birth and through adulthood (Fig. 1B and E). Real-time polymerase chain reaction (PCR) assay on tissue from gastrocnemius muscle of Tg-rmd, Tg-WT, rmd and +/rmd mice was performed to assess the levels of Chkb cDNA expression. We observed 18-fold higher expression of Chkb mRNA levels in the Tg-rmd muscles, compared to C57BL/6J controls, with no detectable levels in mutant rmd muscle, a 0.21-fold expression in heterozygous +/rmd muscles and a 14-fold higher expression of Chkb in the Tg-WT mice (Fig. 1C). Body weights of Tg − +/rmd mice were not significantly different from Tg-rmd and +/rmd, indicating absence of a detrimental effect of Chkb overexpression (Fig. 1D). It was also observed that Tg-rmd and Tg-WT mice were physically indistinguishable from each other (Fig. 1D).
Figure 1.
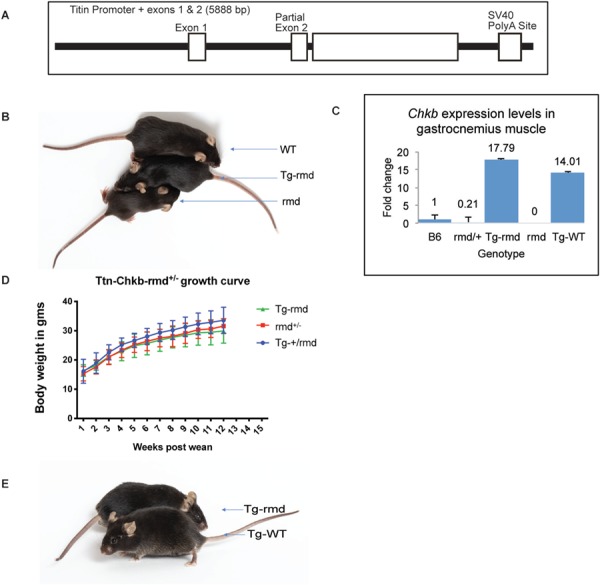
(A) demonstrates the Chkb transgene construct with a functional copy of Chkb cDNA-inserted downstream of a complete exon 1 and partial exon 2 structure followed by a SV40Poly A site downstream of the Chkb cDNA. (B) shows a WT (+/+), Tg-rmd and rmd (rmd/rmd) mouse from top to bottom with the Tg-rmd mouse being physically comparable to the WT. Chkb gene expression in muscle is 18-fold higher in Tg-rmd and 14-fold higher in Tg-WT mice as seen from real-time PCR analysis, whereas expression in rmd mice was not detectable and that in +/rmd mice was 0.21 times higher than in rmd mice (C). Tg-rmd, +/rmd (unaffected littermates) and Tg+/rmd mice do not show significant differences in their body weights when measured starting at 3 weeks of age (post-wean; D). The Tg-rmd and Tg-WT mice are physically indistinguishable from each other (E). N = 4 mice/sex/genotype.
To determine if our transgene functionally rescued motor performance, we tested for rescue of motor strength in 8- to 10-week-old Tg-rmd mice (n = 10 mice/sex/genotype) using behavioral assays including open field, rotarod, grip strength and the Erasmus ladder tests. Mutant rmd mice showed megamitochondria with decreased mitochondrial numbers and increased mitochondrial areas at 2 weeks of age (Supplementary Material, Fig. S2A–D) accompanied by significant motor deficits at 4–5 weeks of age, rendering them unsuitable for testing at 8–10 weeks as with the Tg-rmd mice (Supplementary Material, Fig. S2E–K).
The open field test was used to assess basic locomotor function (ambulation) (17,18). Tg-rmd male and female mice travelled an equal distance (Fig. 2A and B) and showed similar vertical activity (Fig. 2C and D) relative to age- and sex-matched WT littermate controls, suggesting normal ambulation and rearing ability.
Figure 2.
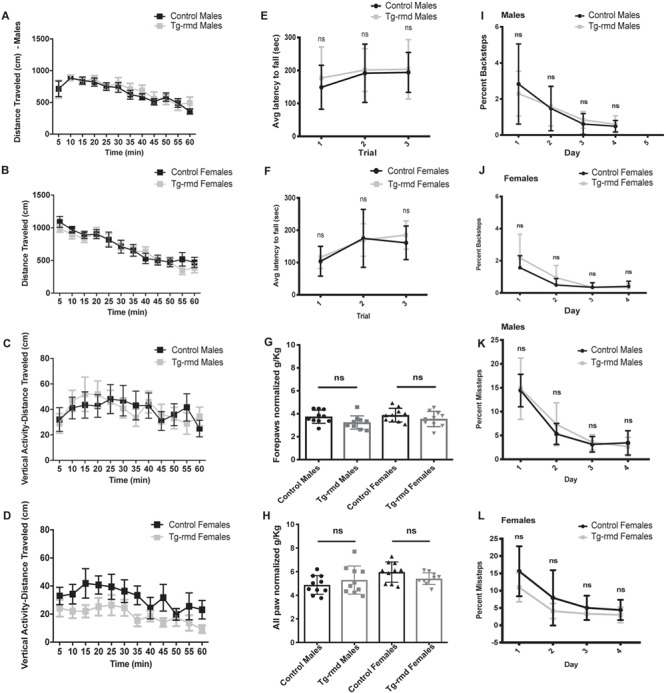
Evaluation of motor activity and coordination in Tg-rmd mice. Open field results for spontaneous locomotion suggest normal ambulation in the Tg-rmd mice, with distance travelled not significantly different from that of the controls tested in males (P = 0.96) and females (P = 0.66; A and B). Tg-rmd male (P = 0.31) and female (P = 0.67) mice show no significant differences in their vertical activity indicative of normal rearing behavior (C and D). Latency to fall from three individual trials on the rotarod assay shows normal motor coordination in the Tg-rmd male (P = 0.41) and female (P = 0.74) mice relative to age- and sex-matched WT littermate controls (E and F). Grip strength assay results suggest normal forepaw and all paw normalized grip strength in the Tg-rmd mice compared to the unaffected controls tested. Males showed a P = 0.07 and females, a P = 0.25 in forepaw grip strength (G), whereas males showed a P = 0.37 and females, a P = 0.09 in all paw grip strength (H). Erasmus assay for fine motor assessment and coordination in Tg-rmd mice shows no significant difference in percent backsteps (males, P = 0.59; females, P = 0.54; I and J) and percent missteps (males, P = 14; females, P = 0.56; K and L), suggesting no perturbance in motor coordination when compared to controls. N = 10 mice/sex/genotype, aged 8–10 weeks. Error bars represent mean with standard deviation (SD).
The accelerating rotarod is a forced performance test used to examine motor impairments (17,19). When tested for their ability to perform on the rotarod, our Tg-rmd mice were not different from their age- and sex-matched WT littermate controls, indicating normal motor endurance and performance levels (Fig. 2E and F).
We tested muscle function using a grip strength assay (20). Grip strength of forepaws and all paws were tested and normalized to body weight in order to gauge the severity of the dystrophy phenotype. The Tg-rmd males and females showed a grip strength similar to their age- and sex-matched WT littermate controls, indicating normal forepaw and all paw grip strength (Fig. 2G and H).
Fine motor coordination was tested using the Erasmus ladder, which specifically tests cerebellar motor coordination related to the precision and accurate timing of movement. The Erasmus ladder assay is sensitive to perturbations or disorders in fine movement, equilibrium, posture and fine learning and can distinguish between motor learning and motor coordination problems, making it a suitable device to test for subtle motor coordination disabilities in rodents (19,21,22). In this assay, motor and coordination deficiencies would be indicated by a greater percentage of missteps and backsteps on the ladder rungs. In the present studies, Tg-rmd males and females were not significantly different from age- and sex-matched WT littermate controls in percent backsteps or percent missteps.
Mitochondrial area measurements (n = 4 mice/sex/genotype) from sections of the gastrocnemius muscle analyzed by transmission electron (TE) microscopy showed that while rmd mutant mice had mitochondria that averaged five times larger than those of WT mice (P < 10−4), mitochondrial size was restored to normal in Tg-rmd mice (P = 0.1355; Fig. 3A–D). This can also be seen in the frequency distribution of mitochondrial areas where Tg-rmd mice restore the percentage of small mitochondria while reducing the frequency of mitochondria exceeding 1 nm2, whereas the rmd mice showed few mitochondria as large as 6 nm2 (Fig. 3E). Mitochondrial numbers are reduced by half in the affected rmd mice, whereas Tg-rmd mice show similar mitochondrial numbers to their WT littermates (Fig. 3F). Therefore, restoration of Chkb gene expression in skeletal muscle provides a cell-type specific rescue of Chkb expression and rescues the mutant rmd phenotype.
Figure 3.
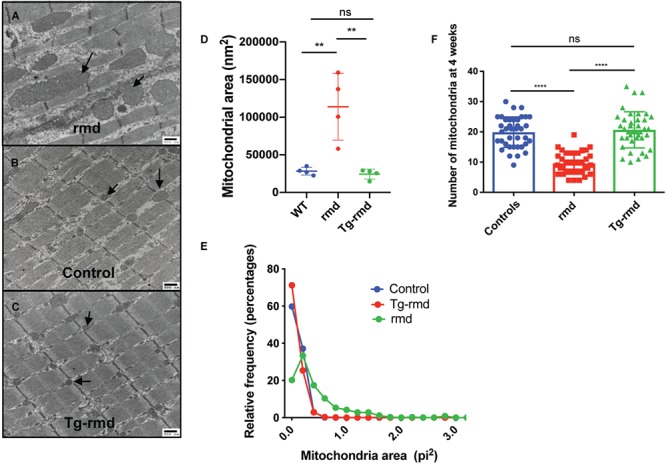
Mitochondrial phenotype in Tg-rmd mice. Demonstration of megamitochondria in rmd mice (A), normal-sized mitochondria in WT littermates (B) and rescued, normal-sized mitochondria in Tg-rmd mice (C). Mitochondrial areas are rescued in Tg-rmd (rescue) mice as compared to WT (control) and rmd (affected) littermates with Tg-rmd mice having mitochondrial areas closer in value to control mice (D). Tg-rmd mice show a higher percent of larger fibers (area of fibers) when compared to rmd mice (E). Mitochondrial numbers are restored in Tg-rmd mice compared to the decreased numbers in rmd mice, P < 0.0001 (D). N = 4 mice/sex/genotype, aged 4 weeks. Error bars represent mean with SD.
Together, these results indicate restoration of muscle strength and coordination in Tg-rmd mice and hence a prevention of the rmd disease phenotype. No aberrant mouse behavior or deaths were observed in any transgenic mice indicating an absence of toxic effects from overexpression of the Chkb transgene. However, in order to test this in a more thorough manner, we examined WT C57BL/6J mice carrying the Ttn-Chkb transgene (Tg-WT) and assessed levels of Chkb expression and motor strength. For these studies, Tg-WT mice were evaluated in the open field, rotarod and grip strength assays to confirm absence of any detrimental effects of transgene overexpression on motor performance.
Open field tests performed on n = 10 mice/sex/genotype revealed similar activity levels in Tg-WT and WT mice as measured by distance travelled (Fig. 4A and B). The Tg-WT male and female mice showed a similar latency to fall compared to the WT controls on the rotarod assay (Fig. 4C and D). In the grip strength assay, the Tg-WT males were not significantly different from WT male controls for forepaw and all paw grip strength. While the Tg-WT females did not show any difference in forepaw grip strength, there was a modest increase in all paw grip strength compared to WT controls (P = 0.01; Fig. 4E and F). Importantly, these results confirm that overexpression of Chkb gene does not result in any immediate detrimental effects in mice.
Figure 4.
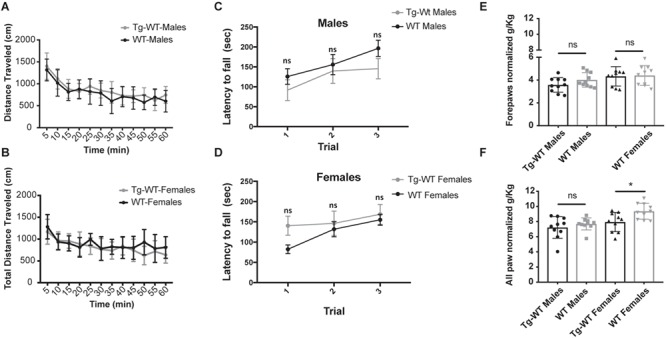
When tested for detrimental effects of overexpression of Chkb transgene in a WT mouse, it was observed that Tg-WT mice did not show significant differences in locomotor activity in both male (P = 0.45) and female (P = 0.10) groups when compared to WT mice on the open field assay (A and B). Latency to fall on the rotarod assay in males (P = 0.25; C) and females (P = 0.25; D) and forepaw and all paw grip strength in male (P = 0.13 and 0.39, respectively; E) Tg-WT mice were not significantly different compared to WT controls. The female Tg-WT mice did not show difference in forepaw grip strength (P = 0.86) but showed slight difference in all paw grip strength (P = 0.01), when compared to WT controls. These tests indicate lack of immediate toxic effects in transgene expression. N = 10 mice/sex/genotype, aged 8–10 weeks. Error bars represent mean and SD.
Dietary circumvention of defects in Kennedy pathway fail to rescue rmd phenotype
While the restoration of Chkb gene expression with a muscle-specific transgene was capable of preventing the onset and progression of dystrophic symptoms, we also sought to test whether we could bypass the biochemical defect by providing a downstream metabolic product of the Kennedy pathway via dietary supplementation. Previous studies have shown that intravenous injections of CDP-choline, a metabolite in the Kennedy pathway that is downstream of the action of CHKB, can help to prevent acute muscle damage in rmd mutant mice (23). We tested whether dietary supplementation with CDP-choline could also decrease dystrophy in the absence of Chkb. For the study, 500 mg/kg/day of CDP-choline was provided in the diet for a period of 3 months beginning at 3 weeks of age (24–26). Mutant rmd mice (n = 8 mice/sex) on dietary supplementation were weighed daily for changes in growth rate and effects of supplementation of the overt dystrophic symptoms compared to their non-supplemented controls (n = 4 mice/sex) and supplemented WT mice (n = 4 mice/sex). However, no improvements were observed in the growth rate or behavior of treated mice over the study duration (Supplementary Material, Fig. S1A and B). At the end of this study, two mice/sex/genotype were randomly selected to perform mass spectrometry analysis for components of the Kennedy pathway in muscle tissues of the treated mice, and body weights were analyzed. Mass spectrometry analyses showed no significant differences in the levels of CDP-choline in the gastrocnemius muscle between the control and test groups (Supplementary Material, Fig. S1C). These results indicate that a dietary supplementation of CDP-choline does not lead to improvement of MD in adult rmd mice.
Introduction of Chkb post-disease onset improves dystrophy phenotype in rmd mice
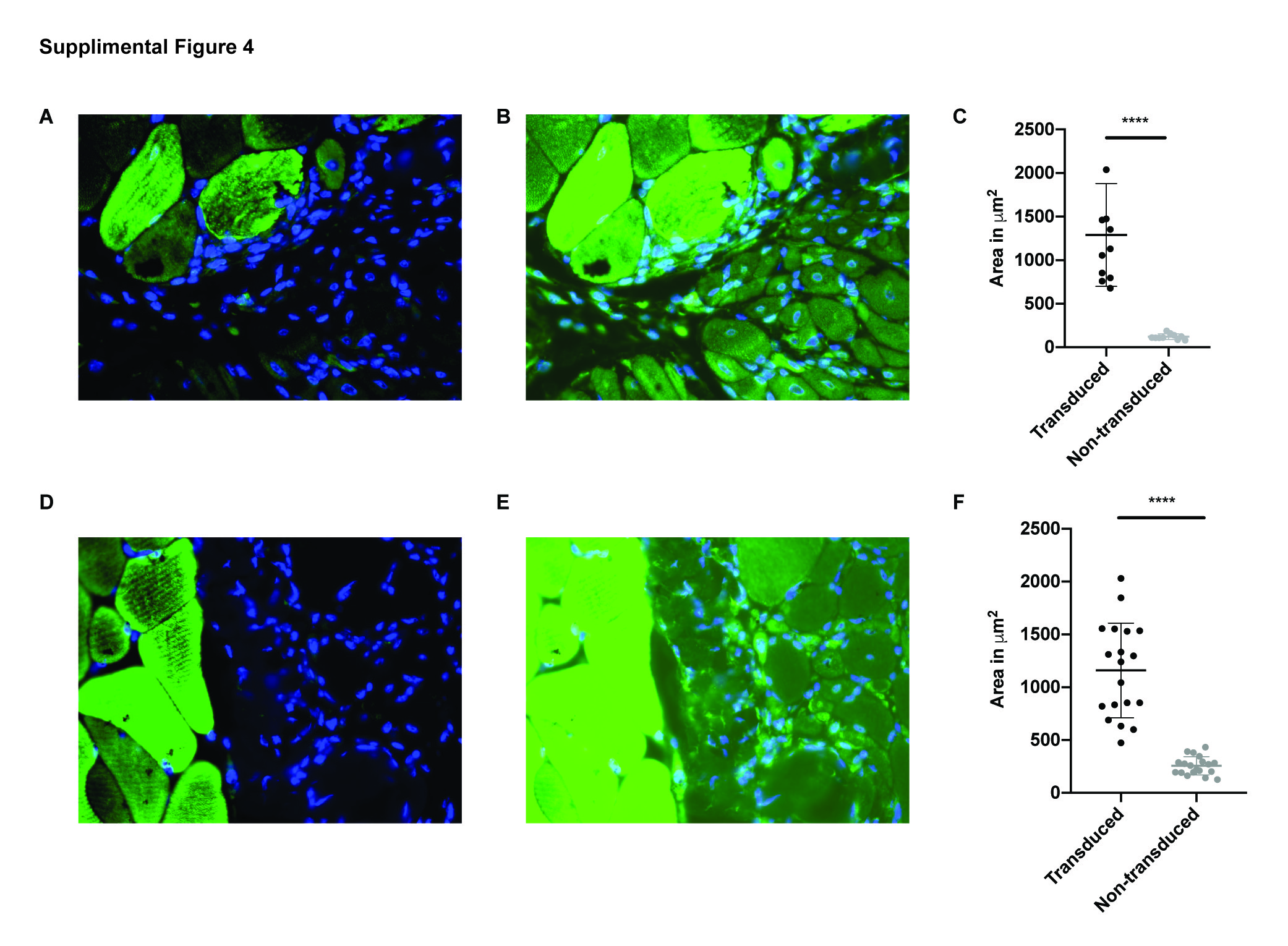
The mutant rmd disease phenotype is observed as early as 2 weeks of age, with the mice showing megamitochondria with decreased mitochondrial numbers and decreased muscle strength (Supplementary Material, Fig. S2A–D). Hence, in order to test the effects of upregulation of Chkb expression in post-disease onset adult skeletal muscle, we performed intramuscular injections of rmd mutant mice at 3 weeks of age with an AAV6 carrying a 3× Flag-tagged functional copy of the mouse Chkb cDNA and a self-cleaving p2A peptide-enhanced green fluorescent protein (EGFP) reporter gene into the gastrocnemius muscle. Each rmd mouse served as its own control with the left leg being injected with 25 μl of 2 × 1010 vg of the AAV vector and the right leg being sham injected with an equal volume of saline. Seven weeks post-injection, their gastrocnemius muscles were analyzed for changes in muscle weight, fiber area and percent of centralized nuclei. Gross observation showed an increase in muscle size in the AAV-injected muscle (Fig. 5A). In vivo fluorescence images of injected rmd and unaffected WT mice showed that EGFP fluorescence was localized to the site of injection indicating localized muscle transduction. No EGFP fluorescence was detected in the saline-injected leg (Fig. 5B). Gross hematoxylin and eosin (H&E)-stained histological sections (Supplementary Material, Fig. S3A and B) revealed a significant improvement in muscle fiber morphology in the AAV-Chkb injected muscles compared with the saline-injected muscles. H&E-stained montage images of the medial and lateral gastrocnemius muscle show a rescue of muscle fiber morphology in AAV-Chkb-injected muscles (Fig. 5C) when compared to the saline-injected muscle (Fig. 5D). We describe this as a partial rescue with the majority of muscle fibers in the injected rmd leg having cross-sectional fiber areas comparable to that of unaffected muscle fibers and show no centralized nuclei. However, there remain a percentage of muscle fibers with significantly smaller cross-sectional areas comparable to those of rmd mutant muscle and that show centralized nuclei. The partial nature of rescue is not unexpected, with intramuscular injections often resulting in a mosaic pattern of transduction. Evidence of mosaicism is observed by immunofluorescent staining for EGFP in the AAV-Chkb-injected muscles. Transduced fibers stain positive for EGFP and have larger fiber areas, whereas non-transduced fibers (visible upon overexposure of the tissue) do not stain positive for EGFP and have significantly smaller (P < 0.0001) fiber areas (Supplementary Material, Fig. S4A–C). The AAV-injected muscles of rmd mice showed a significant increase in muscle weight in both males (P = 0.004) and females (P = 0.006) compared to the saline-injected muscles, whereas the AAV-injected muscles of the unaffected WT mice showed no difference in muscle weight (P > 0.99 in males and P = 0.65 in females) suggesting that overexpression of CHKB rescues muscle in dystrophic mice but does not induce muscle hypertrophy in control mice (Fig. 6A and B). AAV-injected rmd muscles showed a reduced percent of centralized nuclei compared to saline-injected muscles (P = 0.001 in males and P = 0.06 in females), while those from AAV-injected WT muscles did not show any significant differences when compared to the saline-injected contralateral muscles (P = 0.35 and 0.29 in males and females, respectively; Fig. 6C and D), reflective of the specificity of rescue of the disease phenotype without alteration of normal muscle. Analysis of cross-sectional myofiber areas showed a significant increase (P = 0.0001) in the average fiber areas of AAV-Chkb-injected rmd muscles. Classifying fiber areas in four quartiles into small (S = 0–307 nm2), medium 1 (M1 = 307.01–795 nm2), medium (M2 = 795.01–1391 nm2) and large (L = 1391.01–14 093 nm2) showed a significant increase in the percent of M1, M2 and L fiber sizes and a significant decrease in the percent of S fiber sizes in the AAV-injected rmd muscle compared with the saline-injected muscle (Fig. 6E and F).
Figure 5.
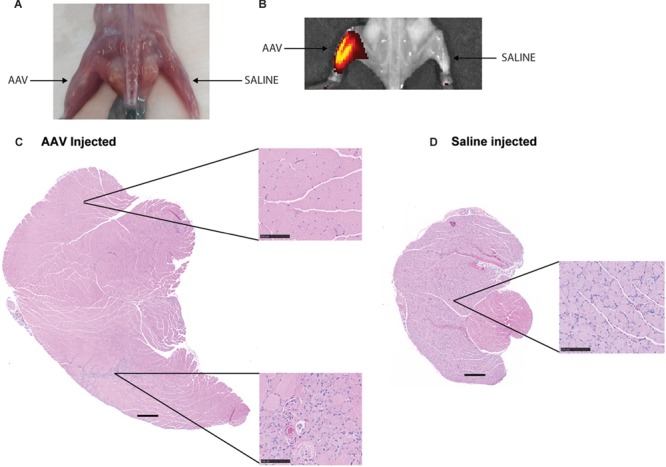
AAV-Chkb-injected gastrocnemius muscles in males (A) and females (B) show significantly higher and restored muscle weights compared to sham-injected muscles, whereas there is no difference in the AAV-Chkb and sham-injected gastrocnemius muscles of unaffected mice. Percent of centralized nuclei, an indicator of poor muscle health, was observed to be significantly reduced in AAV-Chkb-injected rmd muscles compared to the sham-injected muscles in both males (C) and females (D). Injected unaffected muscles did not show any significant differences when compared to sham-injected unaffected muscles in both males and females (C and D). Injected rmd muscles show a significant (P = 0.0001) increase in average fiber areas, in males and females, compared to sham-injected rmd muscle with a significant increase in percentage of large (L) and medium (M1 and M2) fibers and a decrease in the percent of small (S) muscle fibers (E and F). N = 8 mice/sex/genotype. Mice were injected at 3 weeks of age, and their gastrocnemius muscles were harvested at 7 weeks of age. Error bars represent mean and SD. Scale bars on (C) and (D) represent 500 μm; those in insets represent 100 μm at 20× magnification.
Figure 6.
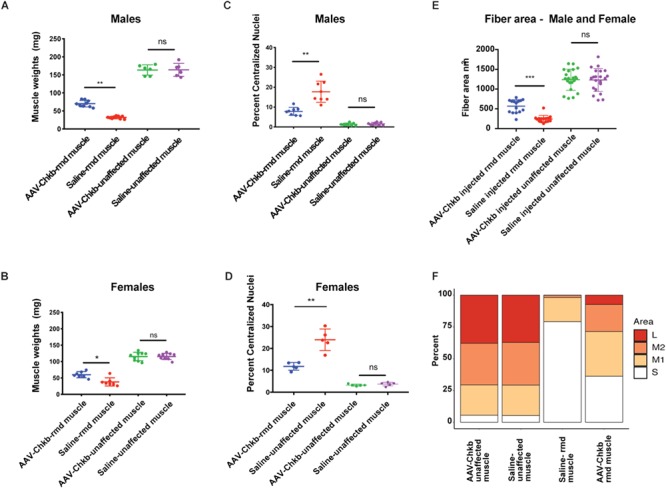
AAV-Chkb-injected gastrocnemius muscles in males (A) and females (B) show significantly higher and restored muscle weights compared to sham-injected muscles, whereas there is no difference in the AAV-Chkb and sham-injected gastrocnemius muscles of unaffected mice. Percent of centralized nuclei, an indicator of poor muscle health, was observed to be significantly reduced in AAV-Chkb-injected rmd muscles compared to the sham-injected muscles in both males (C) and females (D). Injected unaffected muscles did not show any significant differences when compared to sham-injected unaffected muscles in both males and females (C and D). Injected rmd muscles show a significant (P = 0.0001) increase in average fiber areas, in males and females, compared to sham-injected rmd muscle with a significant increase in percentage of large (L) and medium (M1 and M2) fibers and a decrease in the percent of small (S) muscle fibers (E and F). N = 8 mice/sex/genotype. Mice were injected at 3 weeks of age, and their gastrocnemius muscles were harvested at 7 weeks of age. Error bars represent mean and SD.
Chka can compensate for lack of Chkb in rmd mutant mice
The normal developmental loss of Chka gene expression in mouse caudal muscles during postnatal muscle differentiation makes skeletal muscles of the hindlimb particularly susceptible to disease in the case of Chkb gene mutations in mice. In order to test the hypothesis that the paralogous CHKA protein can functionally compensate for CHKB deficiency in rmd mice, we injected rmd muscles with an AAV vector expressing a human CHKA cDNA. The AAV-injected muscles were observed to be larger than the saline-injected muscles seven weeks post-injection (Supplementary Material, Fig. S3C). In vivo fluorescence imaging of rmd mice showed that EGFP florescence was localized to the site of injection whereas the saline-injected site did not display any fluorescence (Supplementary Material, Fig. S3D). Like the Chkb gene therapy experiment, H&E staining of AAV-injected muscles showed major regions of restored muscle structure (Supplementary Material, Fig. S3E and F). Montage images stained with Gordon and Sweet’s to highlight the circumference of each myofiber showed significant regions with normal morphology and regions of partial rescue in the AAV-CHKA-injected rmd muscles (Supplementary Material, Fig. S4E and F). The AAV-injected muscles of rmd mice weighed significantly more than the saline-injected contralateral muscles with a P = 0.008 and 0.006 in males and females, respectively. The difference in muscle weights between the AAV- and sham-injected WT control mice was not significant (P = 0.31 in both males and females; Fig. 7A and B). AAV-injected rmd muscles showed a decrease in the percent of centralized nuclei in comparison to saline-injected rmd muscle (P = 0.16 and 0.008 in males and females, respectively), while the AAV-injected control muscles were not significantly different from the saline-injected control muscles (P = 0.06 and 0.16 in males and females, respectively; Fig. 7C and D). Similar to our AAV-Chkb injections, treatment with the AAV-CHKA virus rescues the disease phenotype in dystrophic muscles without inducing hypertrophy of normal muscle in WT mice. AAV-injected muscles showed a significant increase (P < 0.0001) in average muscle fiber area in males and females. Fiber areas were classified into four quartiles, as in case of Chkb injections, into small (S = 0–209 nm2), medium 1(M1 = 209.01–630 nm2), medium 2 (M2 = 630.01–1159 nm2) and large (L = 1159.01–8387 nm2). AAV-injected rmd muscles showed a significant increase in the percent of M1, M2 and L fiber sizes and a significant decrease in the percent of S fiber sizes (Fig. 7E and F). Transduced AAV-Chka muscles stain positive for EGFP and show larger fiber areas (P < 0.0001) when compared to non-transduced fibers in the AAV-Chka-injected muscles (Supplementary Material, Fig. S4C and D).
Figure 7.
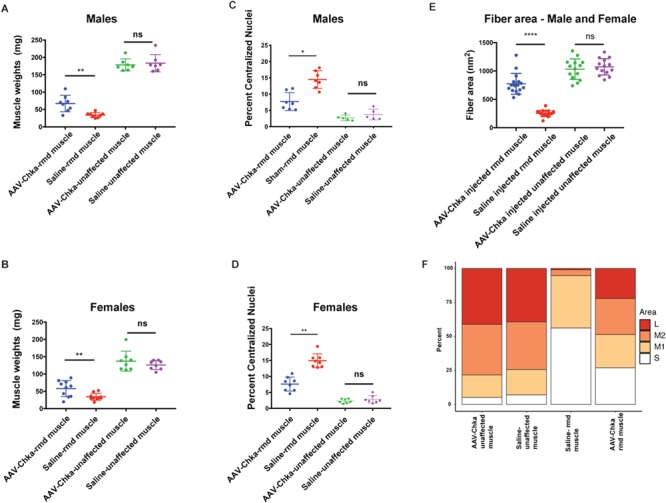
AAV-Chka-injected gastrocnemius muscles in males (A) and females (B) show significantly higher and restored muscle weights compared to sham-injected muscles, whereas there is no difference in the AAV-Chkb and sham-injected gastrocnemius muscles of unaffected mice. Percent of centralized nuclei was observed to be significantly reduced in AAV-Chka-injected rmd muscles compared to the sham-injected muscles in both males (C) and females (D). Injected unaffected muscles did not show any significant differences when compared to sham-injected unaffected muscles in both males and females (C and D). Injected rmd muscles show a significant (P < 0.0001) increase in average fiber areas, in males and females, compared to sham-injected rmd muscle with a significant increase in percentage of large (L) and medium (M1 and M2) fibers and a decrease in the percent of small (S) muscle fibers (E and F). N = 8 mice/sex/genotype. Mice were injected at 3 weeks of age, and their gastrocnemius muscles were harvested at 7 weeks of age. Error bars represent mean and SD.
Discussion
The discovery and characterization of the spontaneous rmd mutant mouse led to the discovery of CHKB as the gene underlying MDCMC (2,4), establishing the validity of the rmd mouse as a model of the human disease and opening up avenues for mechanistic and therapeutic preclinical studies. MDCMC is caused by recessive loss of function mutations in the Chkb gene. We have shown that the ubiquitous loss of Chkb in rmd mutant mice leads to a cell and tissue-specific disease which can be restored by upregulating expression of Chkb in skeletal muscle tissues only, indicating that regeneration of skeletal muscles in a Chkb-deficient environment is cell-autonomous. Intramuscular AAV injections of Chkb and Chka genes also help restore muscle morphology in rmd mutant mice, while dietary supplementation of Kennedy pathway intermediates does not have a regenerative or rescue effect.
Gene replacement of Chkb by transgenesis or by AAV6 delivery rescues the MD phenotype of the rmd mouse. Introduction of a functional copy of the Chkb transgene under a muscle-specific promoter prevented disease onset, with the Tg-rmd mice showing normal mitochondrial dimensions and muscle strength. This indicates that although the Chkb gene is ubiquitously expressed, the enzyme deficiency results in a muscle-specific defect in PC biosynthesis that can only be rescued by a cell-autonomous increase of CHKB activity within muscle cells themselves. Overexpression of the Chkb gene up to a 14-fold magnitude does not cause a negative impact on the Tg-WT animals, with mice showing behavioral and muscle strength phenotypes comparable to their non-transgenic littermates. Further, introduction of the Chkb gene using AAV6 post-disease onset in young adult rmd mice can help reverse the degenerative muscle disease. Intramuscular AAV6 vector injections containing a functional copy of Chkb gene in adult skeletal muscle resulted in improved muscle regeneration capacity and subsequent increased muscle weights and fiber areas with a decreased number of centralized nuclei compared to untreated rmd muscles (27–29). A detailed analysis of fiber areas shows an increase in the percent of large and medium-sized fibers in AAV-injected muscles with a significant decrease in the percent of small-sized fibers. The mosaic pattern of rescue in AAV-injected muscles correlates with the transduction efficiency of intramuscular delivery and further supports the cell-autonomous nature of this dystrophy (Supplementary Material, Fig. S4). These results suggest that replacing Chkb gene post disease-onset and in adult mice helps reverse MD by increasing muscle regeneration and maintenance of transduced fibers in dystrophic rmd mice. Injecting and thus upregulating Chkb expression in unaffected muscles did not have any detrimental effects as indicated by lack of hypertrophy in AAV-Chkb-injected skeletal muscles. Our results support a strategy of reintroducing the CHKB gene in MDCMC patients via AAV-mediated gene therapy as a possible therapeutic measure. Future preclinical studies focused on systemic injection of vectors will need to be performed to support moving this into the clinic.
Biochemical studies suggest that Chka and Chkb can function either as homodimers or heterodimers in the phosphorylation of choline to phosphocholine for the production of PC (30). In rmd mice, total choline kinase activity in the hindlimbs is absent whereas in the forelimbs, choline kinase activity is attenuated by only 50% (15). These results led to the hypothesis that there must be an age-dependent reliance on the activity of CHKB in WT hindlimb skeletal muscles of mice as Chka gene expression decreases. The partial retention of choline kinase activity in the forelimbs of adult rmd mice (due to residual Chka expression) provides a possible explanation for the decreased severity of disease in forelimb muscles versus the much more severe and progressive dystrophy in hindlimb muscles. This led us to test the possibility that CHKA can functionally compensate for CHKB deficiency in the severely affected hindlimb muscles of the rmd mutant mouse. The mechanism by which the endogenous expression of Chka shuts down in adult mouse skeletal muscles is still unclear. A study of CHKA expression in human skeletal muscle over time has not been tested; however, GTEx expression data across 53 adult tissues shows that CHKA transcript levels are the lowest in adult skeletal muscles (GETx Portal version V7) and likely explains why both human and mouse skeletal muscles are particularly sensitive to loss of CHKB activity. Our data indicated that increasing the expression of CHKA in post-disease onset, adult skeletal muscles results in improved muscle regeneration, increased muscle weights and fiber areas and a decrease in the percent of centralized nuclei with no immediate observable toxic effects. This suggests that even though CHKA expression is normally shut down in adult skeletal muscles, upregulation of its expression can compensate for lack of CHKB activity. Thus, strategies that could reactivate endogenous CHKA expression in adult skeletal muscles could provide a complementary avenue for therapeutic intervention.
One potential caveat to manipulating CHKA expression is that increased levels of Chka, but not beta, has been implicated in tumorigenesis, with CHKA overexpression detected in 40–60% of human tumors (31). Transfecting human (Hek293T) cells with CHKA was found to result in anchorage-independent growth activity similar to that demonstrated by Rho-A activation (32). It can be inferred that CHKA upregulation via AAV injections could potentially cause non-cancer cells to take on a cancerous phenotype, while siRNA downregulation could lead to cancer cell death (33). Immediate evidence (7 weeks post-injection) for this was not found in our experiments utilizing intramuscular injections of CHKA in rmd skeletal muscles. CHKA-injected mice will need to be aged and studied for potential tumorigenic effects of gene upregulation in order to determine whether the association between CHKA and cancer is directly causative of disease or whether it is instead a biomarker of the cancerous state. Additionally, off-target effects of AAV-CHKA injected systemically could be reduced by using muscle-specific promoters like muscle creatinine kinase (MCK) (34–36).
Chkb deficiency causes MD in a rostral to caudal gradient in mice as Chka expression is shut down in caudal adult skeletal muscles. One might expect that there is a functional reason for the observed decrease in normal Chka gene expression in adult skeletal muscles; however, our data suggest that overexpression of CHKA via an AAV gene therapy approach is not deleterious in skeletal muscles. In the CHKB-deficient condition, CHKA can compensate for the lack of Chkb to form functional α–α homodimers for the phosphorylation of choline to phosphocholine in skeletal muscles. Our results support that both the upregulation of either CHKB or CHKA can be used as a potential therapy for the rescue of MDCMC symptoms. Interestingly, the upregulation of CHKB in our transgenic experiments also reduces the presence of megamitochondria, indicating that the two phenotypes are related and cannot be rescued independent of each other. Megamitochondria are thought to result in a disruption of normal myofiber structure, with altered cellular architecture including disordered sarcomeric and muscle triad structures that are necessary for the orderly distribution of energy-producing mitochondria. Whether the generation of megamitochondria in skeletal muscle directly causes the MD or is just a biomarker of the disease remains to be tested.
Materials and Methods
Mouse colonies
All mice were bred and maintained at the Jackson Laboratory (Bar Harbor, ME, USA) following procedures and protocols approved by our Institutional Animal Care and Use Committee (IACUC). For breeding, mice were kept in humidity and temperature-controlled rooms with a 12:12 dark:light cycle. They were given an extruded pellet 5K52 chow diet with 6% fat (LabDiet, Brentwood, MO) ad libitum with free access to water (HCl-acidified, pH 2.8–3.2). For all motor and behavioral tests, the mice were moved to a separate room and singly housed where they were maintained under IACUC-approved conditions until completion of the test procedures. In this facility, the mice were kept on a 4% extruded grain diet and were provided with clean acid water unless mentioned otherwise.
Generation of transgenic (rescue) mouse
Expression of Chkb transgene in transgenic mice, for the generation of rescues, was achieved by using the Ttn promoter to express the Chkb cDNA transgene specifically in skeletal and cardiac muscles (16). This promoter reproduces the endogenous pattern of Ttn expression in muscles prior to the onset of the rmd disease symptoms. The Ttn promoter includes 3.5 kb of sequence upstream of the non-coding Ttn exon 1; the entire 2.1 kb of intron 1 and exon 2 is truncated just before the start codon. The inclusion of an intron in the construct is beneficial for proper long-term expression in transgenic mice and to take advantage of any possible control elements that might be located in the first intron. The polyadenylation signal is a 200 bp fragment derived from the SV40 viral genome that we have successfully used in numerous transgenic lines. A 1589 bp Chkb cDNA fragment, including 234 bp upstream from the exon 1 ATG start codon (Ch15 reverse strand, 89 429 834 bp; Ensembl GRCm38) to 145 bp downstream from the exon 11 TGA stop codon (89 426 584 bp) was PCR-cloned and blunt-ligated into the Ttn plasmid backbone immediately after the truncated exon 2. The completed pTtn–Chkb constructs were injected into fertilized C57BL/6J eggs by the microinjection. Transgenic founders were bred to C57BL/6J mice to generate a stable colony and maintain the transgene on an inbred background. Transgenic mice were identified through PCR of tail DNA with a forward primer in the 3′ end of the TTN promoter sequences, TTN100F (5′-TCTCCACCAAGAAGACGCTG-3′) together with a reverse primer in the third exon of Chkb, Chkb-e3R (5′-CTTTCTAATACCAAGGAGTCTACACC-3′).
Mitochondrial area
Specimen preparation
For TE microscopy, four mice per genotype (rmd, WT and Tg-rmd) were used to analyze mitochondrial structure. The medial and lateral gastrocnemius muscle were isolated and fixed in a solution of 2% paraformaldehyde and 2% glutaraldehyde, in 0.1 M cacodylate buffer (pH 7.2) at 4 °C overnight. Tissues were then washed, dehydrated in graded series of ethanol and processed for 812 resin embedding. Samples were then cured at 70 °C for 48 h followed by thin sectioning (90 nm) with a Leica EM UC6 ultramicrotome (Leica Microsystems, Buffalo Grove, IL) on a diamond knife. The sections were then placed on 300 mesh copper grids and stained using 2% uranyl acetate and Reynolds lead citrate. Samples were evaluated at 80 kV using a JEOL JM-1230 TE microscope (JEOL, Tokyo, Japan) and images collected with an AMT 2 K digital camera (Advanced Microscopy Techniques, Woburn, MA).
Mitochondrial area calculations
Ten pictures per sample (n = 4) were taken using the TE microscope. Mitochondrial areas were measured using FiJi (version 2.0) and analysis of variance (ANOVA) calculations were performed on Prism software (version 7.0c for Mac OS X).
Behavioral and motor assays
Adult mice (n = 10 sex/genotype) were evaluated in a battery of behavioral tests for motor function. All technicians were blinded to genotype for the duration of the entire battery of assays. The time of the day at which each test was performed was kept constant for each assay performed. The mice were allowed a minimum resting period of two days between every test. All equipment was sanitized and wiped with 70% ethanol before and in between testing mice. The test animals were habituated for 60 min in the testing room before each test. The order of testing was as follows; open field, rotarod, grip strength and Erasmus ladder.
Open field test
mice were evaluated in the open field as previously described by Sukoff et al. (35). Briefly, mice were individually placed in clear polycarbonate open field arenas (40 × 40 × 40 cm) housed in sound attenuated, ventilated chambers illuminated to a light level consistent with lighting in the housing and procedure rooms (~500 lux). A 16 × 16 grid of infrared beams were used to detect total distance traveled (cm) and vertical activity. Data were recorded for 60 min with data collection in 5 min bins and analyzed by Versamax software (Omnitech, Columbus, OH).
Rotarod
The Ugo Basile rotarod was used to analyze motor coordination and balance. The rotarod drum was steadily accelerated from 4 to 40 rpm over a 300 s duration. Each animal was evaluated in three consecutive trials with an inter-trial interval of ~ 1 min, and the average latency to fall was recorded for each trial as well as the average latency across all three trials (17,19,37).
Grip strength test
Mice were individually tested using the force gauge grip strength meter (BIOSEB, Kyoto, Japan). Each subject was tested for three consecutive forepaw trials followed by three consecutive all paw trials (six total trials). Grip strength was measured in grams and normalized to body weight (19,20,37).
Erasmus ladder
The Erasmus ladder (Noldus, Wageningen, Netherlands) was used to study locomotion and motor coordination in mice as previously described (21, 38). Briefly, each subject was tested for 4 consecutive days, during which no audible tone stimulus or perturbation (hurdles) were used. Data for percent missed steps and percent backsteps were recorded and later analyzed (22).
CDP-choline diet supplementation
For this study, four WT mice/sex and eight rmd mice/sex were provided with CDP-choline-supplemented 76A laboratory diet gel from ClearH2O (Portland, ME), and four rmd mice/sex were used as controls and were fed plain diet gel. Same-sex mice were housed in doublets in each side of a duplex mouse cage. 87.5 mg of CDP-choline was mixed in 56 gms of standard 76A laboratory diet gel cups. On an average a mouse eats 8 gms of diet gel per day giving them a dose of 500 mg/kg/day. Diet gel cups were changed three times a week (26). The WT mice were administered diet gel cups spiked with CDP-choline to assess to possibility of toxic effects due to increased consumption of CDP-choline. Eight rmd mice/sex were administered CDP-choline-spiked diet gel cups, these are referred to as Test mice; the remaining four rmd mice/sex were administered standard diet gel cups with no CDP-choline added, these are referred to as control mice. The total length of this study was 3 months. Mice were weighed and assessed for general health every week (24,39,40).
Adeno-associated viral vector design
An AAV vector design: an AAV subtype 6 was selected as it shows higher gene expression and tropism in skeletal muscles (27,28). AAV vector plasmid AAV-Chkb was derived from AAV–CAG–GFP plasmid (Addgene #28014). This plasmid contains mouse Chkb cDNA tagged by a 3× Flag tag (5-GACTACAAAGACCATGACGGTGATTATAAAGATCATGACATCGACTACAAGGATGACGATGACAAG-3′) at the 5′ end in order to distinguish transduced Chkb expression from endogenous expression, all under the control of a CAG (Cytomegalovirus (CMV) early enhancer/chicken beta actin) promoter. The plasmid also contains EGFP as a gene expression reporter protein, the expression of which is driven by the CAG promoter through P2A ribosomal skipping sequence downstream of the Chkb coding sequence (41). Recombinant AAV6 vectors were produced by triple transfection of HEK293T/17 cells with Chkb plasmid DNA, pAAV2/6 packaging plasmid and pAd delta F6 helper plasmid (Penn Vector Core, Philadelphia, PA) using linear polyethylenimine. Transfected cells were incubated at 37°C, 5% CO2 in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 2.5% fetal bovine serum. Three days after transfection, the crude vector fraction was obtained by combining the precipitated products from culture medium with final 8% polyethylene glycol and the cell extracts lysed by repeated freeze–thaw cycles. The crude vectors were then purified by iodixanol density-gradient ultracentrifugation. Purified AAV6 vector was dialyzed against 1× Phosphate-buffered saline (PBS) with 5% sorbitol. Viral titer was measured using real-time PCR with primers for the inverted terminal repeats (ITR) region (5′-GGAACCCCTAGTGATGGAGTT-3′ and 5′-CGGCCTCAGTGAGCGA-3′). A final volume of 300 μl with a concentration of 1.84 × 1013 vg/ml was obtained for AAV-Chkb injections and 150 μl of 2 × 1014 vg/ml of AAV-Chka.
Transduction of muscle fibers and tissue processing
The left gastrocnemius muscle of 3-week-old rmd mice and unaffected controls were injected locally with 2 × 1010 vg of AAV vector solution in 25 μl PBS. The right gastrocnemius muscle was treated as control and injected with 25 μl of sterile PBS solution only (42,43). The injected animals were aged on shelf for 7 weeks post-injection, and the gastrocnemius muscles were harvested at 10 weeks of age and fixed in 10% neutral-buffered formalin (NBF) for 24 h before embedding in paraffin.
Muscle fiber staining
Gordon and Sweets stain for reticular fibers
Injected gastrocnemius muscles were harvested 7 weeks post-injection and fixed in 10% NBF. Samples were then oxidized in 1% potassium permanganate solution, bleached in oxalic acid, sensitized in 2.5% ferric ammonium sulfate and impregnated with ammoniacal silver solution. Following a rinse and a 2 min fix in formalin, the samples are toned in 0.2% gold chloride solution and washed with 5% sodium thiosulphate. Samples are rinsed, dehydrated and clear mounted for microscopy analysis.
Immunofluorescent staining of AAV-injected muscle
AAV-injected gastrocnemius muscles were harvested 7 weeks post-injection and fixed in 10% NBF. Samples were then paraffin-embedded, sectioned and mounted on microscope slides for staining to visualize AAV-encoded GFP expression. Samples were incubated overnight at 4 °C in primary antibody (abcam290; diluted 1:1000), washed in PBS, blocked in a solution of 2% bovine serum albumin (BSA) and 0.2% Triton in PBS. Samples were then incubated in secondary antibody (ab150081) for 2 h at room temperature (RT), washed in PBS and stained with 4',6'-diamidino-2-phenylindole (DAPI). Coverslips were mounted in ProLong Gold Anti-Fade by Thermo Fischer Scientific (Waltham, MA).
H&E staining
Injected muscle fibers were fixed in 10% NBF and embedded. Post-embedding, the samples were processed in a Leica automated stainer by Histology Core Services at the Jackson Laboratory.
Muscle fiber area analysis
Cross sections of AAV and saline-injected muscles, stained using Gordon and Sweets stain, were imaged using the Nanozoomer (Hamamatsu, Shizuoka, Japan) and analyzed on the NDP view 2 software. Multiple images were collected at 20× magnification and were then stitched using Fiji (version 2.0). Fiji was then used to count fiber areas and numbers. Centralized nuclei were manually counted using ImageJ-generated stitched images.
Statistical analysis
All the experiments were performed at the Jackson Laboratory. All mouse handling, testing and analysis were performed blinded for mouse genotype. Where appropriate, statistical significance was calculated using Student’s t-test or two-way ANOVA with post-hoc Bonferroni corrections unless otherwise noted. Calculations were performed using Prism 7.0c software for Mac OS X, and any significant differences (P < 0.05) between test and control strains are denoted by an asterisk symbol.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We would like to thank David G. Schroeder for assistance with general laboratory procedures and in colony maintenance, Jennifer Stauffer for assistance in laboratory procedures, Pete Finger for assistance with electron microscopy, Nick Gott for assistance with histology procedures and Gaylynn Wells and Traci McGarr for assistance with behavioral procedures.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health (AR054170 to G.A.C.).
References:
- 1. Mercuri E., Sewry C., Brown S.C. and Muntoni F. (2002) Congenital muscular dystrophies. Semin. Pediatr. Neurol., 9, 120–131. [DOI] [PubMed] [Google Scholar]
- 2. Sher R.B., Aoyama C., Huebsch K.A., Ji S., Kerner J., Yang Y., Frankel W.N., Hoppel C.L., Wood P.A., Vance D.E. and Cox G.A. (2006) A rostrocaudal muscular dystrophy caused by a defect in choline kinase beta, the first enzyme in phosphatidylcholine biosynthesis. J. Biol. Chem., 281, 4938–4948. [DOI] [PubMed] [Google Scholar]
- 3. Gibellini F. and Smith T.K. (2010) The Kennedy pathway—de novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life, 62, 414–428. [DOI] [PubMed] [Google Scholar]
- 4. Mitsuhashi S., Hatakeyama H., Karahashi M., Koumura T., Nonaka I., Hayashi Y.K., Noguchi S., Sher R.B., Nakagawa Y., Manfredi G. et al. (2011) Muscle choline kinase beta defect causes mitochondrial dysfunction and increased mitophagy. Hum. Mol. Genet., 20, 3841–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nishino I., Kobayashi O., Goto Y.I., Kurihara M., Kumagai K., Fujita T., Hashimoto K., Horai S. and Nonaka I. (1998) A new congenital muscular dystrophy with mitochondrial structural abnormalities. Muscle Nerve, 21, 40–47. [DOI] [PubMed] [Google Scholar]
- 6. Mitsuhashi S., Ohkuma A., Talim B., Karahashi M., Koumura T., Aoyama C., Kurihara M., Quinlivan R., Sewry C., Mitsuhashi H. et al. (2011) A congenital muscular dystrophy with mitochondrial structural abnormalities caused by defective de novo phosphatidylcholine biosynthesis. Am. J. Hum. Genet., 88, 845–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Quinlivan R., Mitsuahashi S., Sewry C., Cirak S., Aoyama C., Mooore D., Abbs S., Robb S., Newton T., Moss C. et al. (2013) Muscular dystrophy with large mitochondria associated with mutations in the CHKB gene in three British patients: extending the clinical and pathological phenotype. Neuromuscul. Disord., 23, 549–556. [DOI] [PubMed] [Google Scholar]
- 8. Castro-Gago M., Dacruz-Alvarez D., Pintos-Martínez E., Beiras-Iglesias A., Delmiro A., Arenas J., Martín M.Á. and Martínez-Azorín F. (2014) Exome sequencing identifies a CHKB mutation in Spanish patient with megaconial congenital muscular dystrophy and mtDNA depletion. Eur. J. Paediatr. Neurol., 18, 796–800. [DOI] [PubMed] [Google Scholar]
- 9. Oliveira J., Negrão L., Fineza I., Taipa R., Melo-Pires M., Fortuna A.M., Gonçalves A.R., Froufe H., Egas C., Santos R. and Sousa M. (2015) New splicing mutation in the choline kinase beta (CHKB) gene causing a muscular dystrophy detected by whole-exome sequencing. J. Hum. Genet., 60, 305–312. [DOI] [PubMed] [Google Scholar]
- 10. Castro-Gago M., Dacruz-Alvarez D., Pintos-Martínez E., Beiras-Iglesias A., Arenas J., Martín M.Á. and Martínez-Azorín F. (2016) Congenital neurogenic muscular atrophy in megaconial myopathy due to a mutation in CHKB gene. Brain Dev., 38, 167–172. [DOI] [PubMed] [Google Scholar]
- 11. Brady L., Giri M., Provias J., Hoffman E. and Tarnopolsky M. (2016) Proximal myopathy with focal depletion of mitochondria and megaconial congenital muscular dystrophy are allelic conditions caused by mutations in CHKB. Neuromuscul. Disord., 26, 160–164. [DOI] [PubMed] [Google Scholar]
- 12. Gutiérrez Ríos P., Kalra A.A., Wilson J.D., Tanji K., Akman H.O., Area Gómez E., Schon E.A. and DiMauro S. (2012) Congenital megaconial myopathy due to a novel defect in the choline kinase beta gene. Arch. Neurol., 69, 657–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cabrera-Serrano M., Junckerstorff R.C., Atkinson V., Sivadorai P., Allcock R.J., Lamont P. and Laing N.G. (2015) Novel CHKB mutation expands the megaconial muscular dystrophy phenotype. Muscle Nerve, 51, 140–143. [DOI] [PubMed] [Google Scholar]
- 14. Haliloglu G., Talim B., Sel C.G. and Topaloglu H. (2015) Clinical characteristics of megaconial congenital muscular dystrophy due to choline kinase beta gene defects in a series of 15 patients. J. Inherit. Metab. Dis., 38, 1099–1108. [DOI] [PubMed] [Google Scholar]
- 15. Wu G., Sher R.B., Cox G.A. and Vance D.E. (2010) Differential expression of choline kinase isoforms in skeletal muscle explains the phenotypic variability in the rostrocaudal muscular dystrophy mouse. Biochim. Biophys. Acta, 1801, 446–454. [DOI] [PubMed] [Google Scholar]
- 16. Maddatu T.P., Garvey S.M., Schroeder D.G., Zhang W., Kim S.Y., Nicholson A.I., Davis C.J. and Cox G.A. (2005) Dilated cardiomyopathy in the nmd mouse: transgenic rescue and QTLs that improve cardiac function and survival. Hum. Mol. Genet., 14, 3179–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Paumier K.L., Rizzo S.J.S., Berger Z., Chen Y., Gonzales C., Kaftan E., Li L., Lotarski S., Monaghan M., Shen W. et al. (2013) Behavioral characterization of A53T mice reveals early and late stage deficits related to Parkinson’s disease. PLOS ONE, 8, e70274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bailey K.R. and Crawley J.N. (2009) Chapter 5 Anxiety-Related Behaviors in Mice. 2nd edition, 21–26. [PubMed] [Google Scholar]
- 19. Vaart T., Woerden G.M., Elgersma Y., Zeeuw C.I. and Schonewille M. (2011) Motor deficits in neurofibromatosis type 1 mice: the role of the cerebellum. Genes Brain Behav., 10, 404–409. [DOI] [PubMed] [Google Scholar]
- 20. Brooks S.P. and Dunnett S.B. (2009) Tests to assess motor phenotype in mice: a user's guide. Nat. Rev. Neurosci., 10, 519–529. [DOI] [PubMed] [Google Scholar]
- 21. Vinueza Veloz M.F., Buijsen R.A.M., Willemsen R., Cupido A., Bosman L.W.J., Koekkoek S.K.E., Potters J.W., Oostra B.A. and De Zeeuw C.I. (2012) The effect of an mGluR5 inhibitor on procedural memory and avoidance discrimination impairments in Fmr1 KO mice. Genes Brain Behav., 11, 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vinueza Veloz M.F., Zhou K., Bosman L.W.J., Potters J.W., Negrello M., Seepers R.M., Strydis C., Koekkoek S.K.E. and De Zeeuw C.I. (2014) Cerebellar control of gait and interlimb coordination. Brain Struct. Funct., 220, 3513–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu G., Sher R.B., Cox G.A. and Vance D.E. (2009) Understanding the muscular dystrophy caused by deletion of choline kinase beta in mice. Biochim. Biophys. Acta, 1791, 347–356. [DOI] [PubMed] [Google Scholar]
- 24. Secades J.J. and Frontera G. (1995) CDP-Choline: pharmacological and clinical review Methods Find. Exp. Clin. Pharmacol., 17, 1–54. [PubMed] [Google Scholar]
- 25. Agut J., Font E., Sacristan A. and Ortiz J.A. (1983) Bioavailability of methyl-14C CDP-choline by oral route. Arzneimittelforschung, 33, 1045–1047. [PubMed] [Google Scholar]
- 26. Bachmanov A.A., Reed D.R., Beauchamp G.K. and Tordoff M.G. (2002) Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behav. Genet., 32, 435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hollinger K. and Chamberlain J.S. (2015) Viral vector-mediated gene therapies. Curr. Opin. Neurol., 28, 522–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zincarelli C., Soltys S., Rengo G. and Rabinowitz J.E. (2008) Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther., 16, 1073–1080. [DOI] [PubMed] [Google Scholar]
- 29. Ramos J., Chamberlain J.S. and Muscular W. (2015) Gene therapy for Duchenne muscular dystrophy. Expert Opin. Orphan Drugs, 3, 1255–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aoyama C., Liao H. and Ishidate K. (2004) Structure and function of choline kinase isoforms in mammalian cells. Prog. Lipid Res., 43, 266–281. [DOI] [PubMed] [Google Scholar]
- 31. Chang C.C., Few L.L., Konrad M. and See Too W.C. (2016) Phosphorylation of human choline kinase beta by protein kinase a: its impact on activity and inhibition. PLoS One, 11, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramírez De Molina A., Gallego-Ortega D., Sarmentero J., Bañez-Coronel M., Martín-Cantalejo Y. and Lacal J.C. (2005) Choline kinase is a novel oncogene that potentiates RhoA-induced carcinogenesis. Cancer Res., 65, 5647–5653. [DOI] [PubMed] [Google Scholar]
- 33. Falcon S.C., Hudson C.S., Huang Y., Mortimore M., Golec J.M., Charlton P.A., Weber P. and Sundaram H. (2013) A non-catalytic role of choline kinase alpha is important in promoting cancer cell survival. Oncogenesis, 2, e38–e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dunant P., Larochelle N., Thirion C., Stucka R., Ursu D., Petrof B.J., Wolf E. and Lochmu H. (2003) Expression of dystrophin driven by the 1.35-kb MCK promoter ameliorates muscular dystrophy in fast, but not in slow muscles of transgenic mdx mice. Mol. Ther., 8, 80–89. [DOI] [PubMed] [Google Scholar]
- 35. Hauser M.A., Robinson A., Connor D.H.O., Gregory W., Buskin J.N., Apone S., Kirk C.J., Hardy S., Hauschka S.D. and Chamberlain J.S. (2000) Analysis of muscle creatine kinase regulatory elements in recombinant adenoviral vectors. Mol Ther., 2, 16–25. [DOI] [PubMed] [Google Scholar]
- 36. Chicoine L.G., Rodino-Klapac L.R., Shao G., Xu R., Bremer W.G., Camboni M., Golden B., Montgomery C.L., Shontz K., Heller K.N. et al. (2014) Vascular delivery of rAAVrh74.MCK.GALGT2 to the gastrocnemius muscle of the rhesus macaque stimulates the expression of dystrophin and laminin α2 surrogates. Mol. Ther., 22, 713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sukoff Rizzo S.J., Anderson L.C., Green T.L., McGarr T., Wells G. and Winter S.S. (2018) Assessing healthspan and lifespan measures in aging mice: optimization of testing protocols, replicability, and rater reliability. Curr. Protoc. Mouse Biol., 8, e45. [DOI] [PubMed] [Google Scholar]
- 38. Van Der Giessen R.S., Koekkoek S.K., van Dorp S., De Gruijl J.R., Cupido A., Khosrovani S., Dortland B., Wellershaus K., Degen J., Deuchars J. et al. (2008) Role of olivary electrical coupling in cerebellar motor learning. Neuron, 58, 599–612. [DOI] [PubMed] [Google Scholar]
- 39. Wu G., Zhang L., Li T., Lopaschuk G., Vance D.E. and Jacobs R.L. (2012) Choline deficiency attenuates body weight gain and improves glucose tolerance in Ob/Ob mice. J. Obes., 2012, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wurtman R.J., Regan M., Ulus I. and Yu L. (2000) Effect of oral CDP-choline on plasma choline and uridine levels in humans. Biochem. Pharmacol., 60, 989–992. [DOI] [PubMed] [Google Scholar]
- 41. Wang Y., Wang F., Wang R., Zhao P. and Xia Q. (2015) 2A self-cleaving peptide-based multi-gene expression system in the silkworm Bombyx mori. Sci. Rep., 5, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lu Y.Y., Wang L.J., Muramatsu S.I., Ikeguchi K., Fujimoto K.I., Okada T., Mizukami H., Matsushita T., Hanazono Y., Kume A. et al. (2003) Intramuscular injection of AAV-GDNF results in sustained expression of transgenic GDNF, and its delivery to spinal motoneurons by retrograde transport. Neurosci. Res., 45, 33–40. [DOI] [PubMed] [Google Scholar]
- 43. Liu M., Yue Y., Harper S.Q., Grange R.W. and Jeffrey S. (2008) Adeno-associated virus-mediated microdystrophin expression protects young mdx muscle from contraction-induced injury. Mol. Ther., 11, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.